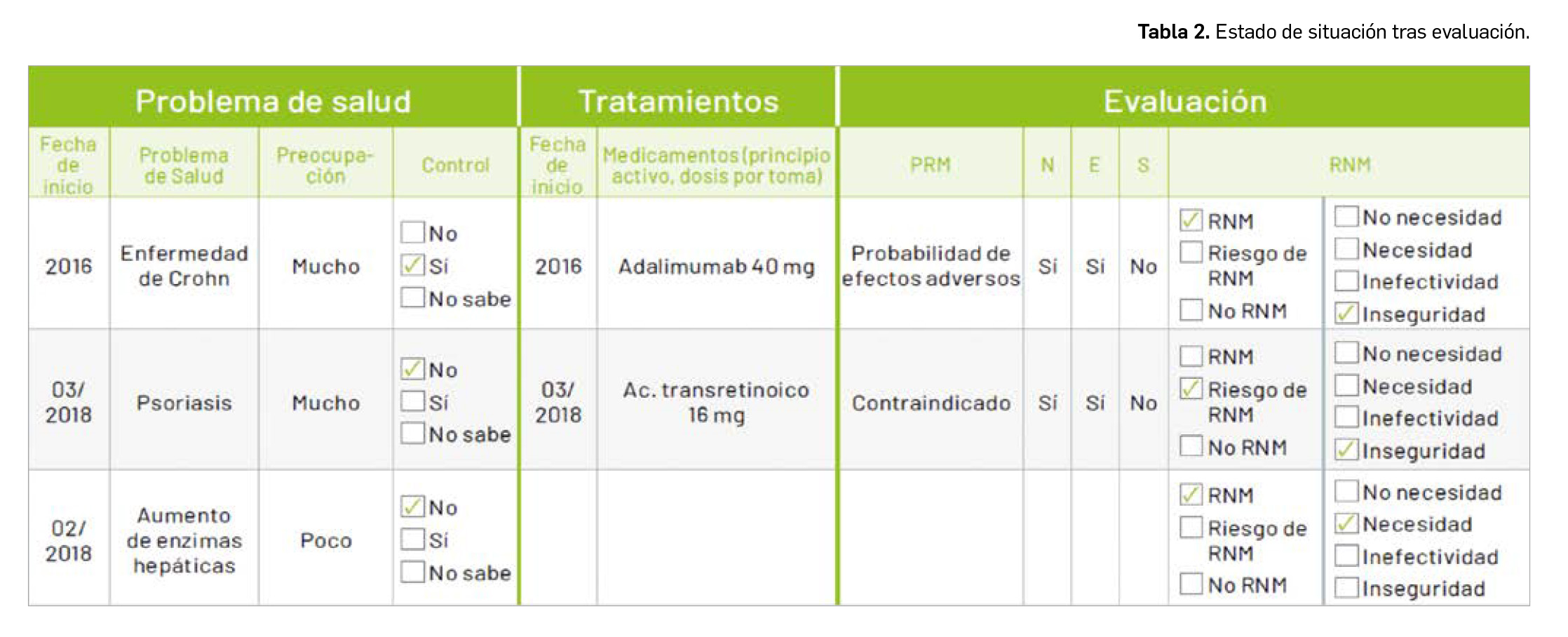
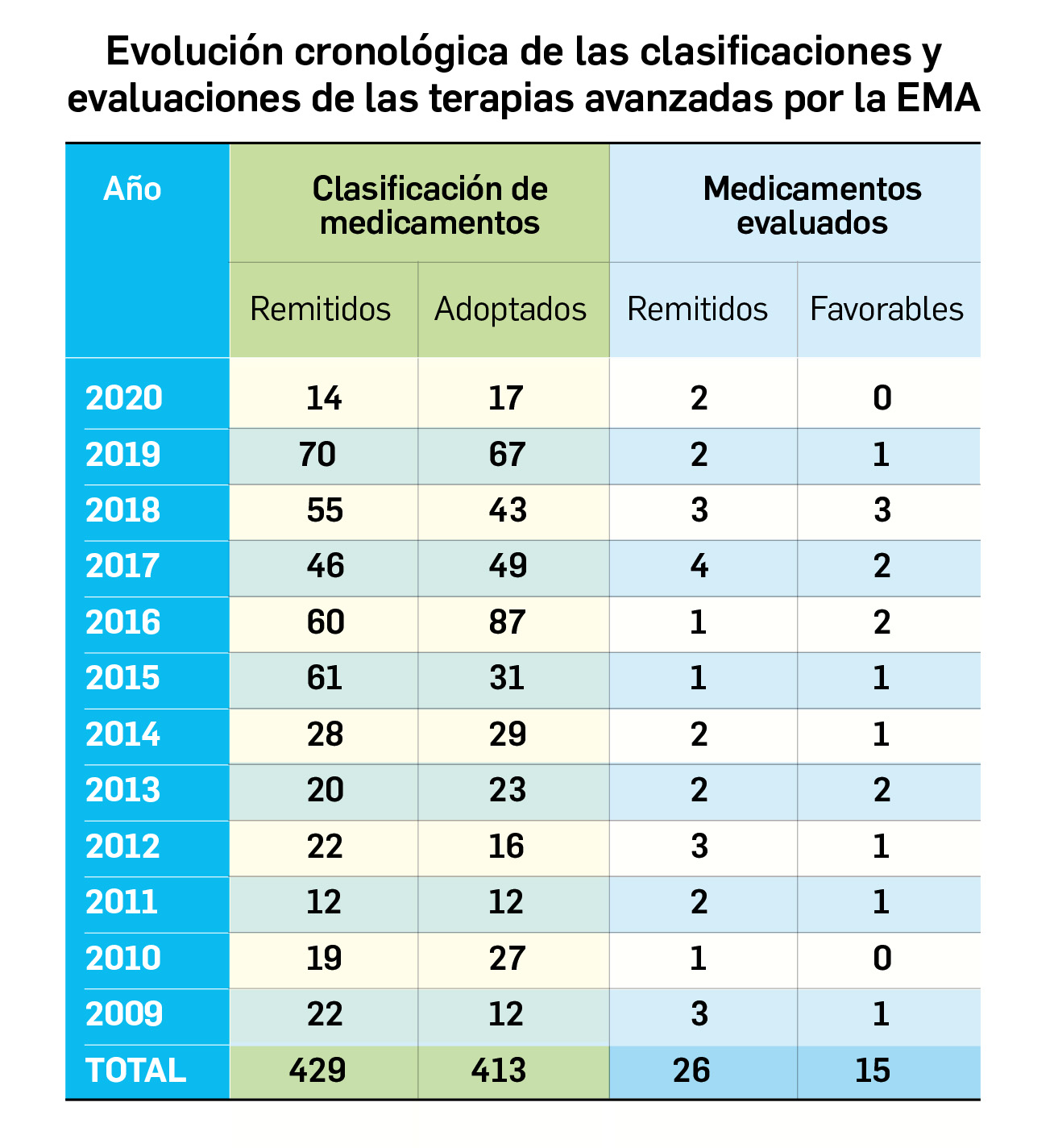
Evidencias que respaldan el uso de mascarillas en la profilaxis
En un contexto de pandemia en que, por ahora, no disponemos de ninguna herramienta farmacológica terapéutica o preventiva con seguridad y eficacia probadas (en Europa no se ha autorizado ningún medicamento para tratar o prevenir la infección por SARS-CoV-2), cobran especial relevancia las medidas no farmacológicas preventivas; entre otras, el uso de mascarillas. La creciente concienciación social sobre el uso de estos elementos como profilácticos útiles frente al contagio por el nuevo coronavirus se ha visto condicionada por la aparición de noticias controvertidas sobre su eficacia protectora y cambios de criterio de las autoridades sanitarias en cuanto a la recomendación de su empleo por la población general; se acepta en mayor medida la conveniencia del uso de ciertos tipos de mascarillas en el entorno hospitalario o ante casos sospechosos de infección. Dejando de lado los problemas de abastecimiento (que eventualmente hagan necesario optimizar o restringir el suministro de mascarillas a los profesionales sanitarios u otros profesionales de riesgo), en la bibliografía científica son numerosos los autores que abogan por un uso generalizado de mascarillas en un escenario de elevada transmisión comunitaria del virus, en parte por los motivos que se exponen a continuación.
La evidencia disponible apunta a una mayoritaria transmisión del coronavirus a través de las gotículas respiratorias (que pueden recorrer hasta 7-8 m de distancia, según estudios de laboratorio1) expulsadas al aire por la tos o estornudos de pacientes infectados, de los cuales una gran proporción (50-75% de pacientes) pueden ser asintomáticos o tener clínica leve, lo que facilitará su movilidad social y el riesgo de transmisión. También se puede transmitir el virus durante el periodo de incubación previo a la aparición de los síntomas e incluso en partículas de aerosol (de < 5 µm, menor tamaño que las gotículas respiratorias) que se pueden mantener en el aire más tiempo; todavía hay dudas del tiempo de persistencia del virus en el aire en espacios cerrados o abiertos. Parece que el uso de mascarillas podría reducir la transmisión de gotículas respiratorias desde personas asintomáticas, pre-sintomáticas o leves, controlando el foco de contagio.
