Resumen
El pie y el tobillo, como los elementos óseo-articulares más distales que soportan el peso del cuerpo y entran en contacto continuo con diversas superficies, están expuestos a lo largo de la vida a numerosos traumatismos y lesiones también de origen reumático. La mayoría de las patologías que afectan a estas estructuras se caracterizan por la presencia de dolor y son un motivo muy frecuente de consulta en Atención Primaria.
Entre la diversidad de trastornos, el esguince de tobillo es la lesión más incidente –y una de las más frecuentes en traumatología–, en muchos casos relacionada con la práctica deportiva. También la fascitis plantar o las lesiones aquíleas implican una importante morbilidad en nuestro medio. Aunque en la mayoría de casos pueden detectarse con una diligente exploración física y clínica, algunas patologías requieren de pruebas de imagen para su diagnóstico diferencial (ecografía, resonancia magnética o radiografía).
En cuanto a su abordaje terapéutico, si bien el tratamiento farmacológico (antiinflamatorios y analgésicos, fundamentalmente) es necesario para el control de los síntomas, en estos trastornos resulta fundamental la rehabilitación propioceptiva y funcional o la corrección ortopédica, recurriendo a cirugía en casos graves o, por ejemplo, ante una rotura del talón de Aquiles. El presente artículo revisa, desde un punto de vista clínico, las principales patologías de pie y tobillo.
Introducción
Si consideramos que el esguince de tobillo es posiblemente una de las lesiones más frecuentes y una de las peor tratadas (se calcula que se produce un esguince diario por cada 10.000 personas en todo el mundo, y que hasta una tercera parte presentan secuelas), se puede afirmar que se trata de un importante problema de salud. Además, es una lesión subestimada tanto por traumatólogos, rehabilitadores y fisioterapeutas, salvo cuando se produce en el ámbito deportivo.
En este artículo trataremos las patologías más relevantes, más frecuentes desde un punto de vista traumatológico, que afectan a pie y tobillo.
Algunas de las definiciones que se deben tener en cuenta para la lectura del artículo son:
- Fractura: la aparición de una solución de continuidad anatómica en el tejido óseo.
- Luxación: la pérdida completa de la congruencia articular; los huesos se encuentran desplazados, y se produce la rotura de la cápsula articular y de los ligamentos que fijan la articulación.
- Subluxación: pérdida de contacto incompleta entre los huesos que componen las superficies articulares, que dejan de ser congruentes.
- Esguince: desgarro incompleto de ligamentos responsables de la estabilidad articular.
Normas generales de manejo inicial de cuadros dolorosos del aparato locomotor
Cuadros dolorosos traumáticos
¿Cuándo actuar? y ¿cuándo derivar?
Es un hecho que el dolor se presenta como uno de los síntomas claves, si no el síntoma guía, en la mayoría de las alteraciones del aparato locomotor, ya sean éstas de etiología traumatológica o reumatológica. Ante un paciente que se presenta en la consulta de Atención Primaria refiriendo manifestaciones dolorosas referidas al aparato locomotor, lo primero que debemos aclarar es la pertinencia o no de derivar de manera urgente o preferente tanto al hospital como a la consulta especializada.
Desde este punto de vista, siempre que se trate de un enfermo que ha sufrido un traumatismo, las preguntas claves son:
- ¿Existe sospecha de fractura o luxación?
- ¿Existen alteraciones vasculares o neurológicas asociadas a la lesión?
- ¿Existe la posibilidad de complicaciones a corto plazo?
- ¿Es necesario el estudio radiográfico urgente?
Si la respuesta a cualquiera de estas cuestiones es afirmativa, se derivará al enfermo, en las mejores condiciones y en el menor tiempo posible, al segundo nivel.
Siempre que exista la sospecha clínica de una posible fractura o luxación, debemos proceder a realizar una exploración neurovascular en la región distal al foco de fractura. Se comprobarán pulsos distales, aspecto del miembro (sonrosado, cianótico, pálido) y si existen o no alteraciones en la sensibilidad (parestesias, anestesia, etc.) o en la fuerza (paresia), aunque este último dato es difícil de valorar, pues el dolor obliga al enfermo a no realizar ningún movimiento en un acto reflejo de protección.
Cuando encontremos signos que nos hagan sospechar la existencia de una fractura (dolor intenso espontáneo y a la palpación ósea, impotencia funcional absoluta –aunque no siempre–, crepitación, movimientos anormales de los fragmentos, deformidad, etc.) o de una luxación (incongruencia articular, deformidad, dolor, mal alineamiento óseo, impotencia funcional, etc.) debemos derivar al enfermo con urgencia.
Si el traumatismo se ha producido en un accidente deportivo, se deben tener muy presentes las necesidades futuras del deportista en cuestión, considerando principalmente si la actuación es la mejor opción para garantizar una precoz recuperación funcional y un adecuado regreso a la competición.
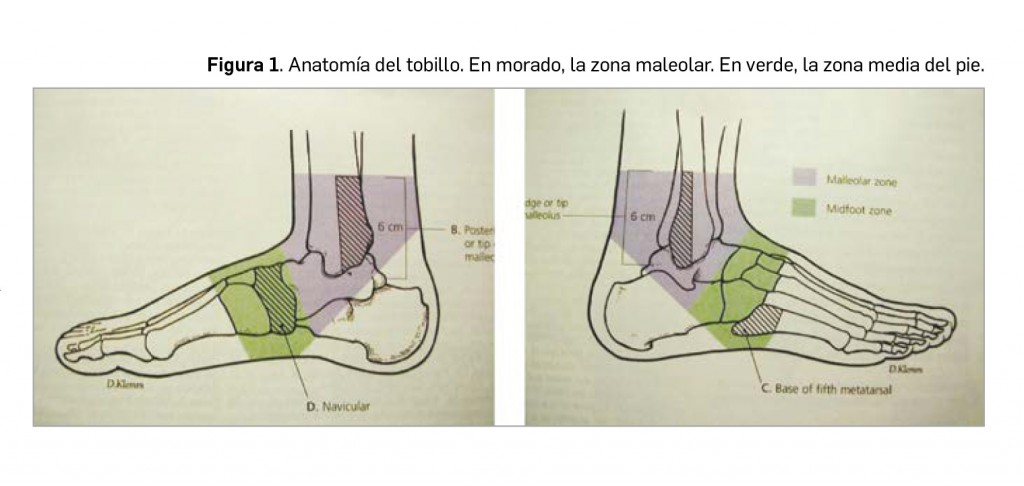
¿Cuándo se justifica el estudio radiológico en un traumatismo? – Evidencias y recomendaciones para la petición racional de estudios radiológicos
En lo referente a la petición de estudio radiográfico, existen una serie de normas valiosas como ayuda en la toma de decisiones clínicas diferentes según la patología de que se trate.
REGLAS DEL TOBILLO DE OTTAWASolo el 15% de los pacientes con traumatismos de tobillo o pie presentan fracturas clínicamente significativas (desplazamiento > 3mm). Exclusivamente se deben pedir radiografías de tobillo o medio pie si encontramos:
- Dolor espontáneo o a la palpación en el extremo distal, o en los últimos 6 cm de cualquiera de los maleolos tibial o peroneo externo o interno.
