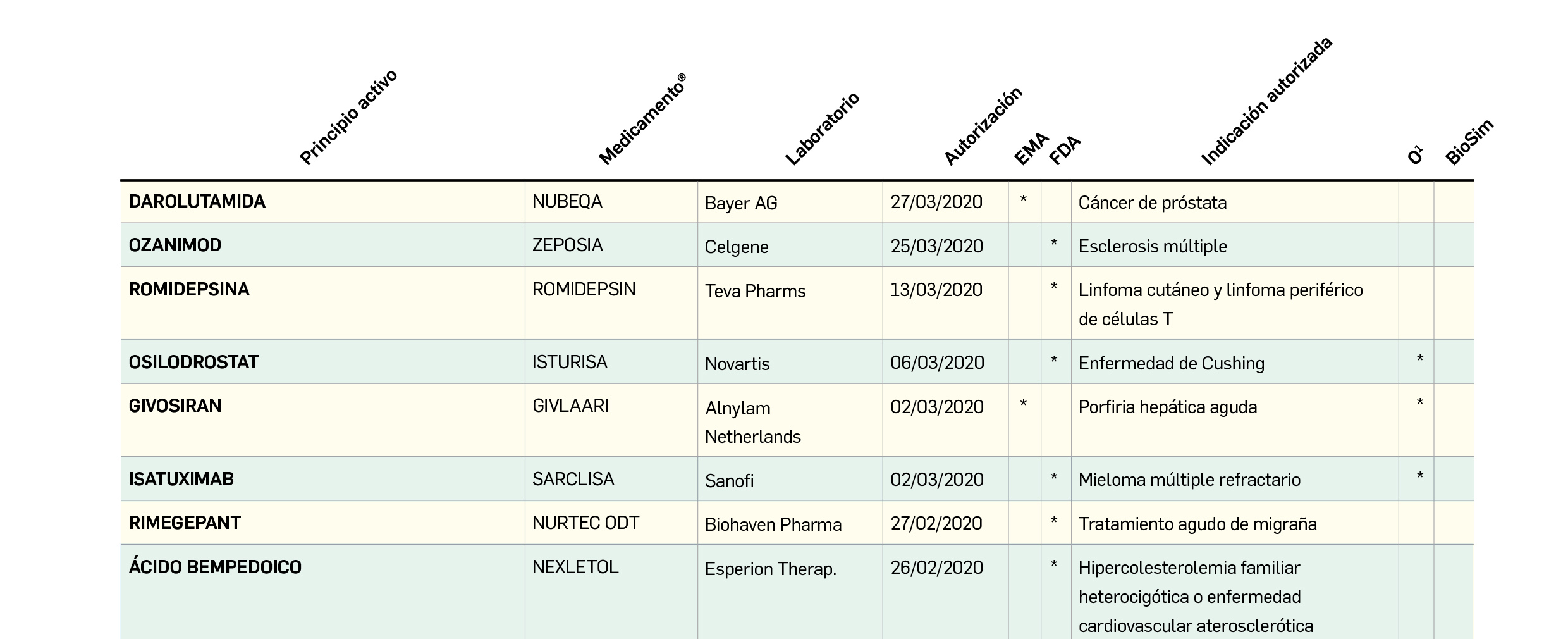
MEDICAMENTOS DE USO HUMANO AUTORIZADOS EN LA UNIÓN EUROPEA (EMA) Y ESTADOS UNIDOS (FDA) DURANTE LOS ÚLTIMOS DOCE MESES, CON NUEVOS PRINCIPIOS ACTIVOS O BIOSIMILARES QUE AÚN NO ESTÁN COMERCIALIZADOS EN ESPAÑA
Número 432, Abril 2020
MEDICAMENTOS DE USO HUMANO AUTORIZADOS EN LA UNIÓN EUROPEA (EMA) Y ESTADOS UNIDOS (FDA) DURANTE LOS ÚLTIMOS DOCE MESES, CON NUEVOS PRINCIPIOS ACTIVOS O BIOSIMILARES QUE AÚN NO ESTÁN COMERCIALIZADOS EN ESPAÑA
Alertas debidas a defectos de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En BOT PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS.