No obstante, aún no se ha realizado ningún estudio específico para examinar la efectividad de las máscaras contra el SARS-CoV-2. Un trabajo reciente en pacientes infectados con coronavirus estacionales ha demostrado que las mascarillas quirúrgicas reducen significativamente la detección de ARN viral en aerosoles y muestran una tendencia a reducir el ARN viral en las gotículas respiratorias. Un meta-análisis de ensayos controlados aleatorios también mostró que las mascarillas quirúrgicas son tan eficaces como las N95 (el equivalente estadounidense de las mascarillas filtrantes FFP2 europeas) para reducir la transmisión de enfermedades víricas, como la gripe. Si tomamos como referencia un estudio de casos y controles realizado durante el brote de SARS-CoV-1 en Hong Kong, los datos sugieren que el uso frecuente de mascarillas (principalmente quirúrgicas) en lugares públicos puede proteger hasta en un 64%.
En relación al uso de las recientemente popularizadas mascarillas higiénicas o incluso “caseras”, que en algunas situaciones pueden ayudar a paliar problemas de suministro, conviene subrayar que la evidencia es más limitada. Estudios en otras infecciones respiratorias, como la gripe aviar y la gripe estacional, indican que las mascarillas caseras confieren cierto grado de eficacia, que oscila entre el 40 y el 95%, siempre inferior a la de mascarillas médicas (quirúrgicas o filtrantes), pero ciertamente mejor que nada. Se han evaluado varios materiales para este tipo de mascarillas con diferentes resultados. Uno de los estudios apuntaba a que mascarillas caseras fabricadas de una funda de almohada o una camiseta 100% de algodón tenían un tercio de la eficacia de las mascarillas médicas, pero significativamente superior en comparación con la ausencia de protección en la reducción de la cantidad de microorganismos expulsados en las gotículas respiratorias. Otro trabajo más reciente comparó unas mascarillas caseras compuestas de cuatro capas de papel de cocina y una capa de tela con mascarillas N952 y quirúrgicas: fueron capaces de reducir en un 95,2%, 99,98% y 97,14%, respectivamente, el paso de virus de la gripe aviar en aerosol. Una investigación realizada en Vietnam en 2015 reportó una eficacia significativamente superior de las mascarillas médicas sobre las higiénicas para prevenir la transmisión de virus similares a los de la gripe, si bien quedan dudas sobre el papel del material –algodón de dos capas– de que estaban fabricadas (por ejemplo, no está claro si el material tenía resistencia a las gotículas).
