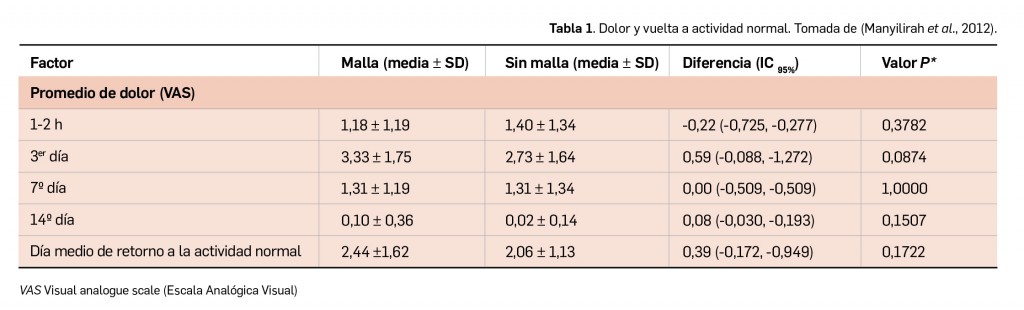
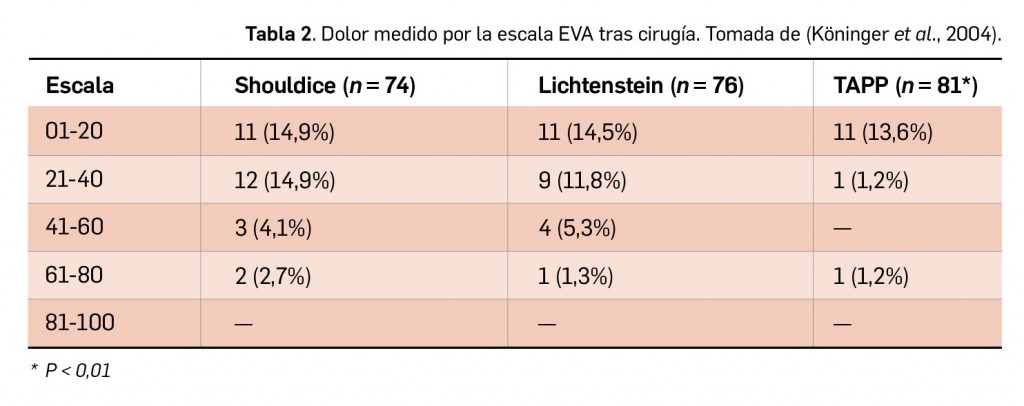
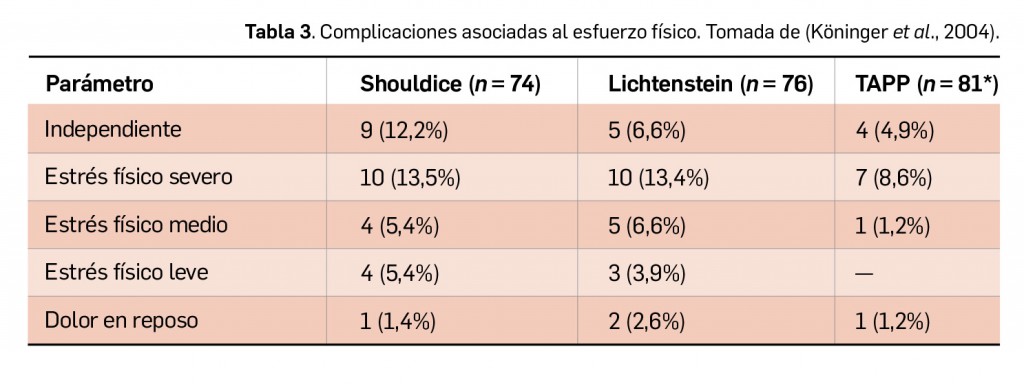
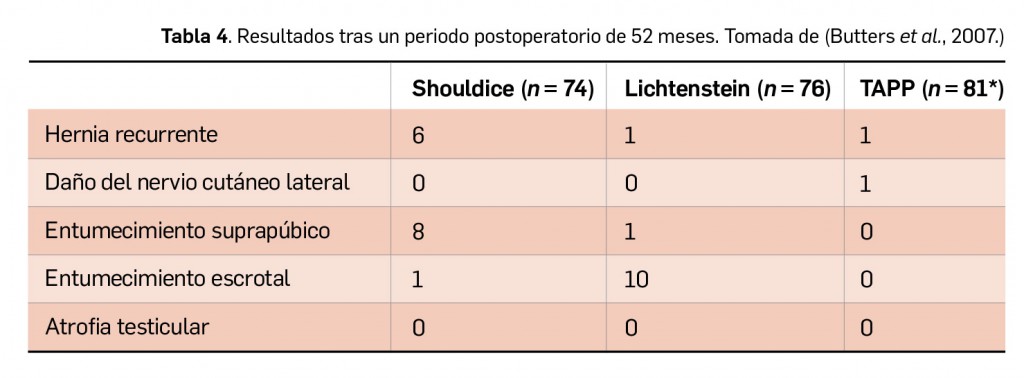
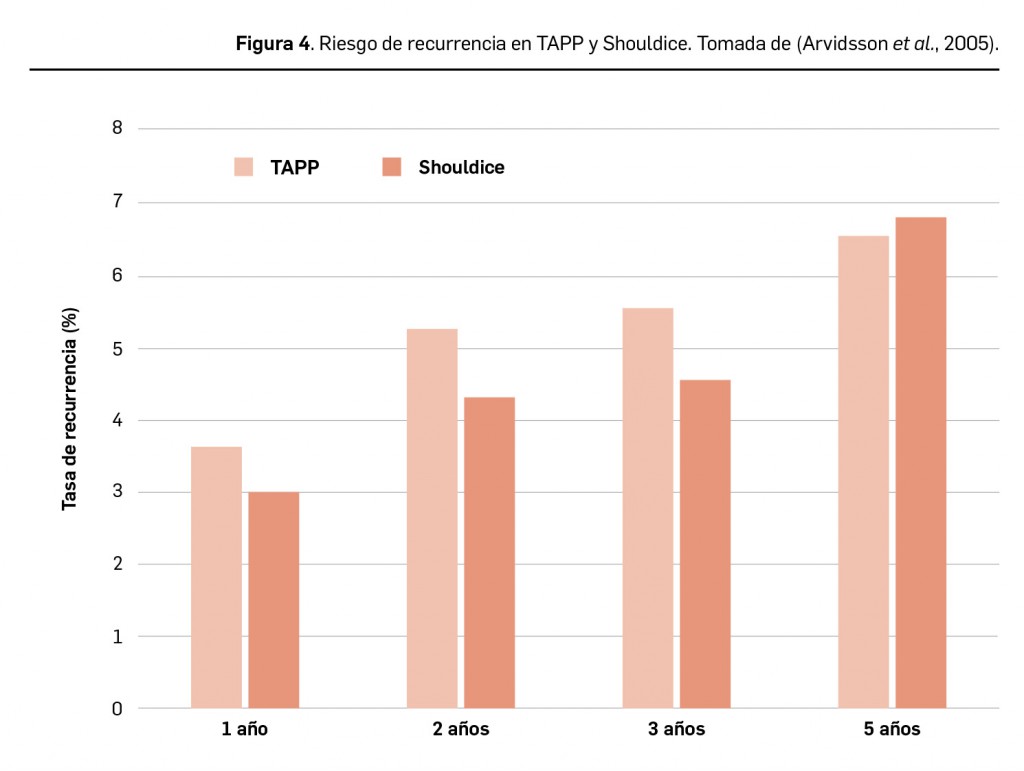
Resumen
La nueva versión de BOT PLUS incrementa la información relativa a la identificación de los medicamentos, con la inclusión de las imágenes del envase y de la forma farmacéutica (información procedente de la AEMPS), así como con la creación de una nueva pestaña específica donde se recoge la descripción física de dicha forma. Por último, se potencian las opciones del buscador de medicamentos por las características físicas de su forma farmacéutica, añadiendo la posibilidad de buscar ahora también formas líquidas, como soluciones o suspensiones orales, o jarabes, y se incorporan nuevas características como la búsqueda por el sabor de la forma farmacéutica.
Como continuación al artículo anterior de esta sección, en el que se enumeraban las diferentes novedades incluidas en la nueva versión de BOT PLUS, activada el pasado 3 de junio de 2020, en esta ocasión nos vamos a centrar en la primera de las novedades: la inclusión de las imágenes de los medicamentos y productos de parafarmacia, y la evolución del buscador Forma/Color/Sabor.