- Incapacidad para permanecer de pie y dar cuatro pasos (dos con el pie sano, dos con el pie lesionado) en la sala de urgencias.
- Las radiografías del medio pie solo son necesarias si existe dolor en el hueso navicular o en la base del 5º metatarsiano, o si se cumple el criterio anterior.
La sensibilidad de estas reglas es cercana al 100% y su especificidad del 50%. No resultan aplicables si el paciente tiene una edad mayor de 55 o menor de 18 años, si el traumatismo ha ocurrido 10 días o más antes de ser visto en consulta, en pacientes embarazadas (valorar beneficio-riesgo de la radiación en una gestante) o si hay una herida asociada al traumatismo.
REGLAS DE RODILLA DE OTTAWASolo el 6% de los pacientes que han sufrido un traumatismo de rodilla presentan fracturas. Exclusivamente se deben pedir radiografías de rodilla si se cumple lo siguiente:
- Edad igual o mayor de 55 años;
- dolor en la cabeza del peroné;
- dolor aislado en la patela;
- incapacidad para flexionar la rodilla más de 90º.
- O incapacidad para caminar 4 pasos o permanecer de pie, bien justo tras el traumatismo o bien en la sala de urgencias.
La sensibilidad de estas reglas es del 92% y su especificidad del 57%.
NORMAS PARA LA PETICIÓN DE RADIOGRAFÍAS EN LUMBALGIA Según la Guía AHCPR (Agency for Health Care Policy Research), se debe solicitar radiografía si existen signos de alarma (“red flags”) que sugieran la existencia de:- Fractura espinal (traumatismo importante, alcoholismo, tratamiento con esteroides, osteoporosis establecida, edad mayor de 70 años), o
- tumor o infección (historia de cáncer, edad menor de 20 o mayor de 50, pérdida de peso no explicada, dolor lumbar que empeora por la noche o en la posición de supino).
Cabe destacar que algunos autores cuestionan la validez de dichos criterios de la AHCPR, pues justifican que incrementan el coste, ocasionan una petición excesiva de radiografías y, aunque estuviera indicado, no influyen claramente en el desarrollo final del proceso.
NORMAS PARA LA PETICIÓN DE RADIOGRAFÍAS EN TRAUMATISMOS CERVICALESNo es necesaria la petición de radiografías –por ser muy poco probable la existencia de fractura– si:
- No existe dolor cervical en la anamnesis ni en la exploración.
- No existen signos o síntomas neurológicos (parestesias o paresia en extremidades).
- No se produjo pérdida de conciencia.
- No existen cambios del estado mental (dependientes del uso de drogas o alcohol).
- No existen otras lesiones estructurales en el resto del organismo.
Estas reglas solo resultan aplicables a adultos y a pacientes sin cambios en su estado mental, ni previo ni posterior al traumatismo.
Procesos reumatológicos dolorosos
¿Cómo iniciar el abordaje?
En el caso de que el motivo de dolor sea una enfermedad reumatológica, debemos valorar la posibilidad de realizar una primera aproximación diagnóstica efectiva la consulta de Atención Primaria. Ello implica, lógicamente, una adecuada anamnesis, con especial atención a las características del cuadro doloroso (inicio, localización, irradiación, calidad, tiempo transcurrido desde su aparición, estímulos desencadenantes, factores que lo alivian o agravan, síntomas acompañantes, etc.).
La semiología del dolor nos ayudará a tomar la decisión adecuada, desde realizar una artrocentesis y procesar el líquido sinovial obtenido, hasta iniciar de entrada un tratamiento sin demora (por ejemplo, en el caso de sospecha fundada de arteritis de la temporal), o derivar con urgencia en el caso de que sea necesario (artritis séptica, importante afectación del estado general, etc.).
Conceptos básicos y normas generales para el tratamiento del dolor en afecciones del aparato locomotor
Lógicamente, siempre que sea posible se realizará un tratamiento etiológico del problema. En el caso de traumatismos importantes, como fracturas y luxaciones, el tratamiento será la reducción ortopédica o quirúrgica de las mismas. Es evidente que en muchas ocasiones será necesaria la intervención multidisciplinar de médicos de familia, rehabilitadores, traumatólogos, reumatólogos, fisioterapeutas, etc., de manera conjunta y coordinada. Aun así, debemos tener presente que el primer contacto del enfermo, la mayoría de las veces será con su médico de cabecera, lo que obliga a instaurar un tratamiento inicial desde el primer nivel asistencial, en ocasiones tan solo para mitigar el dolor (hasta que el enfermo es visto por el especialista correspondiente).
Patrones dolorosos del aparato locomotor
Dentro de los trastornos del aparato locomotor, se definen dos tipos claramente diferenciados de patrones dolorosos (Tabla 1):
- Patrón capsular (dolor inflamatorio). Es aquél dolor que se presenta no solo al movilizar la estructura o articulación afectada, sino también en reposo, que no cede con el mismo e incluso aumenta, que impide el descanso del individuo y que despierta al paciente por la noche. Es muy sugerente de capsulitis y de artritis.
- Patrón periarticular (dolor mecánico). Es el dolor que aparece con la movilización de la estructura afectada (tendones, músculos, ligamentos, bursas sinoviales), mejora o desaparece con el reposo y no despierta al individuo por la noche. Es típico de las tendinitis, entesitis, bursitis y lesiones musculares.
Tampoco se debe olvidar la existencia del “dolor referido”, es decir, un dolor proveniente de estructuras alejadas de la región cutánea donde se percibe, como en los casos de angor pectoris, cervicalgias o tumor de Pancoast, por ejemplo, que pueden manifestarse como dolor en el hombro.