Mientras no se disponga de estudios directamente comparativos de la eficacia de mascarillas médicas frente a las mascarillas higiénicas en la transmisión de SARS-CoV-2, no se debe tampoco asumir su ineficacia; en vista de las actuales recomendaciones de uso de mascarillas en el entorno comunitario (entre otros organismos, por la OMS), es previsible que aparezcan pronto. Será especialmente importante examinar los datos procedentes de países como China, Hong Kong o Singapur, donde la práctica totalidad de la población utiliza mascarillas en público (y donde la epidemia se contuvo en mayor medida que en Europa o en EE.UU.). Hasta entonces, la modesta evidencia que respalda el uso comunitario extendido de mascarillas no médicas indica que sí podrían ser útiles para reducir la transmisión del coronavirus, siempre con la advertencia de que es fundamental su uso correcto (posición, técnica de colocación, etc.) y de que la mascarilla no debe aportar una sensación de falsa seguridad que induzca a una relajación del cumplimiento de otras medidas preventivas eficaces, a las que debe complementar.
- Sunjaya AP, Jenkins C. Rationale for universal face masks in public against COVID-19. Respirology. 2020; DOI: 10.1111/resp.13834.
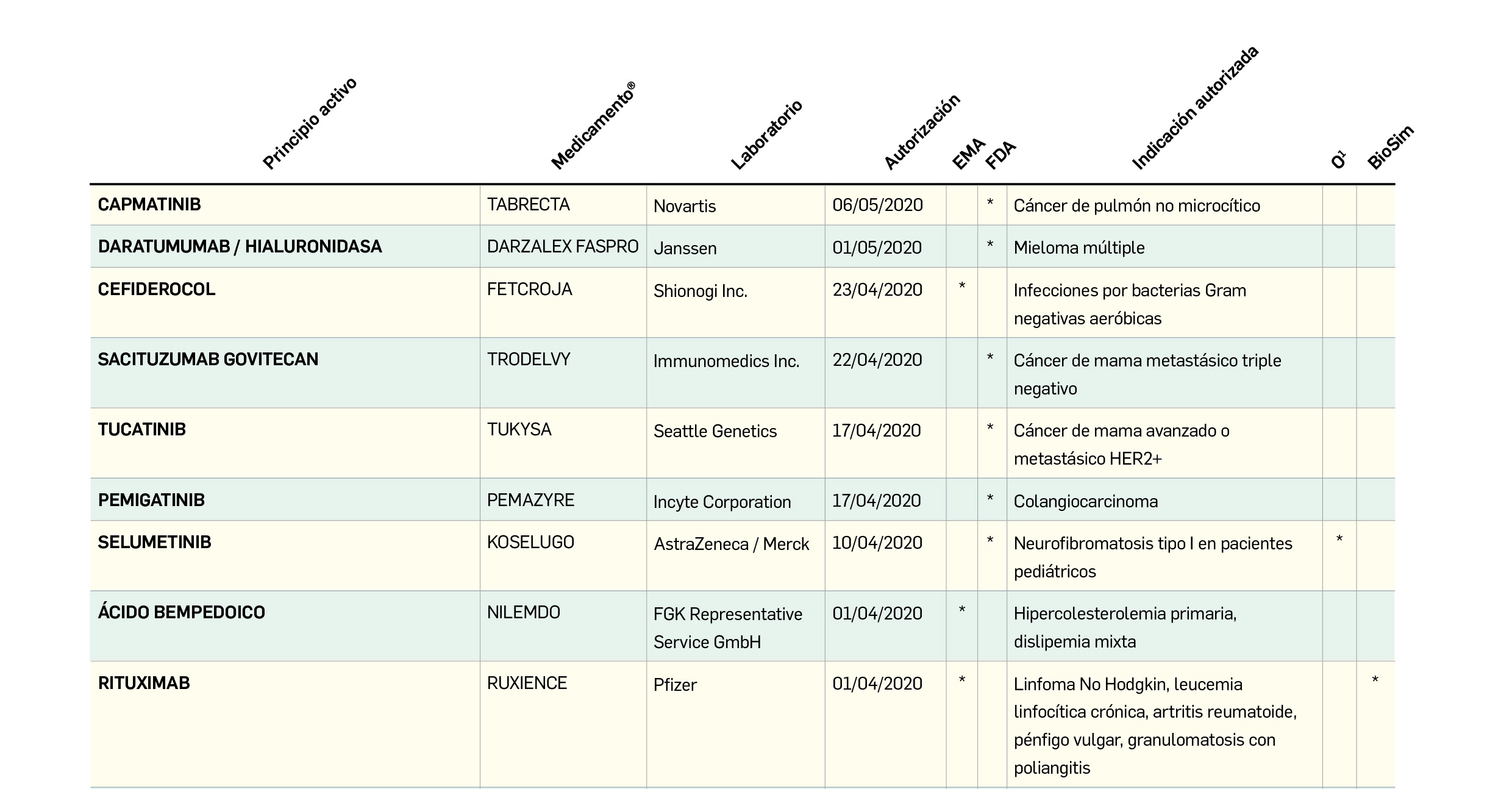
Cloroquina/Hidroxicloroquina: avances en su investigación clínica
Si ya en el número anterior de PAM hacíamos referencia a los estudios preliminares aparecidos sobre estos fármacos antimaláricos y antiinflamatorios, incluidos los primeros datos clínicos para hidroxicloroquina (el llamado estudio de Marsella del profesor Raoult sugería resultados significativamente positivos para la combinación de hidroxicloroquina con azitromicina), que no permiten sacar conclusiones claras, se deben comentar ahora nuevos resultados divulgados al respecto; éstos representan solo una pequeña parte del alto número de estudios en marcha que evalúa las posibilidades de la cloroquina y la hidroxicloroquina frente a la infección por SARS-CoV-2.