En multitud de ocasiones los usuarios consultan al farmacéutico en relación al aspecto de los medicamentos. Con la aprobación de la Directiva 2011/62/UE en la que se establece la obligatoriedad de incluir tanto el identificador único como los dispositivos de seguridad anti-manipulación, resulta imposible para al farmacéutico conocer de primera mano esta información.
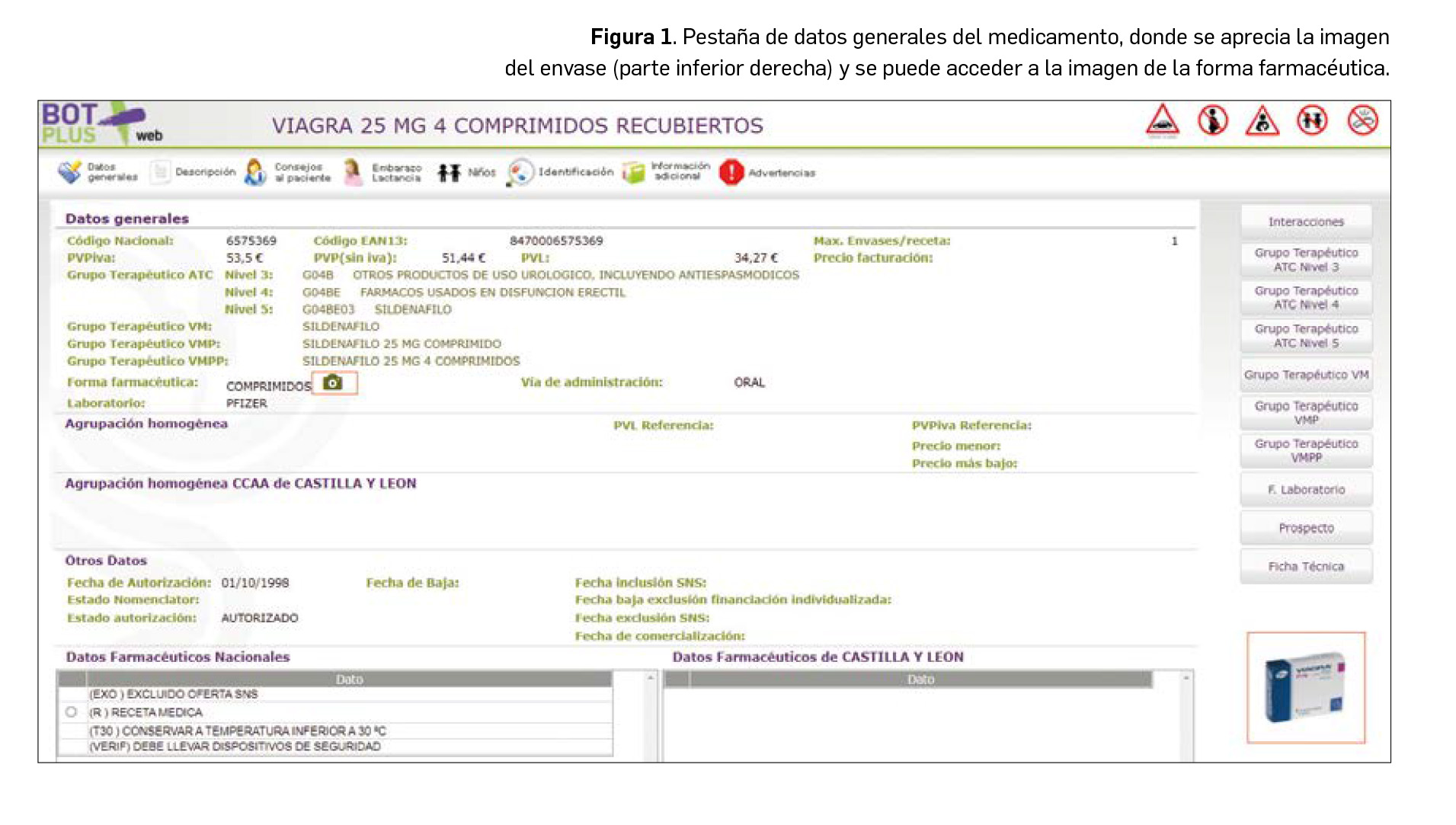
Para paliar esta limitación, BOT PLUS incluye a partir de ahora las imágenes de los medicamentos y de los productos de parafarmacia, tanto del cartonaje externo como de la propia forma farmacéutica. Estas imágenes están accesibles desde la pantalla inicial del medicamento o del producto de parafarmacia, en su caso (Figura 1). Para ampliar la imagen, simplemente hay que pinchar en la foto del envase, o en el caso de la forma farmacéutica, en el icono de la cámara de fotos. Esta información ha sido cedida por la Agencia Española de Medicamentos y Productos Sanitarios, a quien queremos transmitir nuestro mayor agradecimiento por la colaboración prestada.