Cuadros dolorosos del pie
Artropatía de Charcot
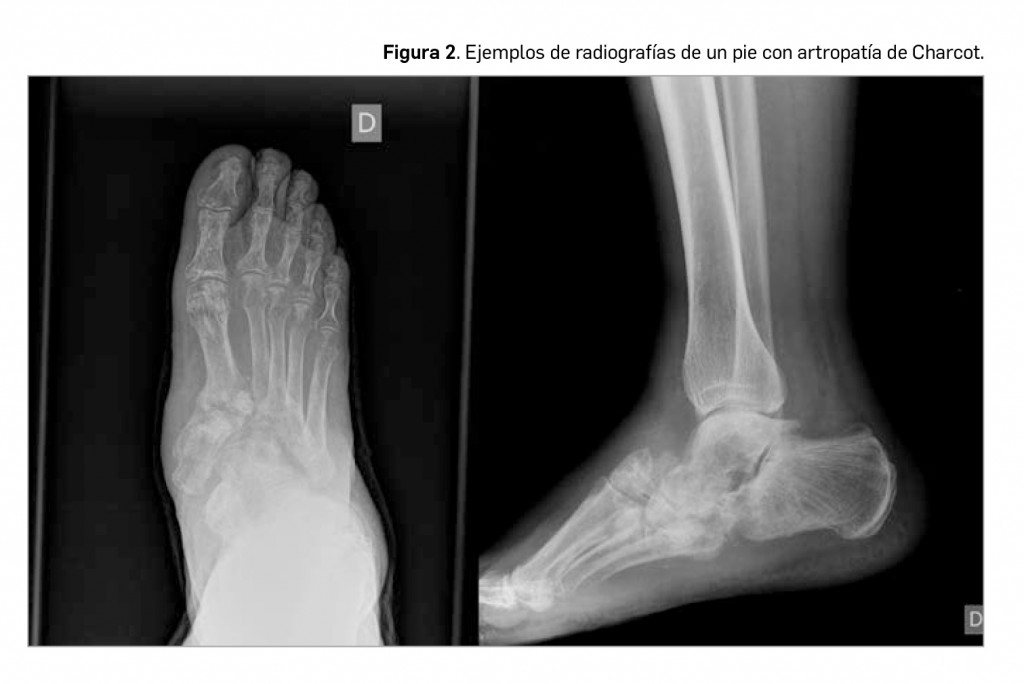
La artropatía de Charcot supone un cuadro clínico, relativamente frecuente y posiblemente infradiagnosticado en multitud de ocasiones. Se traduce en la presentación de una pérdida de sensibilidad en el pie, donde los micro-traumatismos repetidos acaban produciendo una inestabilidad articular, con la aparición de micro fracturas y esclerosis subcondral en los fragmentos óseos afectados como hallazgos típicos de esta patología. Se trata, por tanto, de una forma crónica de artropatía degenerativa asociada a una pérdida de sensibilidad dolorosa térmica y propioceptiva. Aparece con mayor frecuencia en diabéticos, enfermos con siringomielia o en presencia de una tabes dorsal luética. El gran problema es que, debido a su inicio insidioso, con escaso dolor y discretos signos inflamatorios, en muchas ocasiones se producen diagnósticos erróneos (infección, celulitis, distrofia simpático-refleja, artritis séptica, gota, etc.) que implican un retraso en el inicio del tratamiento adecuado.
La fisiopatología de esta entidad no acaba de estar clara. En el diabético posiblemente coexistan varios factores: ante el mal control de la diabetes aparecería una pérdida sensitiva, una neuropatía motora responsable del desequilibrio muscular y una neuropatía autonómica acompañada de osteopenia; esta situación nos presenta un “pie insensible”, incapaz de responder a agresiones por un fallo en la propiocepción. Se debe sospechar la existencia de una artropatía de Charcot en todo diabético mal controlado que presente edema unilateral del pie acompañado de ausencia de dolor y deformidad e inestabilidad articular.
En la radiografía (Figura 2), se pueden observar cambios atróficos (aumento de partes blandas, resorción ósea e incluso pequeñas líneas de fisuración) o hipertróficos (con destrucción articular, subluxaciones o luxaciones francas de los huesos del tarso, destrucción articular y aparición de grandes osteofitos). De cualquier modo, la exploración radiológica es útil cuando la enfermedad ya se encuentra muy avanzada. En estadios precoces se hace necesario la realización de gammagrafía ósea, TAC o resonancia magnética. Estas pruebas de imagen ayudarán a diferenciar esta entidad de otras de presentación clínica similar, como la osteomielitis.
El tratamiento se basa en la descarga articular. En los primeros estadios se puede iniciar un abordaje farmacológico mediante pamidronato. La cirugía tiene escasa cabida en el inicio y se recurre a ella cuando se hace necesario la artrodesis en articulaciones inestables. En conclusión, la neuroartropatía de Charcot es una enfermedad con gran dificultad diagnóstica, ya que no hay criterios definitivos ni pruebas que confirmen su diagnóstico, por lo que, ante la presencia de inflamación y edema del pie en un paciente diabético con neuropatía sensitivo motora en extremidades inferiores, se debe pensar en la posibilidad de neuroartropatía de Charcot.
Esguince de tobillo
El esguince de tobillo es posiblemente una de las lesiones más frecuentes y una de las peor tratadas traumatología. Se calcula que se produce un esguince diario por cada 10.000 personas en todo el mundo y que presentan secuelas la tercera parte. Es una lesión generalmente subestimada tanto por traumatólogos, rehabilitadores y fisioterapeutas, excepto si se produce en el medio deportivo. El esguince de tobillo acontece con mayor frecuencia en individuos entre 21 y 30 años, probablemente relacionado con una mayor práctica deportiva en estas edades. Un esguince previo supone el doble de probabilidades de padecer nuevos esguinces en el futuro.
- Localización:
El 85% de las lesiones afectan al ligamento lateral externo (LLE); de éstas, el 65% afectan exclusivamente al ligamento peroneo-astragalino anterior (PAA), y el 20% a ambos, PAA y peroneo-calcáneo (PC).
El 10% de las lesiones se producen en la sindésmosis tibio-peronea-astragalina, y tan solo el 5% afectan al ligamento lateral interno (LLI).
- Factores predisponentes:
Terreno irregular;
contractura o escasa flexibilidad
del tendón de Aquiles;
exceso de peso;
esguinces previos;
alteraciones propioceptivas;
mal balance muscular: desequilibrio
de agonistas-antagonistas.
Primera consulta
Para una correcta anamnesis, se deben de investigar las siguientes cuestiones:
- Existencia de esguinces anteriores;
- Posición del pie (en apoyo, en el aire, rotado, flexionado, etc.) en el momento de la lesión.
- Mecanismo de producción.
- Valoración del tipo de dolor ¿brusco? ¿intenso? ¿breve? un dolor muy intenso pero breve, acompañado de gran edema de instauración rápida, sugiere rotura ligamentosa completa.
- Crujido, chasquido, equimosis, edema, impotencia funcional.
- ¿Pudo seguir realizando la actividad, la competición, o pudo seguir caminando?
- Valorar el tiempo transcurrido desde el traumatismo.
- Localizar zonas concretas de dolor y el inicio del edema.
- Valorar la capacidad de bipedestación inmediatamente tras la lesión y durante la exploración.
Exploración: debe ser inmediata, ya que la tumefacción, el edema y el espasmo muscular de instauración rápida impiden observar posteriormente la localización exacta de la lesión.
- Inspección: deformidad, edema, equimosis (el grado de equimosis se relaciona con la gravedad de la lesión), aumento del perímetro del tobillo (un aumento >4 cm respecto al tobillo sano sugiere rotura ligamentosa en el 70% de los casos).
- Palpación: sensibilidad dolorosa, edema, crujido, crepitación. Se debe palpar siempre buscando zonas dolorosas el maleolo tibial y peroneo (últimos 6 cm), la cola del 5º metatarsiano, el escafoides, los tendones peroneos, el tendón de Aquiles y tendón del tibial posterior, sin olvidar el tercio proximal del peroné.
Recuerdo anatómico
Los elementos básicos en la biomecánica del tobillo son el complejo ligamentoso: tibio-peroneo distal (TPD), peroneo-astragalino anterior (PAA), peroneo-astragalino posterior (PAP), peroneo-calcáneo (PC) conformando el ligamento lateral externo (LLE), y el poderoso ligamento deltoideo (con sus dos fascículos superficial –de 4 haces– y profundo –1 haz fuerte–), que une el maleolo interno con el astrágalo y el calcáneo, conformando el ligamento lateral interno (LLI).