Los resultados del estudio de Marsella contrastan con los reportados, para el mismo régimen terapéutico, por Molina y colaboradores (Molina et al., 2020), que ponen en duda la efectividad antiviral de esta combinación, al menos en los pacientes más graves. Estos autores realizaron un pequeño estudio prospectivo de un solo brazo en un hospital de París, donde evaluaron los resultados virológicos y clínicos de 11 pacientes hospitalizados (7 hombres y 4 mujeres con una edad media de 59 años) que recibieron hidroxicloroquina (660 mg/día durante 10 días) y azitromicina (500 mg el día 1 y 250 mg en los días 2 a 5); 8 de los pacientes tenían comorbilidades significativas y, al inicio del estudio, 10 pacientes –con presencia de fiebre– recibieron oxigenoterapia nasal. Tras 5 días, se produjo el fallecimiento de un paciente, otros dos ingresaron en UCI y otro paciente interrumpió la pauta de tratamiento por una prolongación del intervalo QT del electrocardiograma. La PCR aún era positiva para el ARN de SARS-CoV-2 en 8 de 10 pacientes (80%) tras 5-6 días desde el inicio del tratamiento.
En esa línea, otro estudio prospectivo realizado en China (Chen et al., 2020) en 30 pacientes con COVID-19 y un pronóstico relativamente favorable tampoco encontró diferencias significativas en la tasa de aclaramiento virológico –negatividad en una PCR de muestra nasofaríngea– a los 7 días desde la aleatorización entre los pacientes que habían recibido hidroxicloroquina (400 mg/día durante 5 días) frente a los que no (grupo control con solo el tratamiento de soporte estándar): la tasa de negatividad era del 87% entre pacientes tratados con hidroxicloroquina vs. 93% en el grupo control (p > 0,05); la mediana de días desde la hospitalización hasta la negativización del ARN viral en la muestra fue de 4 días en el grupo experimental vs. 2 días en el grupo control, sin diferencias estadísticamente significativas por el pequeño tamaño de la muestra y la variabilidad de los resultados. En consistencia con lo anterior, tampoco se detectaron variaciones destacables en resultados clínicos tales como la duración de la hospitalización, la normalización de la fiebre o la progresión radiológica. Un paciente del grupo de hidroxicloroquina desarrolló enfermedad severa durante el tratamiento y, en términos de seguridad, los eventos adversos tuvieron similar incidencia en ambos grupos (4 casos de diarrea y alteración de la función hepática en el grupo de hidroxicloroquina vs. 3 casos en el grupo control).
Un último trabajo (Tang et al,. 2020) del que se han divulgado resultados preliminares –no ha sido publicado aún en revistas científicas tras revisión por pares– es un ensayo clínico aleatorizado y abierto que enroló a 150 pacientes con confirmación microbiológica de COVID-19 (148 con enfermedad leve-moderada y 2 graves), 75 de los cuales recibieron hidroxicloroquina (dosis de carga de 1.200 mg/día durante 3 días y dosis de mantenimiento de 800 mg/día hasta un total de 2 y 3 semanas en pacientes leves-moderados y graves, respectivamente) más el tratamiento de soporte estándar y los otros 75 pacientes solo el tratamiento de soporte estándar. La media de tiempo desde el inicio de los síntomas hasta la aleatorización fue de 16,6 días. Los datos revelan que no hubo diferencias significativas en la conversión a negatividad de la presencia viral en PCR en 28 días (85,4% de pacientes en el grupo de hidroxicloroquina vs. 81,3% en el grupo control). No obstante, sí que se detectó un aumento en la tasa de eventos adversos con hidroxicloroquina (30% vs. 8,8% en el grupo control), siendo la reacción adversa más común la diarrea. No parece, pues, que el beneficio clínico aportado por la hidroxicloroquina sea relevante.
Por otra parte, con el objetivo de comparar los efectos antivirales de dos regímenes posológicos de cloroquina difosfato (dosis alta de 600 mg/12 h durante 10 días vs. dosis baja de 450 mg/12 h el día 1 y 450 mg/día durante 4 días), se ha realizado en Brasil un ensayo clínico aleatorizado (1:1) de fase 2b, de grupos paralelos y doblemente enmascarado, sobre 81 pacientes hospitalizados con infección por SARS-CoV-2, la mayoría hombres (75%) y con una media de edad de 51 años. Los resultados publicados (Borba et al., 2020) apuntan a la ausencia de cambios significativos en la detección del ARN viral por PCR a los días 0 y 4 (positividad en el 77,5% de pacientes en el grupo de dosis alta vs. 75,6% en el grupo de dosis baja), mientras que para la mayor dosis se observó una tasa de letalidad superior hasta el día 13 (39% de fallecimientos vs. 15% en el grupo de dosis baja) y una mayor incidencia de prolongación del intervalo QT hasta > 500 ms (19% vs. 11%). Por ello, los autores concluyen que el régimen de dosis alta de cloroquina no debe ser recomendado para pacientes críticos con COVID-19 por sus riesgos de seguridad, en especial cuando se asocia con el uso de azitromicina y oseltamivir. Sin embargo, estos datos no pueden ser extrapolados a pacientes con COVID-19 leve-moderada.
En definitiva, a pesar de las expectativas generadas en torno a la cloroquina/hidroxicloroquina para el tratamiento de la COVID-19, los primeros resultados disponibles no sugieren que vayan a aportar un beneficio clínico demasiado relevante, si acaso se confirma que aportan algún beneficio. Son necesarios ensayos clínicos controlados, amplios y de calidad para poder concluir al respecto.
Lo que sí queda claro, según advierte la Nota de Seguridad (MUH FV 7/2020) publicada por la AEMPS, es que se debe extremar la precaución con el uso de cloroquina/hidroxicloroquina por sus riesgos de toxicidad, y monitorizar especialmente a pacientes con afecciones médicas crónicas (por ejemplo, insuficiencia renal o hepática) o que reciben medicamentos que pueden aumentar el riesgo de arritmias. Aunque son generalmente bien tolerados, pueden prolongar el intervalo QT del electrocardiograma –principalmente, en pacientes con patologías cardiacas previas, síndrome congénito de intervalo QT largo o desequilibrio hidroelectrolítico no corregido– y dar lugar a arritmias cardiacas potencialmente mortales, sobre todo en casos de sobredosificación. Si el tratamiento con hidroxicloroquina se prolonga más de 5 días, se debería realizar un ECG de control, de especial importancia si concurre alguna de las circunstancias especificadas anteriormente. Así, atendiendo a la falta de evidencia de eficacia y los riesgos potenciales, actualmente no se recomienda la utilización sistemática de azitromicina en combinación con hidroxicloroquina en el tratamiento de la infección por SARS-CoV-2.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). RCloroquina/Hidroxicloroquina: precauciones y vigilancia de posibles reacciones adversas en pacientes con COVID-19. Nota de Seguridad MUF (FV) 7/2020. Disponible en: https://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2020/docs/NI_MUH_FV-7-2020-Hidroxicloroquina.pdf?x92515
- Borba MGS, Val FFA, Sampaio VS, Alexandre MAA, Melo GC, Brito M et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection: A Randomized Clinical Trial. JAMA Netw Open. 2020; 3(4): e208857. DOI: 10.1001/jamanetworkopen.2020.8857.