Además de los listados mensuales que podemos consultar en PAM, en BOT PLUS se incorpora la información que publica la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) relativa a notificaciones sobre seguridad y/o calidad de los medicamentos. Mediante un pictograma específico se pueden visualizar de forma rápida medicamentos afectados por alguna alerta de seguridad o de defectos de calidad, con tan solo entrar en su ficha.
Al acceder a la ficha de un medicamento afectado por una retirada, se visualiza un mensaje con la advertencia “Medicamento con lotes retirados por Alerta AEMPS. Ver más información en “Alertas Calidad”.

Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.

De forma interesante, dicha información se puede explotar a través de la Búsqueda Libre de BOT PLUS para obtener listados de todos los medicamentos afectados por alertas de calidad que implica la retirada (o también la inmovilización) de sus lotes en un momento dado.
Esta codificación de los lotes retirados es una información puesta a disposición de todos los usuarios, con el objetivo de ofrecer una nueva información capaz de integrarse con otros sistemas de información y mejorar la gestión e identificación de estos medicamentos, en los que la labor asistencial y de control del farmacéutico es fundamental.
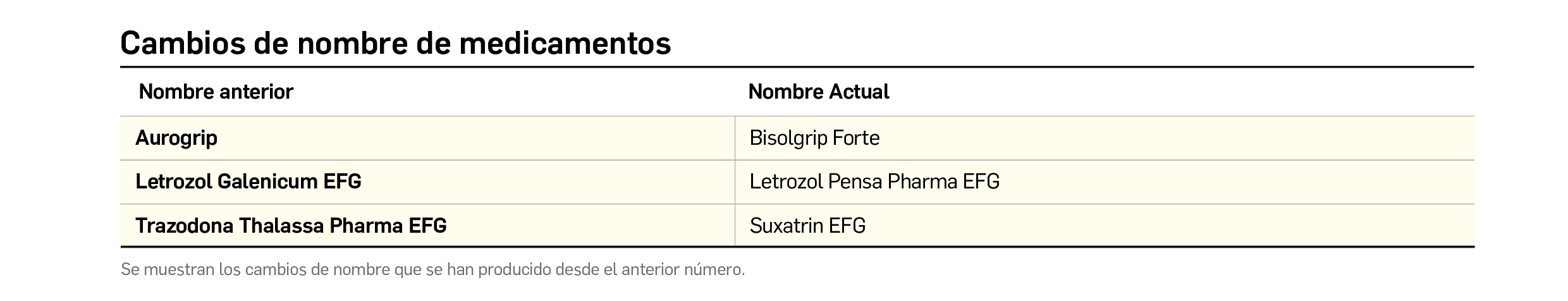
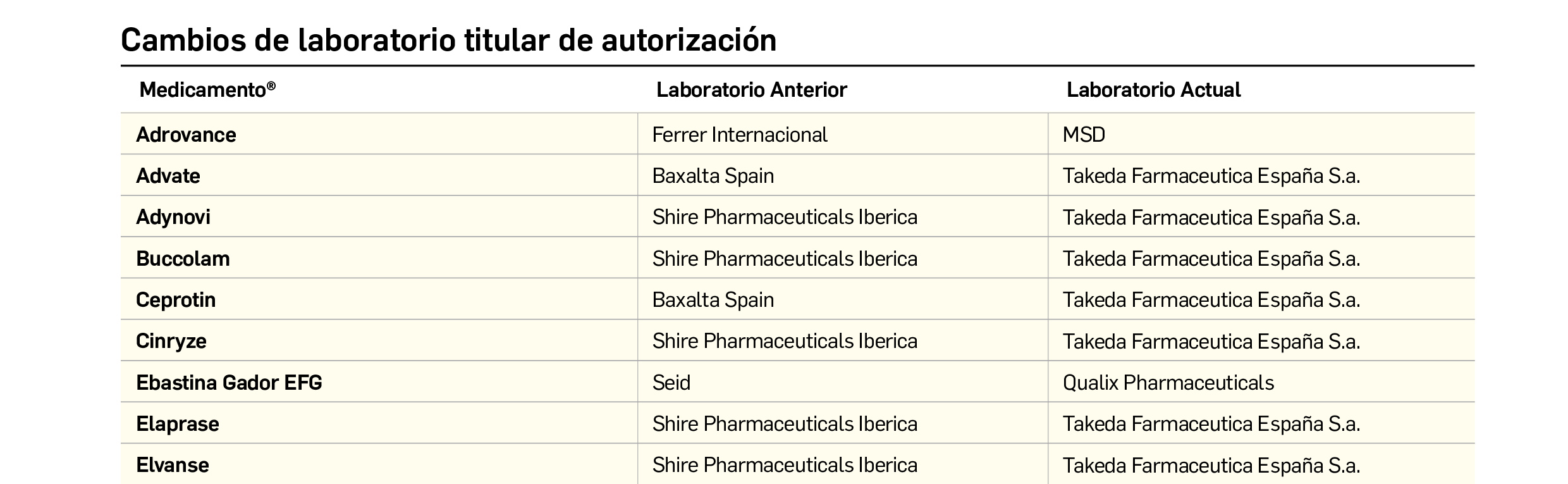
Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
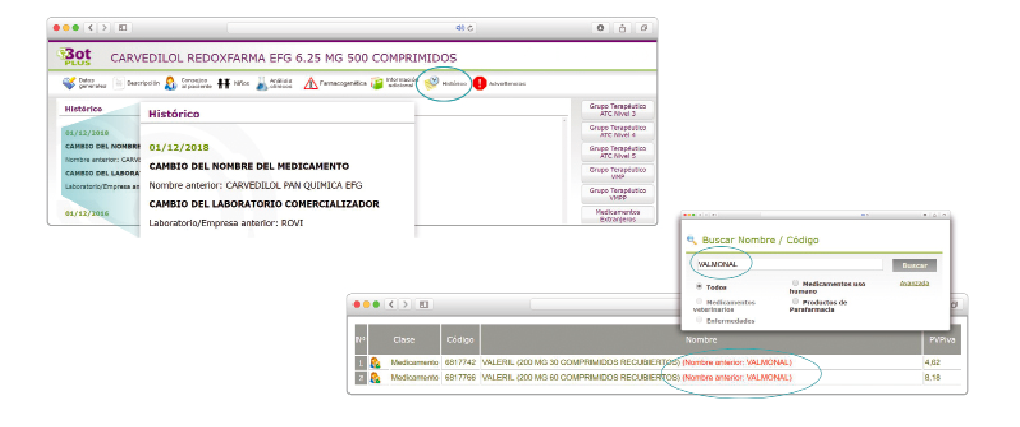
Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.
_______________________________________________________________________________________________
Valoración de la innovación terapéutica en PAM
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
El rucaparib es un nuevo inhibidor selectivo de poli-ADP-ribosa polimerasas (PARP-1, -2 y -3), enzimas implicadas en los mecanismos de reparación del ADN, tanto en células normales como tumorales: mediante su inhibición, la citotoxicidad de rucaparib –que ha demostrado ser independiente de la presencia de mutaciones en los genes BRCA– se debe a un aumento de la formación de complejos PARP-ADN, conducente a un daño del ADN y a la activación de la apoptosis y la muerte celular. Por ello, el medicamento ha sido autorizado en el tratamiento de mantenimiento, en monoterapia, de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, en recidiva, sensible al platino, que responde completa o parcialmente a la quimioterapia con platino. También está indicado en monoterapia para el tratamiento de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, con mutación BRCA (germinal y/o somática), sensible al platino, en recaída o progresión, que hayan sido tratadas con dos o más líneas previas de quimioterapia con platino y que no son capaces de tolerar más quimioterapia a base de platino.