El peroné ejerce una acción estabilizadora (llega a sostener hasta un tercio de la carga total que soporta el miembro inferior), realizando movimientos de vaivén y de arco para estabilizar la mortaja del tobillo ante posiciones forzadas. Por ello, en la flexión plantar el peroné se desplaza hacia distal y hacia adentro.
El haz PAA refuerza una débil cápsula articular anterior, y en flexión plantar se coloca casi verticalmente, actuando en esos instantes como un verdadero ligamento colateral externo, perpendicular al plano de apoyo. Es el más frecuentemente lesionado.
Por su parte, el haz PAP sujeta al astrágalo en su desplazamiento posterior, siendo muy rara su rotura tanto parcial como completa, salvo que se produzca una luxación completa de la mortaja.
El ligamento PC es un ligamento cordonal, vertical, es el auténtico ligamento colateral externo de la articulación, que se sitúa de manera horizontal cuando se produce una flexión plantar. Estabiliza la articulación subastragalina y el tobillo.
Existe también el ligamento calcáneo-astragalino, situado entre el PAA y el PC, entremezclado entre ambos y con la cápsula articular, a la que refuerza.
En las lesiones graves, pueden llegar a afectarse la membrana interósea y el ligamento deltoideo. Ello suele ir asociado a lesión de la sindésmosis, del maleolo peroneo y, en no pocas ocasiones, a luxación o subluxación de los tendones peroneos.
Fisiopatología
El mecanismo fisiopatológico más habitual es la inversión forzada del tobillo, esto es, una situación mixta donde se unen la supinación del tobillo con la flexión plantar. De manera simétrica, puede dañarse el ligamento deltoideo y la sindésmosis peroneo-tibial inferior si se produce una eversión del pie unido a una rotación externa del mismo (lo que implica una rotación interna de la tibia), aunque esta última situación es mucho más infrecuente.
El esguince capsular anterior se produce tras un impacto en flexión plantar (grado I) y aparece dolor a la extensión resistida y a la flexión plantar activa. El tratamiento es rehabilitador.
El esguince del LLI se produce en eversión, mientras que el esguince del LLE es un hecho muy frecuente, que se produce muchas veces durante la carrera. En ella existe de manera fisiológica una ligera aducción del pie, de manera que cualquier obstáculo que implique una desviación del pie lesiona el débil haz PAA. Así pues, el mecanismo es de flexión plantar e inversión, el ligamento PAA se verticaliza y se desgarra. Si en esta situación prosigue la inversión, se verticaliza el ligamento PC y también podría romperse. Aparece tumefacción y equimosis que “gotea” hacia los dedos.
Clasificación
Por lo general, los esguinces se clasifican en tres grados, en función de la magnitud de la lesión:
- GRADO I. Se produce la rotura de < 5% de las fibras del PAA; una distensión del ligamento sin afectación ni laxitud de la cápsula. Se produce dolor y escasa inflamación, con ruptura de propioceptores (muy abundantes en la zona) y la consiguiente respuesta vasomotora, elongación fibrilar, y escaso dolor. La deambulación es perfectamente posible.
- GRADO II. Se produce la rotura del 40-50% de las fibras ligamentosas. Existe una laxitud articular moderada, dolor, hinchazón y dificultad para la marcha “de puntillas”. No se llega a producir la rotura de la cápsula. Se caracteriza por ligera inestabilidad y dolor moderado. La deambulación es posible en posición antiálgica.
- GRADO III. Rotura total y rotura capsular con laxitud articular completa. El enfermo refiere sensación de crujido, unido a la presentación de un dolor fulgurante instantáneo, que después desaparece debido a la rotura de las terminaciones nerviosas de la cápsula. Se presenta una tumefacción importante, con laxitud en equino-varo y cajón anterior. Un tobillo que sufre este tipo de esguince grado III, debería ser explorado precozmente, ya que en fase tardía el edema y el derrame hacen imposibles las maniobras exploratorias. El chasquido audible y el dolor vivo de corta duración deben hacer sospechar de la existencia de una lesión importante (la existencia de un “clic” a la extensión es sugestiva de lesión osteocondral). La bipedestación y la deambulación no son posibles.
Exploración
Antes de iniciar una exploración reglada del tobillo es preceptivo realizar una anamnesis correcta que orientará hacia la existencia o ausencia de lesión grave. Para ello, se deben de tener presentes –y describir de la forma más precisa posible– preguntas como ¿qué estaba haciendo cuando se lesionó (deporte, otros)?, ¿en qué posición tenía el pie cuando notó el dolor?, ¿qué fue lo primero que observó (dolor, crujido, hinchazón, etc.)?, ¿el dolor fue inmediato o tardío?, ¿fue en aumento o desapareció?, ¿pudo andar o continuar la actividad deportiva después de lesionarse?; además, se debe valorar el tiempo transcurrido desde el accidente o la existencia de lesiones anteriores.
James Cyriax describió, ya a mediados del siglo XX, al menos 7 estructuras que deben ser exploradas: LPAA en su inserción peronea, LPC en su inserción peronea (la inserción calcánea se lesiona raramente), LPAA en su inserción calcánea, ligamenteo calcáneo-cuboideo, los tendones de los peroneos, la porción anterior de la cápsula, y el tendón del músculo extensor largo de los dedos.
En cualquier caso, se Debe explorar el tobillo tan pronto como sea posible, ya que la instauración del edema puede impedir obtener hallazgos fiables. Cuando existe hemartros, el derrame ocupa los espacios periaquíleos, mientras que si la lesión es extracapsular, estos se hallan libres; por ello, debemos observar el tobillo también por detrás. Es necesario explorar meticulosamente todas las articulaciones y los puntos óseos para descartar la existencia de fracturas, sobre todo en el peroné y en la cúpula del astrágalo.
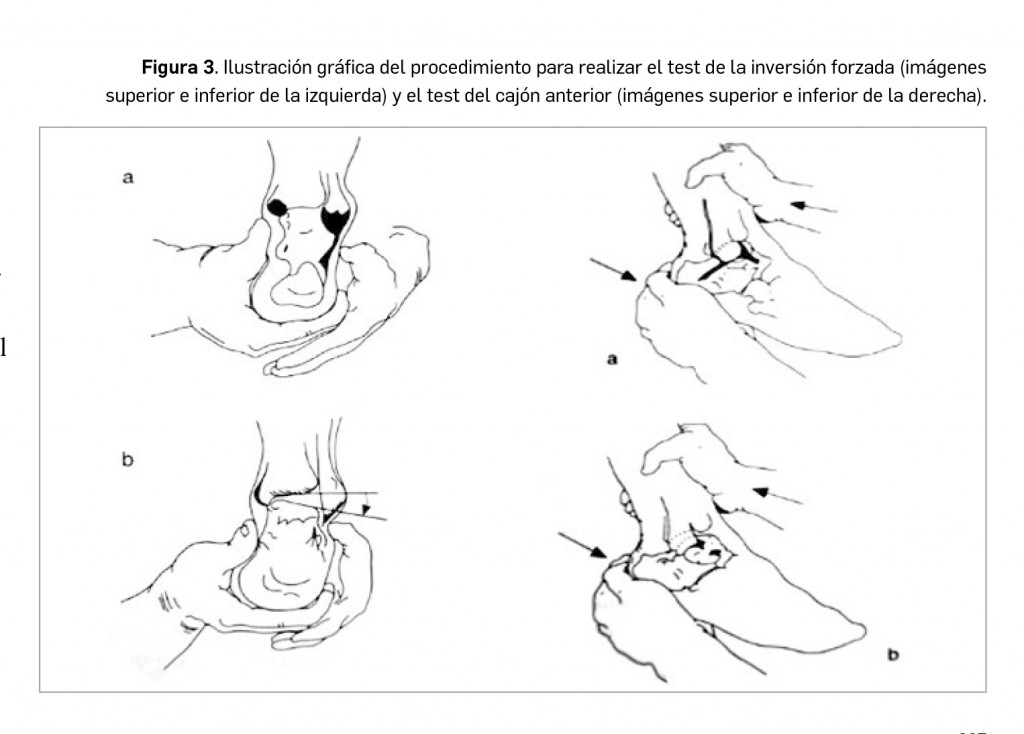
Entre las pruebas más sensibles para una exploración del tobillo, se deben valorar los test del cajón anterior, de la inversión forzada y la exploración de la sindésmosis. Fuerzas de compresión axial combinadas con fuerzas laterales continuas pueden producir la distensión de la sindésmosis.