- Chen J, Liu D, Liu L, Liu P, Xu Q, Xia L, Ling Y et al. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 COVID-19) J Zhejiang Univ Sci. 2020; 49(1). DOI: 10.3785/j.issn.1008-9292.2020.03.03
- Molina JM, Delaugerre C, Le Goff J, Mela-Lima B, Ponscarme D, Goldwirt L et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med Mal Infect. 2020. pii: S0399-077X(20)30085-8. DOI: 10.1016/j.medmal.2020.03.006.
- Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W et al. Hydroxychloroquine in patients mainly with mild to moderate COVID-19: an open-label, randomized, controlled trial. 2020. Disponible en: https://www.medrxiv.org/content/10.1101/2020.04.10.20060558v2; DOI: https://doi.org/10.1101/2020.04.10.20060558.
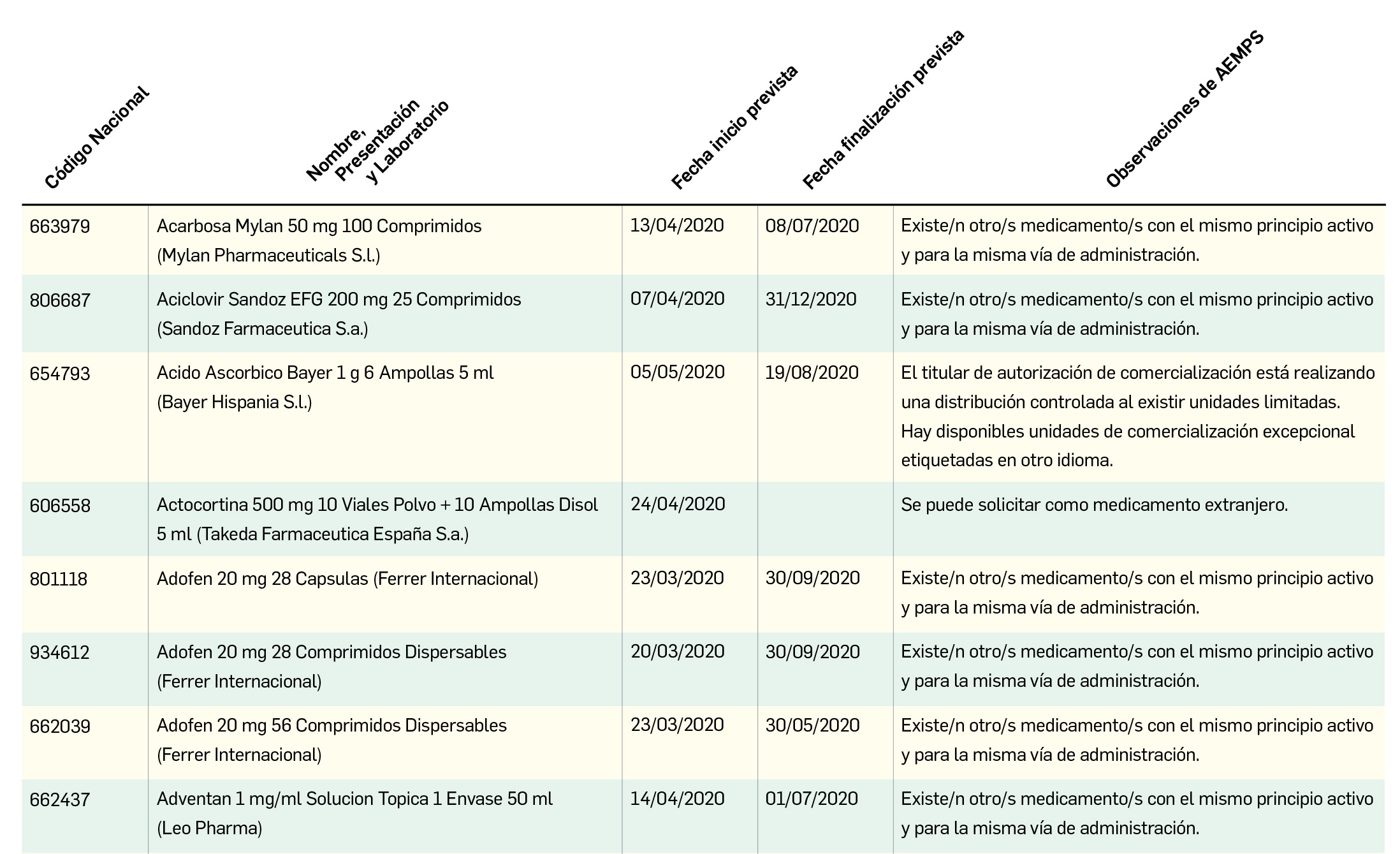
Combinación de Lopinavir con Ritonavir, una de las primeras pautas propuestas
La asociación de lopinavir con ritonavir (L+R) (Kaletra® y EFG), ambos inhibidores de la proteasa del VIH autorizados frente a la infección por dicho virus y empleados frente a la infección por dicho virus en combinación con otros agentes antirretrovirales (ritonavir fundamentalmente por su efecto de potenciador farmacocinético), ha sido uno de los tratamientos recomendados desde el inicio de la epidemia de COVID-19 por las autoridades sanitarias chinas en los protocolos para el manejo de pacientes. Existen numerosos ensayos clínicos en marcha donde se está evaluando la eficacia y seguridad de L+R, de los cuales se han divulgado ya resultados de dos de ellos, no del todo coincidentes ni concluyentes.