En un amplio ensayo pivotal de fase 3, administrado como monoterapia de mantenimiento, rucaparib ha conseguido prolongar la supervivencia libre de progresión en 5,4 meses frente a placebo (hasta más de 8 meses en revisión por comité independiente), reduciendo en un 64% el riesgo de progresión de la patología; ese beneficio, que parece inferior en pacientes sin mutación BRCA, aumentaba hasta los 11,2 meses en la cohorte de pacientes con BRCA mutado. No obstante, la ausencia de datos maduros impide confirmar un beneficio en supervivencia global con el fármaco. Por otra parte, los datos clínicos relativos a su indicación en tratamiento de pacientes pre-tratadas en recaída derivan de 2 ensayos abiertos no controlados de fase 2 y no son confirmatorios. El análisis conjunto de los datos reveló una tasa de respuesta objetiva con el tratamiento de rucaparib del 54,7% (del 44% en revisión por comité independiente), la mayoría (46%) respuestas parciales, y la mediana de la duración de la respuesta fue de 9,7 meses; la eficacia fue mayor en el subgrupo de pacientes sensibles a platino (TRO de 64,6%), el único incluido en la indicación autorizada.
A grandes rasgos, rucaparib muestra una tolerabilidad similar a olaparib y niraparib, con un perfil toxicológico importante pero clínicamente manejable. La mayoría de reacciones adversas son leves-moderadas y destacan, por su frecuencia, los eventos adversos de naturaleza gastrointestinal y hematológica (náuseas, vómitos, anemia, neutropenia); entre los eventos graves, sobresale la anemia, la neutropenia febril y la fatiga. Con los tres fármacos se han descrito casos de síndrome mielodisplásico y/o leucemia mieloide aguda, y se hace necesario considerar este riesgo y conocer, en futuros estudios, más datos sobre los posibles efectos a medio y largo plazo del tratamiento con rucaparib.
En resumen, rucaparib ha demostrado un beneficio relevante frente a placebo en el mantenimiento pos-quimioterapia y parece que viene a representar una alternativa terapéutica más a olaparib y niraparib entre dos intervalos de quimioterapia en pacientes con buen estado funcional (permitirá que las pacientes se beneficien en mayor medida de un nuevo régimen posterior basado en platino); los datos clínicos disponibles no avalan el uso secuencial de los inhibidores de PARP ni permiten establecer entre ellos una superioridad en términos supervivencia (sobre todo en pacientes con BRCA no mutado) o diferencias importantes en el balance beneficio/riesgo. En tratamiento de pacientes pre-tratadas en recaída, los datos de rucaparib son limitados y requieren confirmación, pero sugieren que puede dar lugar a tasas de respuesta similares a otras alternativas de quimioterapia disponibles (doxorubicina liposomal pegilada con o sin trabectedina, o topotecán) con un mejor perfil de seguridad y de adherencia terapéutica. En definitiva, sin incorporar ninguna novedad mecanística respecto a otros inhibidores de PARP usados frente al cáncer de ovario, tampoco parece que vaya a aportar una innovación clínica notable en su tratamiento.
El carcinoma de ovario ocupa el sexto-séptimo lugar entre las neoplasias más frecuentes que afectan a la mujer y constituyó en 2018 el 3,7% de los nuevos diagnósticos de cáncer en las mujeres españolas (con una estimación de 3.645 casos para el año 2020), en línea con la incidencia a nivel europeo (4,0%; mayor en los países del norte de Europa) y mundial (3,6% del total de casos de cáncer). Se estima que en 2018 vivían en España 9.710 mujeres con cáncer de ovario (prevalencia a los 5 años), lo que representa un 2,7% del total de mujeres con cáncer.