Maniobras dinámicas- Test del cajón anterior: con el pie en posición neutra y la rodilla en flexión de 90º, se sujeta el calcáneo y se “empuja” la tibia hacia atrás (Figura 3, derecha). Evalúa la integridad del haz PAA, mediante el desplazamiento anterior del astrágalo en el plano sagital. La sensación de ausencia de “tope final”, con desplazamiento anómalo del calcáneo sugiere rotura ligamentosa.
- Test de la inversión forzada (“talar tilt test”): con el pie a 10-20º de flexión y la rodilla en flexión de 90º, se fija la tibia con una mano y con la otra se realiza una inversión forzada, desplazando el astrágalo en el plano frontal con respecto al techo del pilón tibial (Figura 3, izquierda). Se valora la presencia o no de tope final, asociado a la aparición de una umbilicación en la piel en la zona premaleolar peroneal (signo de la succión) que indica lesión del haz PAA.
- “Clunk test”: con la rodilla a 90º de flexión y la tibia fija evitando cualquier movimiento de inversión o eversión, realizamos movimientos de rotación externa forzada, desplazando la articulación en sentido medial y lateral. Si esta maniobra despierta dolor en el tobillo, se debe sospechar una lesión de la articulación (condral, sindesmal, o membrana interósea).
- “Squeeze test”: con la rodilla a 90º de flexión, comprimimos la musculatura a nivel del tercio medio de la pierna. Este mecanismo provoca la separación de la articulación tibioperonea proximal y de la sindésmosis TPA. Si desencadena dolor, se debe sospechar de afectación de la articulación o fractura de Maissoneauve en el cuello del peroné.
Al forzar el varo del tobillo buscaremos la existencia de un surco palpable en la interlínea articular. Si éste existe, pensaremos en una rotura completa del haz PAA. Contrariamente, si el paciente aguanta bien de puntillas sobre el tobillo lesionado, es un signo de buen pronóstico.
Diagnóstico
El diagnóstico de un esguince de tobillo es fundamentalmente es clínico. Ante la sospecha de rotura parcial o completa del LLE se debe realizar una ecografía, que ofrecerá un diagnóstico certero. Conviene recordar que la capacidad del paciente para mantenerse sobre los dedos de los pies en apoyo monopodal (“de puntillas”) es un signo de buen pronóstico, así como también lo es el mantenimiento en apoyo monopodal sobre el miembro lesionado con los ojos cerrados (prueba de Freeman o Romberg modificado), que indica que no existe una alteración propioceptiva importante.
Es importante realizar un diagnóstico diferencial con:
- Fracturas maleolares.
- Fractura de Maissoneauve (lesión del cuello del peroné). Siempre que se produzca un esguince de tobillo se debe explorar la articulación tibioperonea proximal.
- Fractura de la cola del astrágalo.
- Fracturas de la base del 5º metatarsiano.
Inestabilidad crónica del tobillo
En el caso del tobillo, los ligamentos tienen dos funciones. La primera es puramente mecánica, de sujeción del tobillo, y la segunda es propioceptiva. Los receptores de propiocepción son estimulados por la puesta en tensión del ligamento gracias a la existencia de un arco reflejo simple. Su estimulación provoca la contracción de los músculos peroneos, impidiendo el varismo excesivo de la articulación, protegiendo las diversas estructuras ligamentosas desde las que ha partido la información inicial. Cuando se produce una cicatrización con alargamiento del ligamento, su puesta en tensión en situaciones posteriores se produce de modo tardío, se retarda el estímulo de los propioceptores, con lo que la información llega tarde y el intento de estabilización de los peroneos se produce cuando ya es demasiado tarde.
Por tanto, así se comprende que en pacientes que han sufrido esguinces previos, se produce una inestabilidad crónica de la articulación tibiotarsiana, con frecuentes esguinces posteriores. En estos casos se debe prestar especial atención a la rehabilitación propioceptiva, mediante estímulos desequilibrantes al inicio, y la tabla de Freeman o plato giratorio posteriormente.
Clínicamente podemos encontrar una sensación subjetiva de inestabilidad acompañada o no de inestabilidad mecánica demostrable (cajón anterior >10 mm, o >3-5 mm comparado con el tobillo contralateral; inversión forzada >20º o >10º comparado con el tobillo contralateral). Se manifiesta claramente la inestabilidad si se pide al paciente que realice un “Romberg” mantenido exclusivamente sobre el tobillo lesionado (“Romberg a la pata coja”); ello implica la existencia de una alteración propioceptiva.
Tratamiento
En los esguinces de grado I y II es necesario un tratamiento funcional basado en el vendaje y la rehabilitación propioceptiva. En los esguinces de grado III se discute si es necesaria la intervención quirúrgica o, en ocasiones, puede recurrirse exclusivamente a un buen programa de rehabilitación, sin que ello implique inestabilidades residuales, aunque los defensores de la cirugía argumentan la persistencia de dolor e inestabilidad tras los tratamientos funcionales hasta en el 20-40% de los casos.
El tratamiento funcional pasa por tres fases:
- FASE I. Método PRICE: protección, reposo, hielo, compresión y elevación. No se debe cargar la articulación, siendo recomendable el uso de bastones hasta el 4º-6º día; hasta entonces, reposo sin apoyar el pie. Se recomienda utilizar hielo durante 30 minutos cada 1-2 horas, durante las primeras 24-48 horas. Cuando desaparece el dolor, está indicado la realización de isométricos y el apoyo (mejora la rehabilitación propioceptiva).
- FASE II. Consiste en potenciar la musculatura aquilea y de dorsiflexores mediante ejercicios isométricos de dorsiflexión y eversión del pie (solo cuando haya disminuido de manera importante o desaparecido el dolor). En esta fase también se recomiendan baños de contraste (indicados por su efecto de bombeo, mejora el drenaje linfático y venoso y, con ello, el dolor y la rigidez), que consisten en aplicar frío (a 16ºC) y calor (a 38-41ºC), permaneciendo siempre entre 1,5 y 2 veces más tiempo en calor que en frío (por ejemplo, 1 min en frío y 2-3 min en calor hasta completar series de 20-30 min).