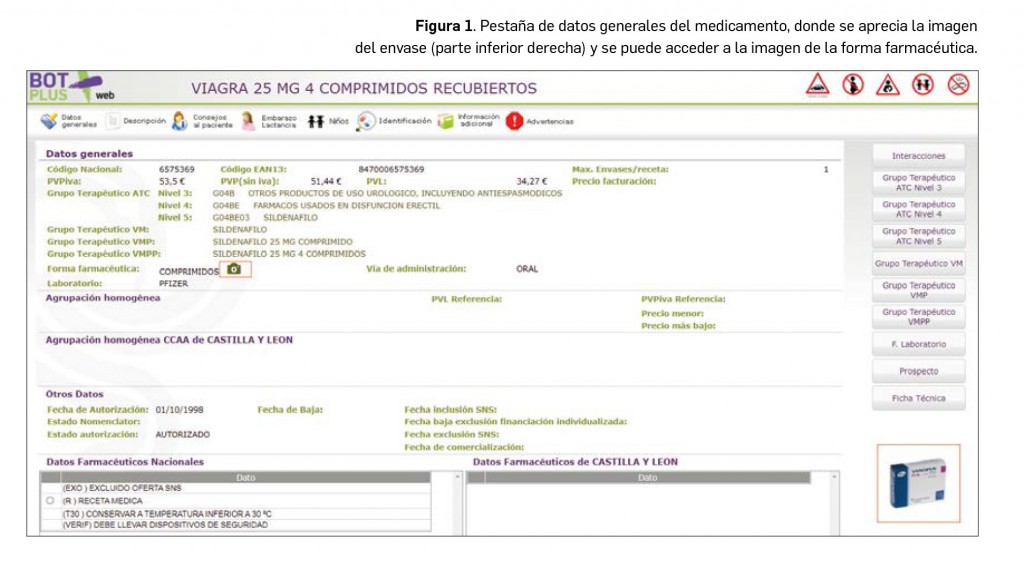
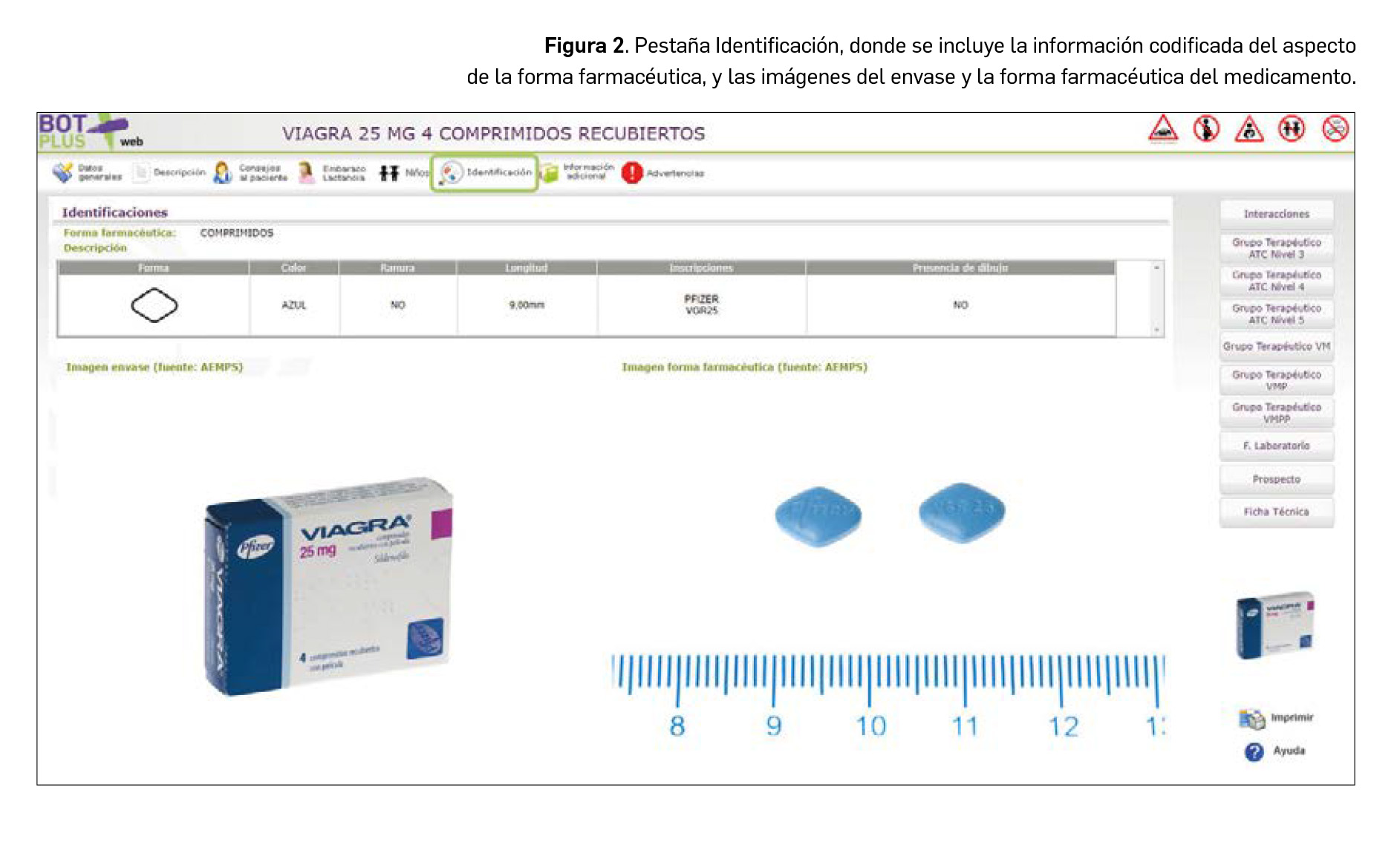
De igual manera, se ha procedido a crear una nueva pestaña denominada Identificación en la ficha de medicamentos y productos de parafarmacia, donde se tiene acceso a estas imágenes. En el caso de medicamentos, además, se incluye en esta pestaña la descripción física de la forma farmacéutica (Figura 2). De tal manera, en el caso de comprimidos, se recogen informaciones tales como: la forma aproximada, su color, sabor, dimensiones, la presencia de ranura o