A nivel global, la OMS informa de que en 2018 se produjeron algo más de 295.400 casos en todo el mundo, y se prevé que esta cifra supere los 430.000 para el año 2040 (IARC, 2018). Además, el carcinoma de ovario es el cuarto tumor más letal entre todos los tumores, y el de mayor tasa de mortalidad entre los que afectan al aparato ginecológico femenino, habiendo supuesto en 2018 en torno a un 4,4% del total de la mortalidad en España respecto al total de tumores en mujeres (1.949 muertes por este tipo de cáncer), con cifras similares en la Unión Europea y a nivel mundial (SEOM, 2020).
En este sentido, se estima que el riesgo de desarrollar cáncer de ovario a lo largo de la vida es de un 1,8%. Dicho riesgo está relacionado con el estado posmenopáusico y aumenta a partir de la quinta década (>80% de casos se diagnostican a partir de los 50 años) con un máximo en la octava; la mediana de edad al diagnóstico se sitúa en 63 años a nivel mundial. La supervivencia global a los 5 años tras el diagnóstico se sitúa en Europa en un 44% y en Estados Unidos en un 47%, reduciéndose al entorno del 30% para las pacientes diagnosticadas en fases avanzadas (Siegel et al., 2019). La mayoría de las pacientes mueren entre los 3 y los 4 años tras el diagnóstico.
Bajo el término cáncer de ovario se designa cualquier tumor maligno que se origina en el ovario. Debido a la localización pélvica de estos órganos, su principal vía de extensión es hasta –y a través de– las estructuras pélvicas adyacentes, como las trompas de Falopio. El desprendimiento de células neoplásicas a la cavidad peritoneal da lugar a su diseminación peritoneal, muy frecuente, con extensión por toda la cavidad abdominal. Por vía linfática se puede diseminar a los ganglios retroperitoneales e iliacos y, finalmente, con menor frecuencia y en estadios avanzados, se puede extender por vía hematógena a hígado, pulmón, hueso y cerebro.
Aproximadamente el 85-90% de los tumores primarios malignos del ovario son de origen epitelial (carcinomas) y derivan del epitelio de superficie del ovario (Figura 1), específicamente de los quistes de inclusión. Los tumores no epiteliales constituyen menos del 10% de los tumores ováricos y, entre ellos, se encuentran los que afectan a los cordones sexuales y los tumores de células germinales.
El carcinoma epitelial de ovario es una enfermedad heterogénea, en la que los distintos subtipos histológicos representan en realidad enfermedades diferentes desde el punto de vista clínico, patológico y molecular. Estas diferencias se han tratado de englobar en dos amplias categorías: los tumores tipo I y los tumores tipo II, grupos que hacen referencia a las vías tumorigénicas y no a la histopatología. En términos histopatológicos, los tumores epiteliales de ovario más comunes son de tipo seroso1 papilar (80%), seguido de los endometrioides (10%), los de células claras (5%) y los mucinoides (2%); los de Brenner (o células transicionales) y el carcinosarcoma (tumor maligno mülleriano mixto) son raros.
Las manifestaciones clínicas iniciales son vagas e inespecíficas y comunes a muchos otros procesos, sobre todo en estadios tempranos, lo cual dificulta su diagnóstico, que se suele hacer en estadios avanzados (III o IV) hasta en el 60-80% de los casos. Las pacientes diagnosticadas de cáncer de ovario localizado pueden tener pocos o ningún síntoma relacionado con su enfermedad, por lo que representan un desafío diagnóstico. Los síntomas que se presentan con mayor frecuencia y gravedad se refieren a clínica gastrointestinal inespecífica (dolor abdominal, distensión abdominal, estreñimiento, pesadez posprandial), sangrado vaginal, alteraciones de la menstruación, dispareunia (coito doloroso), astenia, anorexia, náuseas, dolor de espalda o poliaquiuria (micción frecuente). En la exploración física es común la palpación de una masa pélvica y la presencia de ascitis en la enfermedad avanzada.