- FASE III. Se debe hacer un entrenamiento de resistencia y propiocepción (tabla de Freeman). Posteriormente, pasar a marcha rápida y a carrera continua sin cambios de ritmo ni de dirección; después, carrera en ochos, saltos y cambios de dirección.
La protección posterior se establecerá mediante ortesis o vendaje, durante todo el tiempo que dure la recuperación en casos de esguinces de grado I-II, y durante los 3-6 meses siguientes en casos de esguince de grado III. En líneas generales, se estima que el vendaje debe ponerse durante 3 semanas y que la recuperación completa se produce a las 4-6 semanas de la lesión. Debemos tener la precaución de no poner ningún vendaje inmediatamente después de haber sometido al tobillo a calor o a frío ya que se incrementa mucho el riesgo de lesiones cutáneas.
La reeducación de la propiocepción –entendida como una alteración producida por el desgarro de terminaciones nerviosas (que, como tales, no van a regenerar)– se basa en la reeducación del sentido del equilibrio en el tobillo; esto se consigue, por ejemplo, mediante los ejercicios en una tabla de vaivén (el tobillo se protege ya que la tabla toca el suelo antes de que se produzca la inversión forzada del tobillo). Si es necesario recurrir a rehabilitación, el tobillo debe mantenerse protegido (vendaje funcional) hasta 6-7 meses, ya que ese es el tiempo que tarda el colágeno en madurar.
Complicaciones
Entre las principales complicaciones que pueden producirse a raíz de un esguince de tobillo, podemos destacar las siguientes:
- Fracturas osteocondrales.
- Rotura del retináculo de los peroneos (tras una dorsiflexión súbita, cuando los tendones están contraídos). La luxación o subluxación de los tendones peroneos puede objetivarse haciendo que el enfermo coloque el pie en dorsiflexión y eversión, y realizando resistencia a la inversión del pie. Si el retináculo está lesionado se subluxarán o luxarán los tendones, pasando a situarse por delante del maleolo peroneo.
- Fractura de la base del 5º metatarsiano (por tracción brusca del peroneo lateral corto).
- Fractura del os trigonum.
- Impingement sinovial: pinzamiento capsular que se produce entre el astrágalo, el peroné y la tibia, y que se acentúa en dorsiflexión forzada y en flexión plantar pasiva.
- Síndrome del túnel tarsiano (atrapamiento del nervio tibial posterior entre el maleolo tibial y el ligamento tarsiano). Aparece dolor y disestesias en el arco longitudinal interno del pie, con un signo de Tinel positivo.
Fascitis plantar-espolón calcáneo
La fascitis plantar es un cuadro doloroso en que se produce una sobrecarga de la fascia plantar.
La fascia plantar se localiza insertada en la base del calcáneo y se extiende hasta las cabezas de los metatarsianos, donde presenta dos anclajes muy bien definidos: en la cabeza del primer metatarsiano, formando el arco longitudinal interno, y en la cabeza del quinto, formando el arco longitudinal externo. Existe un tercer arco plantar formado por la fascia, entre el primero y el quinto, que se denomina arco plantar anterior (Figura 4).
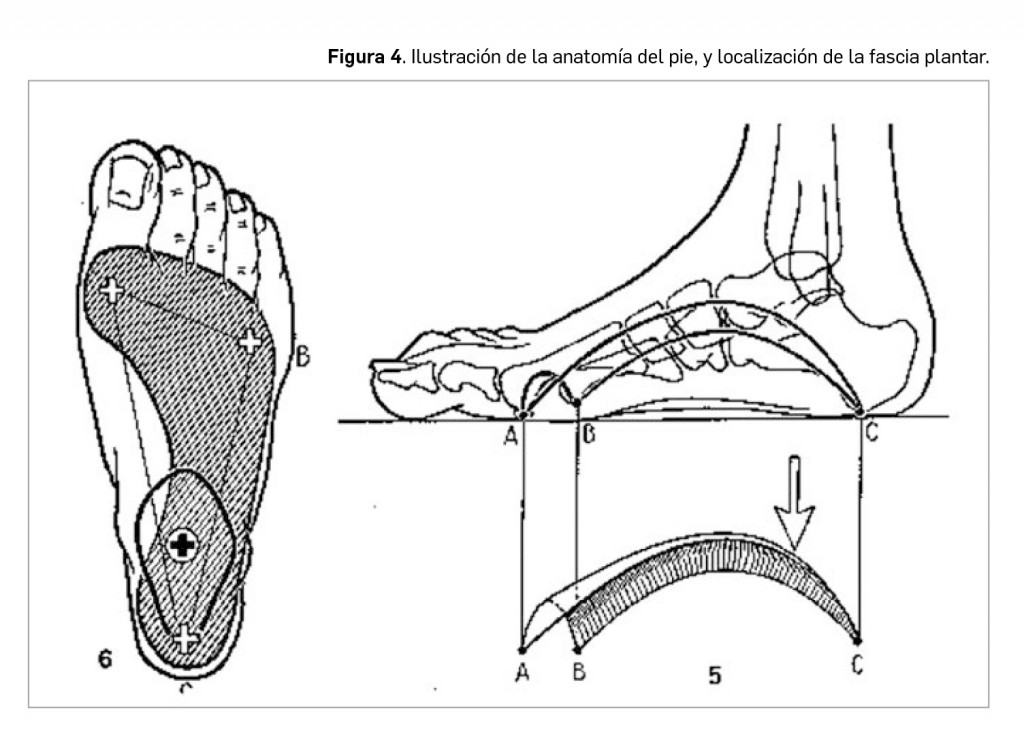
La fascia actúa como una transmisora de fuerzas, una repartidora de las tensiones que provienen del sistema sural de la pierna, además de mantener la forma de la planta del pie. Por ello, en el caso de pies “defectuosos” anatómicamente es más frecuente que aparezca una patología fascial. Así, los pies planos, pies cavos, el pie de Morton (el primer metatarsiano hipermóvil y el segundo radio demasiado largo), pie equino, etc., hacen que determinadas partes de la fascia sufran una mayor tensión que el resto, apareciendo dolor. En cambio, la existencia de un espolón calcáneo en la base no implica nada. Es decir, existen enormes espolones asintomáticos, así como casos de fascitis importantes que no se acompañan de espolón.
La sintomatología clínica es muy sugerente. El enfermo presenta dolor en la planta del pie, en la inserción calcánea-fascial, que empeora por la mañana justo al levantarse y mejora claramente cuando el paciente ya ha dado unos pasos, pero a lo largo del día vuelve a aparecer el dolor, el cual desaparece al tener el pie en reposo. Suele aparecer también rigidez matutina, dolor insidioso, sin historia de traumatismos previos. La incidencia y severidad de la fascitis plantar se correlacionan con la obesidad.
Factores predisponentes:
- Pie de Morton: insuficiencia del primer radio del pie, que es excesivamente móvil, y segundo metatarsiano elongado. Suele asociarse a hiperpronación y a lesiones por sobrecarga (fracturas de estrés).