inscripciones o dibujos en la superficie. Esta información está disponible igualmente para cápsulas y formas líquidas orales, adaptando la misma a las particularidades de cada forma.
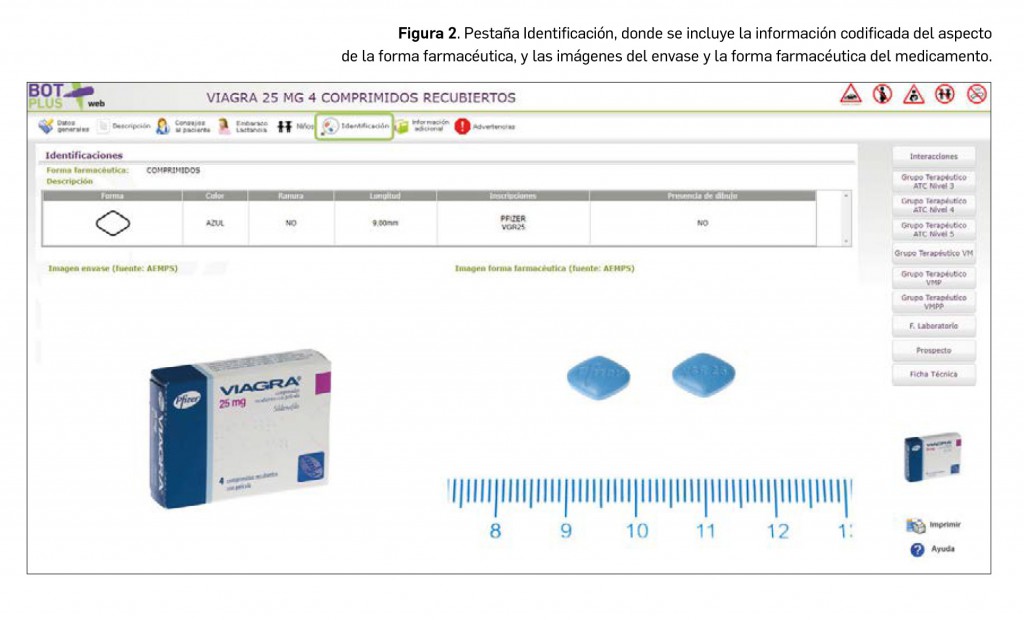
La información relativa al aspecto físico de la forma farmacéutica, que se encuentra en la tabla de esta pestaña de Identificación, se ha codificado en base a la información disponible en las fichas técnicas de estos medicamentos. Para ello, se ha revisado la información de más de 15.000 medicamentos de uso humano.