A finales de marzo de 2020 se publicaron los resultados de un ensayo clínico aleatorizado (Cao et al., 2020), controlado y abierto en pacientes adultos hospitalizados con infección confirmada por SARS-CoV-2 y afectación respiratoria (saturación de O2 ≤ 94%). Un total de 199 pacientes fueron asignados al azar (1:1) a recibir L+R (400+100 mg/12 h por vía oral) durante 14 días más el tratamiento sintomático estándar (N= 99) o solo el tratamiento estándar (N= 101). La variable principal de eficacia fue la mediana del tiempo hasta mejoría clínica (definida como un aumento de 2 puntos en una escala ordinal de 7 o alta hospitalaria), que en ambos brazos fue de 16 días desde la aleatorización, sin diferencias significativas (HR: 1,31; IC95% 0,95-1,80). Un segundo análisis sobre la población ITT modificada (200 pacientes) mostró un día de diferencia a favor de L+R, pero el número de pacientes en cada subgrupo es muy pequeño y la potencia del estudio es baja. Los resultados de algunas variables secundarias mostraron también una tendencia a favor del grupo de L+R, pero sin diferencias significativas entre ambos grupos en términos de mortalidad a los 28 días (19,2% vs. 25% con tratamiento estándar) ni en relación con la carga viral detectable. Si bien se describieron eventos adversos graves con más frecuencia entre los pacientes en tratamiento estándar, las reacciones adversas gastrointestinales (diarrea, náuseas, vómitos, etc.) fueron más comunes en el grupo de L+R, lo que hizo que el tratamiento se interrumpiera en el 14% de pacientes. Los autores concluyeron que no se observaba ninguna ventaja adicional al añadir la combinación de L+R a la atención médica estándar, sin descartar que sí pudiera probarse en otros estudios más amplios o de diferente diseño (por ejemplo, mayor dosis o inicio más precoz del tratamiento).
Las incertidumbres persistentes en torno a la ausencia de beneficio clínico con L+R en etapas más tempranas de la infección (que es actualmente su sitio en terapéutica), unido al riesgo de potencial toxicidad cardiaca y grastrointestinal grave, hicieron crecer las dudas sobre su utilidad; éstas han sido parcialmente esclarecidas, aunque con matices, por un segundo estudio divulgado a principios de mayo de 2020 (Hung et al., 2020).
Se trató de un ensayo clínico de fase 2, aleatorizado, multicéntrico y abierto que incluyó un total de 127 pacientes ingresados en 6 hospitales de Hong Kong, que fueron asignados a recibir durante 14 días un tratamiento combinado de L+R (400+100 mg/12 h), rivabirina (400 mg/12 h) y 3 dosis de 8 millones de UI de interferón β-1b en días alternos (N= 86), o bien solo la combinación de L+R (N= 41) como control; todos los pacientes recibieron el tratamiento de soporte necesario en cada caso (ventilación, diálisis, antibióticos y corticosteroides). La mediana del tiempo desde el inicio de los síntomas hasta el inicio del tratamiento fue de 5 días. Los resultados evidencian que el tratamiento con la combinación de 4 fármacos redujo significativamente el tiempo desde el inicio del tratamiento hasta alcanzar la negatividad en la PCR de una muestra de exudado nasofaríngeo (variable primaria), con una mediana de 7 días (rango intercuartil de 5-11) frente a los 12 días (rango 8-15) en el grupo control (HR: 4,37; IC95% 1,86-10,24; p= 0,001). Los datos de algunas variables secundarias respaldan lo anterior; se observó, por ejemplo, una reducción a la mitad (4 vs. 8 días) del tiempo para alcanzar el alivio completo de la sintomatología de la COVID-19. El estudio no halló diferencias destacables entre ambos grupos en términos de incidencia de náuseas y diarrea.
Así pues, parece que la administración precoz de la terapia cuádruple (combinación de lopinavir/ritonavir, rivabirina e interferón β-1b) –adicionalmente a las medidas de soporte estándar– puede ser una opción terapéutica eficaz, superior a la administración única de lopinavir+ritonavir, para conseguir aliviar las manifestaciones clínicas y acortar la duración de la diseminación viral y la estancia hospitalaria en pacientes con COVID-19 leve-moderada. Eso podría suponer una ventaja para disminuir también el riesgo de contagio por parte de los profesionales sanitarios. No obstante, los propios autores de este último trabajo recuerdan la necesidad de realizar ensayos de fase 3 más amplios, que incluyan un grupo placebo y doble cegamiento, y que confirmen o refuten estos hallazgos con mayor robustez estadística.
- Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G Z et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med. 2020; 382(19): 1787-99. DOI: 10.1056/NEJMoa2001282.
- Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu M et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020; DOI: https://doi.org/10.1016/S0140-6736(20)31042-4.