- Pie pronado (pie plano): hiperflexible, asociado a movimiento subastragalino excesivo.
- Pie cavo: bóveda plantar muy elevada y rígida. Es un pie que no prona lo suficiente, lo que implica que no absorbe bien los impactos ni se adapta bien a las irregularidades del terreno.
—La sintomatología de la fascitis plantar es muy sugerente, destacando el dolor en la planta del pie que empeora al levantarse por la mañana y mejora al dar unos pasos—
Diagnóstico
El diagnóstico es fundamentalmente clínico. Las radiografías son útiles para descartar otras patologías, y en ocasiones muestran la presencia del espolón calcáneo. De forma complementaria, la resonancia magnética nuclear (RMN) ofrece imágenes de la integridad de la fascia, y la ecografía puede poner de relieve la existencia de un edema importante o bien un engrosamiento hipoecoico en la región de inserción (que puede llegar a ser mayor de 4 mm).
Para alcanzar un adecuado diagnóstico, se debe hacer un diagnóstico diferencial con los siguientes cuadros:
- Síndrome ciático (lesión de L5-S1).
- Síndrome del túnel tarsiano.
- Pinzamiento del nervio plantar lateral.
- Rotura de la fascia plantar (tras traumatismo severo o inyecciones repetidas de esteroides).
- Fractura por estrés del calcáneo (dolor a la compresión lateral del calcáneo, antecedentes de sobrecarga).
- Apofisitis calcánea (enfermedad de Sever): afecta a niños entre 6-10 años, generalmente obesos y muy activos. El dolor es más severo tras la actividad deportiva, y aparecen en la región posterior del calcáneo, alrededor de la inserción del tendón de Aquiles.
- Enfermedades sistémicas (artritis reumatoide, espondilitis anquilopoyética, artritis psoriásica, síndrome de Reiter, etc.).
Tratamiento
El tratamiento se basa en la corrección ortopédica adecuada, mediante taloneras semirrígidas de silicona con un orificio de descarga y el uso de calzado que sujete pero no aprisione el talón. Los vendajes funcionales pueden ser de utilidad, y las infiltraciones, aunque dolorosas, consiguen buenos resultados. En tal caso, se debe infiltrar el punto de máximo dolor, habitualmente situado junto a la inserción calcánea. En ocasiones, puede romperse la fascia tras infiltraciones repetidas, lo que haría que se perdiera la forma de la bóveda plantar, pero también conllevaría la desaparición del dolor. El tratamiento quirúrgico no suele ofrecer grandes resultados.
Además, se recomienda instaurar un plan de cuidados complementario, basado en estiramientos del aquileo y de la musculatura intrínseca del pie durante 6-8 semanas, realizando a diario los ejercicios durante al menos 10-20 minutos. Además, cada noche, es conveniente aplicar hielo (masaje o “cold pack”) en la zona plantar del talón durante 15-20 min, durante dos semanas. La recomendación general puede implicar: reposo relativo en lo referente a actividades físicas, administración de AINEs como coadyuvantes para la sintomatología, vendajes funcionales (solo producen alivio momentáneo) y férulas de reposo nocturno (que mantienen el pie en flexión de 90º para prevenir la contracción de la fascia durante el sueño, y son capaces de aliviar el dolor en el 80% de los casos).
Por último, se debe tener en cuenta varias consideraciones:
- La iontoforesis parece eficaz en el inicio (mejoría a las 2 semanas) pero no hay diferencias a medio plazo (6 semanas).
- Las infiltraciones con esteroides tampoco son útiles a medio plazo.
- La cirugía mediante fasciotomía plantar endoscópica debe reservarse para casos sin respuesta a un tratamiento conservador completo, amplio y duradero (≥ 6-8 meses).
En general, se debe iniciar el tratamiento corrigiendo errores de entrenamiento del paciente deportista, con la recomendación de reposo relativo y la evaluación de las actividades del paciente y del calzado empleado; se busca corregir errores biomecánicos e iniciar un programa de estiramientos. Si no existe mejoría, se puede comenzar con las férulas de reposo nocturno. Los AINEs deben ser considerados a lo largo del proceso, aunque solo van a ofrecer alivio sintomático.
Lesiones en el tendón de Aquiles
Tendinopatía aquílea
El tendón de Aquiles es el más poderoso del organismo, pudiendo llegar a transmitir hasta 900 kg de carga. No obstante, en él existe un punto débil, una zona hipovascularizada situada a 2-6 cm de la inserción calcánea donde con mayor frecuencia se producen las roturas y las tendinitis. La tendinopatía aquílea supone una inflamación dolorosa que afecta también con frecuencia a la unión músculo-tendinosa del gemelo interno, a la porción tendinosa que se corresponde con la articulación del tobillo y a la inserción calcánea.
La etiología es la sobrecarga del tendón y los microtraumatismos repetidos, así como la realización de determinados movimientos anormales durante la carrera. Entre sus factores predisponentes, encontramos:
- Envejecimiento tendinoso (degeneración del tendón).
- Mala alineación pierna/pie o talón/antepié.
- Sobre-pronación dinámica (movimiento rotacional anormal del tendón en la fase de apoyo plantar).
- Mala flexibilidad de gemelos y sóleo.
- Traumatismos repetidos.
- Aumento desmesurado del ritmo de entrenamiento.
- Cambios de terreno (arena, terrenos duros, mala absorción de impactos).
- Carrera sobre superficies irregulares.
- Calzado inadecuado o excesivamente desgastado.
Se ha descrito que un calentamiento demasiado breve o un incremento rápido de las distancias durante el entrenamiento son factores de riesgo para las roturas tendinosas, especialmente en “deportistas de fin de semana” mayores de 35 años.
Sintomatología
La afectación del tendón de Aquiles cursa normalmente con dolor al inicio de la actividad física, también en reposo cuando está más avanzada. En los estadios finales el dolor es constante, no tiene relación con el ejercicio e impide cualquier actividad del paciente. Obviamente, el dolor aumenta al tensar el tendón (ponerse “de puntillas”), al palpar el mismo o al estiramiento pasivo. La aparición de un dolor extraordinariamente agudo en la cara posterior y tercio distal de la pierna (sensación de haber recibido el impacto de una pedrada), acompañado de equimosis inmediata e imposibilidad para la deambulación, es muy sugerente de rotura.
En esta tendinopatía puede observarse una zona inflamada en la inserción calcánea, dolorosa, a 4-5 cm de la inserción. Se observa crepitación (ruidos o chasquidos) a la palpación del tendón y la existencia de nódulos en el cuerpo del tendón debe hacernos sospechar una rotura parcial. En ocasiones, se puede palpar un surco (“signo del hachazo”) en la zona de la rotura. También se observa que la compresión de los gemelos, estando el paciente en decúbito prono, no aumenta la flexión del pie (signo de Thompson), es decir, hay una pérdida del equinismo o capacidad de modificar el ángulo del pie debido a que la rotura tendinosa impide la transmisión de la fuerza desde los gemelos –a través del tendón– al pie (Figura 5).