La pestaña de Identificación incluye igualmente la imagen del cartonaje y/o del producto de parafarmacia (Figura 3), lo que constituye una ayuda importante al farmacéutico para conocer al producto.

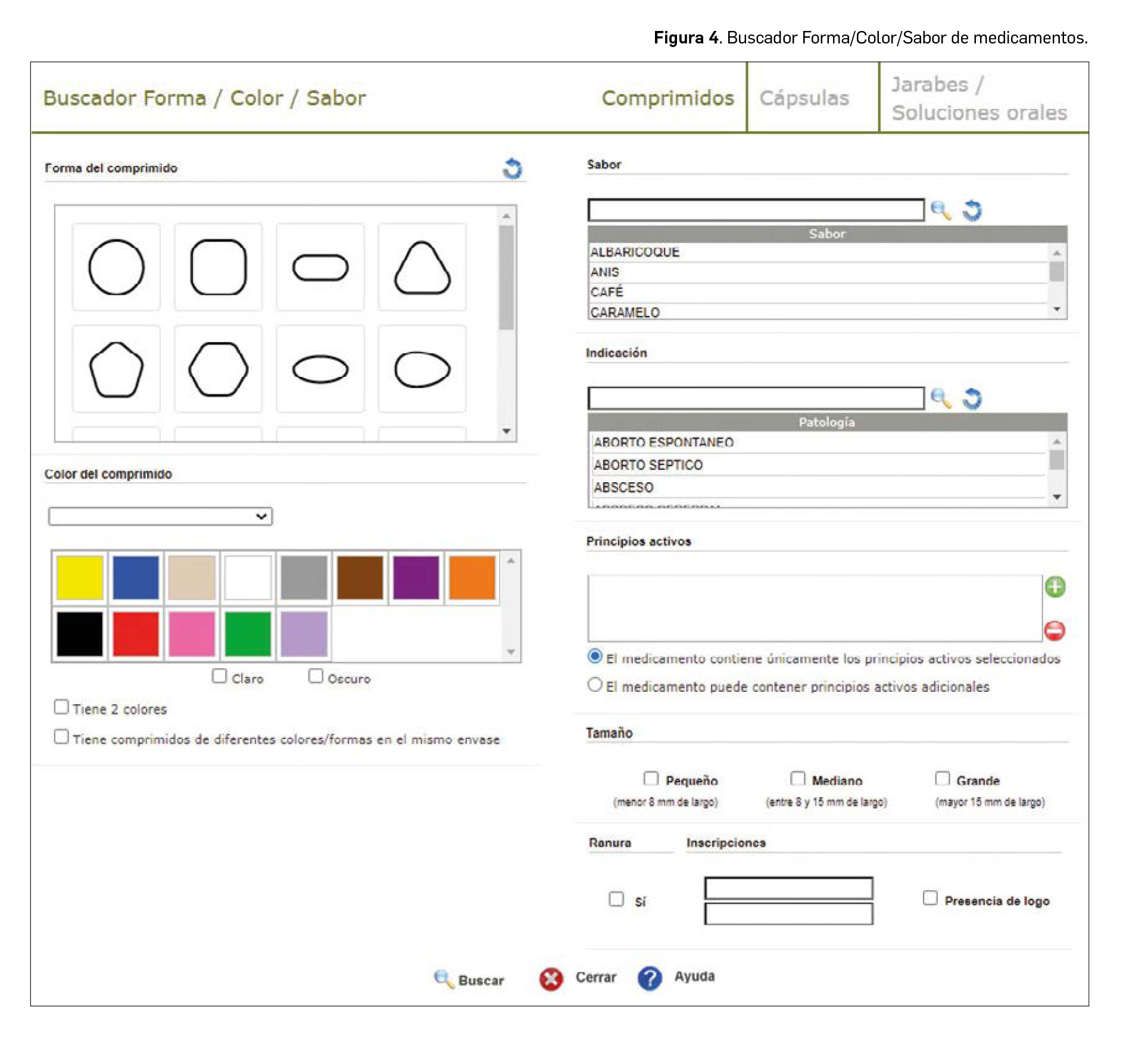
De forma interesante, la codificación de la información del aspecto de la forma farmacéutica del medicamento permite buscar medicamentos en base a estos datos. Para ello, desde 2019 BOT PLUS disponía de un buscador específico denominado Forma/Color para encontrar medicamentos incluyendo