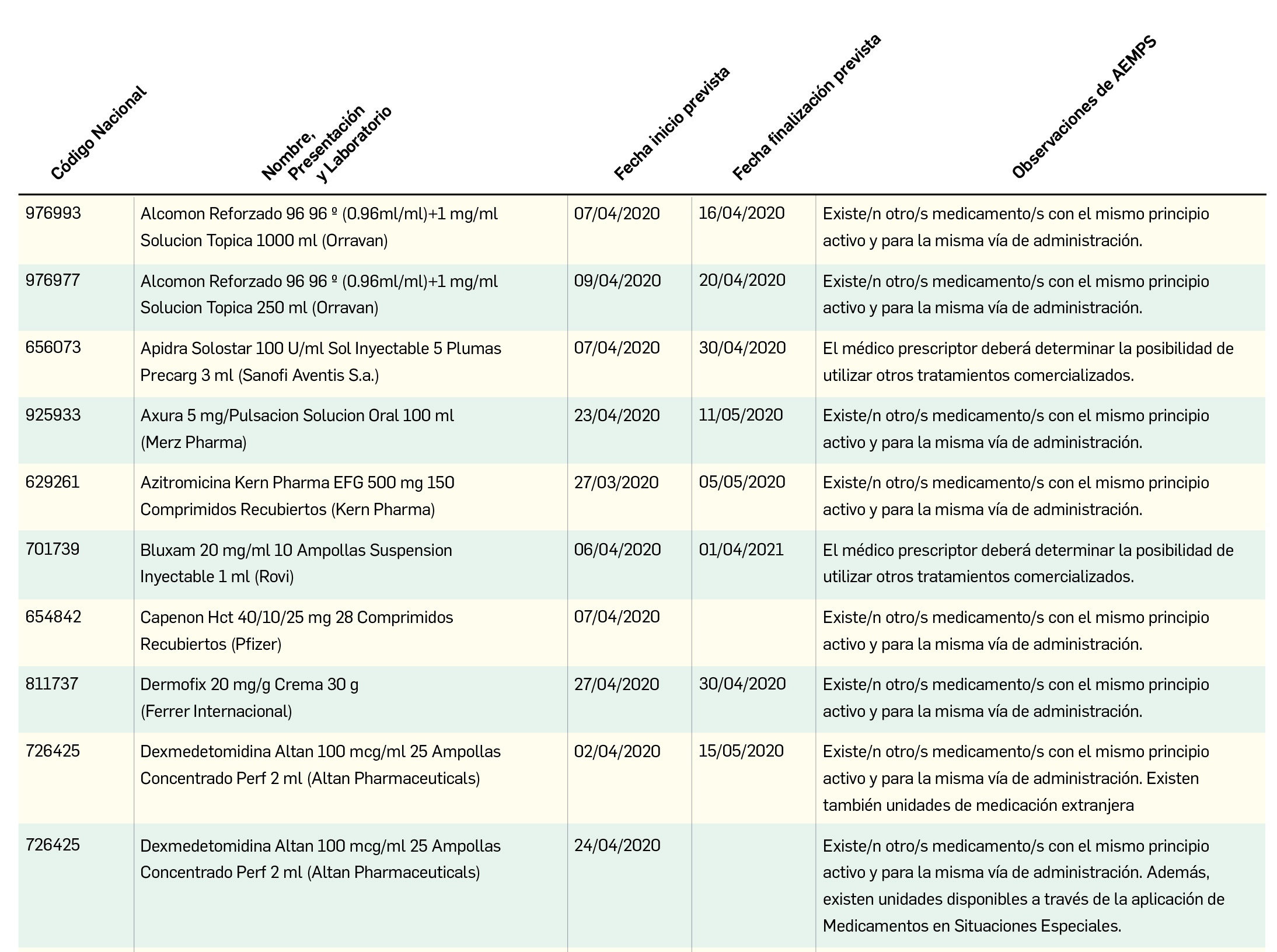
Remdesivir, ¿la mejor opción terapéutica?
Remdesivir es un análogo de nucleótido que interfiere con la polimerización del ARN del virus, y que no ha sido aún autorizado en España. Desarrollado inicialmente como tratamiento para la infección por el virus del Ébola, ha mostrado actividad in vitro e in vivo también frente a otros virus, incluyendo el coronavirus SARS-CoV-2 y el MERS-CoV causante del MERS (frente al cual se mostró más eficaz que el tratamiento con lopinavir/ritonavir más interferón β1b).
En el contexto de la pandemia por COVID-19, los primeros datos clínicos sobre su uso fueron los del primer caso de infección confirmado en EE.UU. (Holsue et al., 2020). Posteriormente, se han publicado los resultados de un primer estudio abierto multicéntrico y multinacional en el que 53 pacientes infectados y con patología respiratoria (saturación de O2 ≤ 94% o necesidad de oxigenoterapia) recibieron remdesivir (vía intravenosa, 200 mg el primer día y 100 mg/día los 9 días siguientes) por uso compasivo. En esta serie de casos, el tratamiento se asoció con una mejoría clínicamente significativa de la necesidad de oxigenoterapia en el 68% de los pacientes (incluyendo la extubación de 17 pacientes de los 30 que necesitaban ventilación mecánica al inicio) tras una mediana de seguimiento de 18 días desde la primera dosis. Sin embargo, son datos que deben interpretarse con cautela por el escaso tiempo de seguimiento, el pequeño tamaño de la cohorte analizada, la ausencia de un grupo control aleatorizado y la limitada recogida de datos en los programas de uso compasivo.
Más información aporta un artículo publicado a finales del mes de abril (Wang et al., 2020) de un estudio aleatorizado de fase 3, doblemente ciego y controlado por placebo desarrollado en 10 hospitales de Wuhan (China), que incluyó un total de 237 pacientes adultos –158 tratados con remdesivir (200 mg el día 1 y 100 mg/día los 9 días siguientes) y 79 con placebo– con diagnóstico microbiológico de COVID-19, saturación de O2 ≤ 94%, un tiempo desde el inicio de los síntomas de ≤ 12 días y neumonía radiológicamente confirmada (enfermedad grave); estos pacientes presentaban, en su conjunto, una media de edad de 65 años y eran mayoritariamente hombres (59%). Durante el estudio se permitió el uso concomitante de lopinavir/ritonavir, interferones y corticoides. Los resultados del análisis por intención de tratar ponen de manifiesto que, en los 28 días de seguimiento máximo, el tratamiento con remdesivir no supuso una diferencia significativa frente a placebo en el tiempo hasta la mejoría clínica (HR: 1,23 a favor de remdesivir; IC95% 0,87-1,75), definida ésta como el número de días hasta el alta hospitalaria o la reducción de 2 puntos en una escala ordinal de 6 (desde 1= alta, hasta 6= muerte). Aunque sin significación estadística, un análisis exploratorio mostró una mejoría clínica más rápida en los pacientes tratados con remdesivir en los primeros 10 días desde el inicio de los síntomas (HR: 1,52; IC95% 0,95-2,43); la confirmación de este beneficio con el tratamiento precoz requiere ensayos clínicos más amplios. En términos de seguridad, la incidencia de eventos adversos fue similar con remdesivir y con placebo (66% vs. 64%), si bien las interrupciones del tratamiento por problemas de seguridad sí fueron más frecuentes con remdesivir (12% vs. 5%). La tasa de mortalidad también fue muy pareja en ambos grupos (14% en el grupo de remdesivir vs. 13% con placebo).