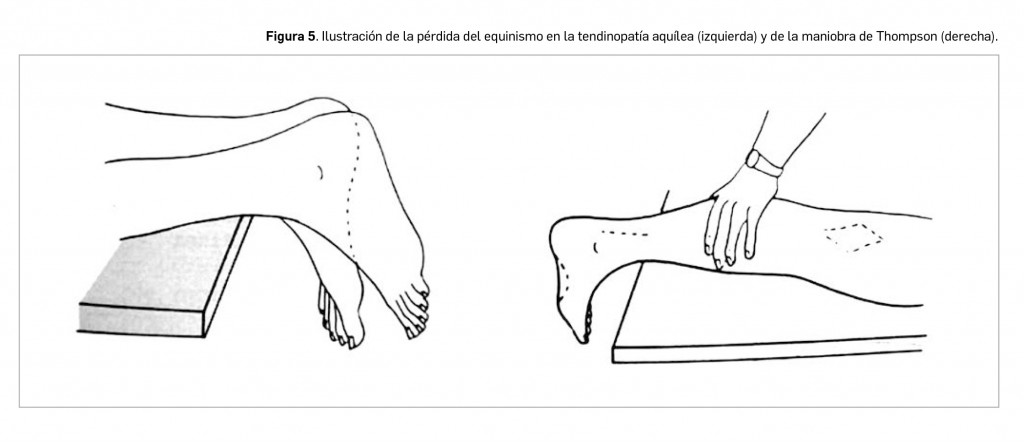
Además, se puede producir también la inflamación de la bursa preaquílea (retrocalcánea), generalmente por micro-traumatismos repetidos en dorsiflexión del tobillo o acompañando a roturas parciales del tendón. Aparece entonces dolor selectivo en la región pre-aquílea, con edema y tumefacción de la misma, que aumenta con los movimientos laterales y la dorsiflexión del tobillo.
Diagnóstico
En esta patología es muy importante el diagnóstico diferencial, entre bursitis aquilea, tendinitis y rotura del tendón, pues orientará a las diferentes opciones terapéuticas. Además, se deberá descartar lesiones musculares de gemelos y sóleo, enfermedad de Sever (apofisitis posterior del calcáneo, en niños) y enfermedad de Haglund (exostosis de la parte superoposterior del calcáneo).
Entre las pruebas complementarias a la exploración física, la radiología simple puede mostrar un aumento del grosor del tendón, calcificaciones de partes blandas, o bien lesiones óseas. La ecografía puede definir lesiones de roturas parciales o bursitis; y la RMN permite una mayor definición de partes blandas.
Tratamiento y plan de cuidados
La rotura –parcial o completa– del talón de Aquiles debe ser siempre evaluada por un especialista, y generalmente el tratamiento es quirúrgico.
En las tendinitis es preceptiva la interrupción de la actividad física durante 2-4 semanas, asociando crioterapia en el inicio, la administración de AINES por vía oral, estiramientos de los grupos musculares de la pierna y, desde luego, la revisión del calzado y el plan de entrenamiento. En el retorno a la actividad, puede ser de utilidad el uso de vendajes funcionales que mantengan el pie en ligero equinismo, de manera que el tendón no se sobrecargue. Siempre que sea posible se atenderá a la corrección de los problemas biomecánicos preexistentes (taloneras adecuadas, cuñas, plantillas de descarga, alineación de segmentos, etc.). Los ultrasonidos, láser y otros tipos de terapia física también han demostrado su utilidad.
En el caso de la bursitis, estaría indicada la infiltración con esteroides, siempre que se respete de manera adecuada el tendón. En los casos más complicados, se recurre a la exéresis (extirpación) de la bursa.
Bursitis retrocalcánea
La bursitis retrocalcánea puede aparecer en el seno de una enfermedad generalizada, aunque también es la consecuencia del sobreuso. La sobrecarga en la región aquílea conducente a una bursitis retrocalcánea puede deberse al uso de un calzado inapropiado en deportistas que entrenan mal (con muchos cambios de terreno, con un programa donde la progresión no sea la adecuada, aumentan demasiado la carga de trabajo en tiempo o kilómetros) o incluso al simple cambio de zapatillas.
Aparece entonces dolor selectivo en la región situada por debajo del tendón de Aquiles, región que puede aparecer inflamada y dolorosa a la palpación. En la lesión del tendón de Aquiles existe dolor a la contracción isométrica del tendón, a la palpación y al estiramiento pasivo, signos éstos que no aparecen en la bursitis; en cambio, este cuadro se caracteriza por la palpación de una bursa dolorosa e inflamada, con dolor localizado en la región retrocalcánea anterior al tendón, y no sobre el tendón. En el caso de que exista una lesión del tendón, están contraindicadas las infiltraciones en el cuerpo del mismo, ya que favorecerían su ruptura.
Metatarsalgia de Morton
Neuroma de Morton
Se trata de un cuadro que aparece debido a la lesión del nervio interdigital, bien por la compresión del mismo entre las cabezas metatarsianas, por luxación de los tendones flexores que pueden comprimir una rama nerviosa, o bien por un neuroma, higroma, ganglión o tumoración que puede comprimir el nervio. Es más frecuente entre las cabezas del tercer y cuarto metatarsiano, a veces entre el segundo y el tercero. Así pues, la denominación correcta sería la de metatarsalgia de Morton, ya que el neuroma es una de las causas de metatarsalgia, pero no la única. De hecho, la causa más frecuente de metatarsalgia es la mecánica, que provoca el atrapamiento del nervio y su opresión en la flexión dorsal del pie, entre la cabeza de dos metatarsianos.
Cursa con dolor agudo, intenso, acompañado de parestesias importantes en los dedos afectados, que obliga al enfermo a detener la marcha, quitarse el zapato, y darse un masaje manual que hace ceder el dolor. En cambio, el dolor aumenta al caminar por terrenos irregulares y se despierta cuando se aprietan las cabezas de los metatarsianos.
Con respecto a su diagnóstico, puede realizarse a través de RMN, si bien una RMN negativa no descarta la existencia de un neurofibroma. También puede ser útil la ecografía, que mostraría formaciones hipoecoicas entre las cabezas de los metatarsianos siempre que sean mayores de 5 mm. El diagnóstico diferencial se debe establecer con los higromas simples (hacen relieve blando bajo las cabezas de los metatarsianos), la osteonecrosis de la cabeza metatarsiana (el dolor es menor o está ausente, y se puede palpar la deformidad de la cabeza en los casos muy evolucionados) o una fractura de estrés en su estado inicial.
El tratamiento es quirúrgico si se demuestra la existencia de un neurofibroma. El calzado debe ser de suela blanda con una plantilla delgada de silicona blanda para amortiguar el impacto del pie sobre el suelo, o bien plantearse la utilización de una plantilla de descarga para los metatarsianos. Las infiltraciones1 están indicadas para aliviar el dolor y mejorar la funcionalidad y son una buena opción terapéutica, aunque no definitiva, que se realiza por la cara dorsal del pie, en el espacio intermetatarsiano (localizando el espacio intermetatarso-falángico), en la zona de máximo dolor, con la aguja perpendicular al plano cutáneo. El paciente no precisará ningún cuidado especial tras la infiltración, pero no es prudente poner más de tres inyecciones, separadas 10-15 días entre sí.
{kind=link}
{kind=link}