estos criterios. En esta nueva versión de BOT PLUS se ha producido una evolución de este buscador Forma/Color/Sabor (Figura 4) para incluir las sugerencias planteadas por los más de 1.100 farmacéuticos que asistieron durante 2019 a los talleres de BOT PLUS realizados en los diferentes Colegios Oficiales de Farmacéuticos.
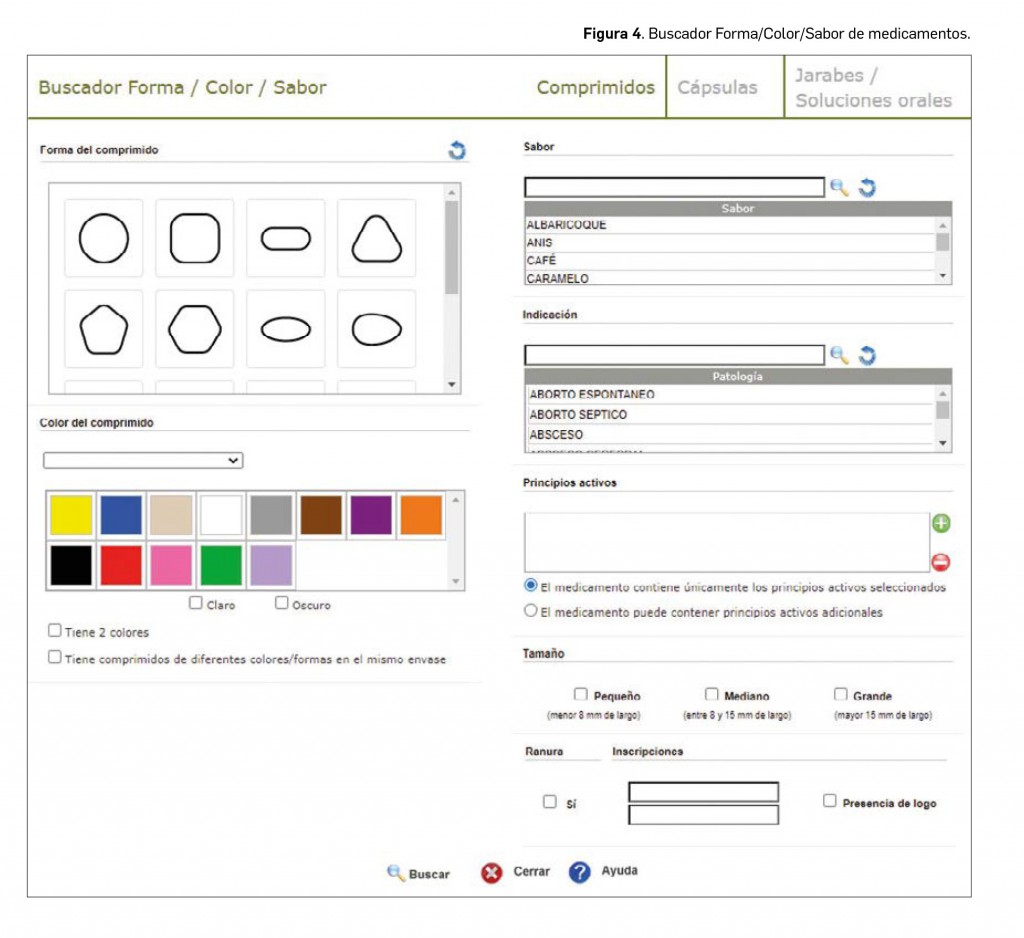
Entre las modificaciones incluidas en este buscador, destacamos las siguientes:
- Posibilidad de especificar el principio activo del medicamento en la búsqueda.