El adecuado diseño y realización (alta adherencia, sin pérdida de seguimiento) de este primer ensayo aleatorizado plantea, por tanto, dudas sobre la utilidad real de remdesivir en la práctica clínica, que contrastan con las altas expectativas generadas por los resultados anteriormente comentados. No obstante, el estudio se detuvo antes de lo previsto (sin llegar a los 453 pacientes que era la muestra pre-establecida3), lo que resta poder estadístico a los datos. Surgen dudas sobre si, aunque se hubiera alcanzado ese tamaño muestral, el estudio habría tenido la robustez necesaria para confirmar o descartar un beneficio clínico estadísticamente significativo con el tratamiento con remdesivir; todo ello unido a que la escasa evidencia disponible dificulta establecer el límite de la diferencia mínima con relevancia clínica (Norrie et al., 2020).
Estos resultados relativamente negativos se contraponen con una noticia publicada el 29 de abril de 2020 por el Instituto Nacional de Enfermedades Infecciosas de EE.UU. (NIAID)4, relativa a los datos provisionales –aún no publicados– de un ensayo clínico aleatorizado y controlado que ha evaluado la seguridad y eficacia del remdesivir en 1.063 pacientes hospitalizados por neumonía por SARS-CoV-2. Se trata de un análisis preliminar realizado por el Comité de seguimiento de seguridad del estudio, que indica que los pacientes tratados con remdesivir se recuperan significativamente más rápido, con un periodo de tiempo hasta mejoría clínica un 31% más corto que los que recibieron placebo (11 días vs. 15 días con placebo; p< 0,001), y con un posible beneficio en la supervivencia (tasa de mortalidad del 8% en el grupo de remdesivir vs. 11,6% en el grupo placebo; p= 0,059). En todo caso, se trata de un análisis preliminar que debe interpretarse con prudencia, sin poder extraer conclusiones sólidas hasta que se puedan analizar todos los datos del estudio.
En este lado de la balanza también hay que subrayar que la compañía farmacéutica que desarrolla el fármaco (Gilead) ha informado de que dispone ya de esperanzadores resultados de los ensayos de fase 3. Se trata de uno de los dos estudios SIMPLE, ensayos abiertos multicéntricos y multinacionales que evalúan los resultados clínicos con remdesivir en pautas de tratamiento de 5 y 10 días en paciente hospitalizados con manifestaciones graves de COVID-19. Según la información divulgada, en la primera parte del estudio se trataron 397 pacientes (se prevé que el estudio enrole en global hasta casi 6.000 pacientes y que los resultados finales de los primeros 600 pacientes estén disponibles a lo largo del mes de mayo), incluidos pacientes con ventilación mecánica, cuyos datos apuntan a que la mediana del tiempo hasta mejoría clínica en el grupo de pauta corta fue de 10 días y de 11 días en el grupo de pacientes tratados durante de 10 días. Más de la mitad de los pacientes en ambos grupos de tratamiento habían recibido el alta hospitalaria el día 14 desde la aleatorización (60,0% de los tratados durante 5 días vs. 52,3% con la pauta de 10 días). Además, en ese momento, el 64,5% de los pacientes en el grupo de tratamiento de 5 días y el 53,8% en el grupo de tratamiento de 10 días lograron la recuperación clínica. No se identificaron eventos adversos nuevos5 en ninguno de los dos grupos de tratamiento.
Aunque no han sido publicados todavía e indudablemente se requieren datos adicionales que ayuden a comprender mejor si remdesivir es seguro y eficaz, así como la optimización de su pauta posológica, hay que destacar que tras conocerse estos resultados favorables, a fecha de 1 de mayo de 2020, la FDA ha otorgado a remdesivir una autorización de uso de emergencia para pacientes hospitalizados con COVID-19 grave. Además, el fármaco ha sido aprobado en Japón siguiendo un procedimiento de aprobación excepcional (primera aprobación oficial completa en el mundo). En Europa, estos datos están bajo evaluación desde el 30 de abril y, habida cuenta de que remdesivir está siendo sometido a numerosos ensayos clínicos en diferentes países, incluyendo 5 en España, es previsible que tengamos novedades en las próximas semanas (tanto del estudio SIMPLE como de otros), de las cuales informaremos diligentemente en esta u otras secciones de PAM.
- Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N Engl J Med. 2020; DOI: 10.1056/NEJMoa2007016.
- Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H et al. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med. 2020; 382(10): 929-36. DOI: 10.1056/NEJMoa2001191.
- Norrie JD. Remdesivir for COVID-19: challenges of underpowered studies. Lancet. 2020; DOI: https://doi.org/10.1016/S0140-6736(20)31023-0
- Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020; DOI: https://doi.org/10.1016/S0140-6736(20)31022-9
_____________________________________________________________________________________
3 El estudio planteó como objetivo pre-establecido de eficacia una razón de riesgo (hazard ratio, HR) de 1,4, que se traduciría en una reducción de la mediana del tiempo para la mejora clínica de 15 días frente a 21 días. Por tanto, con el HR de 1,23 finalmente observado, el beneficio clínico con remdesivir, si existe, es menor de lo esperado en ese estudio.
5 La AEMPS considera que, por ahora, se trata de un fármaco con un perfil de seguridad no bien caracterizado aún. La principal reacción adversa es la hipotensión infusional. Otras posibles reacciones adversas afectan al tracto gastrointestinal (náuseas, vómitos, diarrea, estreñimiento, dolor abdominal etc.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}