- Inclusión de sabores para ciertos comprimidos (efervescentes, masticables, bucodispersables).
- Posibilidad de buscar medicamentos en forma oral líquida (jarabes, soluciones o suspensiones orales) en base al color o sabor del medicamento.
Para proceder a buscar un medicamento concreto, en primer lugar hay que seleccionar la forma farmacéutica.
A continuación, se incluye la información que nos transmita el paciente. Existe una gran cantidad de información que puede incluirse en la búsqueda, teniendo en cuenta que ninguno de estos campos es obligatorio, pero que cuanta más información aportemos al buscador, más va a restringirse la búsqueda. No obstante, sobre la forma de uso de este buscador, y debido a la peculiaridad de esta búsqueda (a fin de cuentas vamos a incluir información que nos transmite el paciente y que tiene cierto grado de subjetividad, como el color, el sabor o el tamaño), se aconseja ser lo más general posible. Para ello, a continuación se indican algunas pautas a ser tenidas en cuenta.
A la hora de indicar el color de la forma farmacéutica, se han establecido una serie de colores básicos, que por supuesto no tienen por qué ser exactamente iguales a los del medicamento en cuestión. De tal manera, si el paciente nos indica que el medicamento es de un color amarillo, marcaremos la opción “AMARILLO”, aunque el comprimido no sea exactamente del color de la paleta.
En ocasiones nos encontramos con formas farmacéuticas de varios colores, como cápsulas con cuerpo y tapa de diferente color, o comprimidos con dos capas. Para indicar el segundo color se debe marcar en la opción “Tiene 2 colores”, abriéndose una segunda paleta donde especificaremos dicho color. En el buscador existe también una opción “Tiene comprimidos de diferentes colores/formas en el mismo envase”. Esta opción es útil para encontrar medicamentos (como los anticonceptivos orales con comprimidos placebo) en los que existen comprimidos de varios colores. En este caso concreto, se incluiría el color o la forma de uno de ellos, y se marcaría esta opción para indicar que existen más colores y/o formas, aunque no se especifiquen cuáles.
Otro aspecto a tener en cuenta es el relativo al tamaño, ya que difícilmente el paciente nos va a transmitir las dimensiones exactas, y la diferencia entre un tamaño y otro puede estar en milímetros (es decir, “pequeño” es menor de 8 mm y “mediano” es entre 8 y 15 mm). En caso de que el paciente no tenga claro el tamaño, para comprimidos que estén en el umbral, pueden elegirse dos opciones, como “pequeño” y “mediano”.
En el campo de Inscripciones se incluirán las letras o números que recuerde el paciente. Cada uno de los huecos está reservado para cada cara del comprimido, o en el caso de las cápsulas, para el cuerpo y la tapa. La opción “Presencia de logo” debe marcarse cuando aparezca un dibujo grabado que no sea una letra o número.
Esta información relativa al aspecto físico de los medicamentos, y en especial la explotación de la misma, supone una importante ayuda para el farmacéutico, con el objetivo de encontrar medicamentos concretos de los que el paciente no recuerde su nombre, pero sí el aspecto que tiene su forma farmacéutica. El acceso a la misma a través de la pestaña de Identificación, así como la inclusión de las imágenes de las formas farmacéuticas permite igualmente al farmacéutico conocer el aspecto del medicamento, y confirmar que el medicamento es el que está utilizando su paciente.
Por último, esta funcionalidad para buscar medicamentos con una forma y color determinado, unido a la posibilidad de incluir la composición exacta del medicamento, puede resultar útil al farmacéutico en la sustitución de medicamentos, especialmente en pacientes mayores, en los que interese buscar medicamentos que tengan el mismo aspecto externo (por ejemplo, dos medicamentos con misma composición y mismo color y forma), reduciendo los riesgos de error en la toma de la medicación de estos pacientes concretos. No obstante, siempre se aconseja consultar los listados oficiales respecto a la sustitución.





{kind=link}
{kind=link}
{kind=link}
{kind=link}