Se han identificado diversos factores de riesgo asociados al desarrollo del carcinoma de ovario, como la raza blanca, la nuliparidad, la infertilidad, la menarquia precoz y la menopausia tardía, el uso de terapia hormonal sustitutiva, algunos factores ambientales (como el talco y el tabaco), la endometriosis y la obesidad.
Mención especial merecen los factores genéticos, que son responsables directos de un 10-15% de los carcinomas de ovario. Alrededor del 75% de las familias con predisposición genética tiene el síndrome de cáncer de mama y ovario familiar, con pérdida de la función de los genes supresores de tumores de cáncer de mama (BRCA) BRCA1 y BRCA2, involucrados en la reparación del ADN; se estima que 1 de cada 400-1.000 mujeres son portadoras de una mutación germinal heredada en los genes BRCA1 y BRCA2, en cuyo caso presentan un mayor riesgo de desarrollar cáncer de mama y ovario (el riesgo de este último aumenta a un 27-44%). Estas mutaciones constituyen, pues, el factor de riesgo más importante para el cáncer de ovario. Se estima que una mujer portadora de una mutación en el gen BRCA1 presenta un riesgo de desarrollar el cáncer de ovario a los 70 años del 59% y del 14,5% si el gen mutado es el BRCA2; además, las mujeres portadoras pueden presentar la enfermedad más precozmente (frecuentemente en la década de los 50).
El síndrome de cáncer de colon hereditario no polipósico (HNPCC, síndrome de Lynch tipo II) es también responsable del 10-15% del resto de los casos de carcinomas de ovario hereditarios y está asociado a una mutación en los genes MSH-2, MLH-1 o MSH-6, por la que además de incrementar el riesgo de cáncer de colon o de endometrio, aumenta el riesgo de carcinoma de ovario.
En el lado contrario, entre los factores protectores se han descrito la paridad, el uso de anticonceptivos orales, la lactancia materna y la realización de histerectomía y ligadura de trompas.
Los principales factores pronósticos son la edad, el estadio tumoral, el volumen residual del tumor tras la cirugía, el subtipo histológico y el grado y los niveles de CA-125 (una glucoproteína). El subtipo histológico de células claras y el mucinoso son los de peor pronóstico; por su parte, el endometrioide presenta mejor pronóstico que el seroso. El grado histológico es de particular importancia en las pacientes con estadios iniciales, ya que en aquellos con estadio I pobremente diferenciado (grado 3) tienen peor supervivencia que los de alto grado. La presencia de mutación de los genes BRCA1 y BRCA2 –y, sobre todo, de BRCA2– es un factor pronóstico favorable, incrementando la supervivencia libre de progresión tumoral y la supervivencia global de forma significativa.
Con respecto al tratamiento, la cirugía es el abordaje terapéutico inicial del carcinoma de ovario en estadios tempranos de desarrollo (I-II). Se inicia con una laparotomía reglada, diagnóstica, de estadiaje y terapéutica. Para ello, la cirugía ha de incluir inspección y palpación de toda la superficie peritoneal, histerectomía total, doble anexectomía bilateral, omentectomía, linfadenectomía pélvica y paraaórtica bilateral hasta las venas renales, biopsias del peritoneo pélvico y goteras paracólicas, y recogida de muestra de líquido peritoneal; la apendicectomía se recomienda en los tumores mucinosos.
La quimioterapia adyuvante es el tratamiento aplicado tras la intervención quirúrgica en la mayoría de casos. Se administra en ausencia de enfermedad detectable con el objetivo de eliminar la enfermedad microscópica y disminuir significativamente las tasas de recaída de la enfermedad, excepto en los pacientes con estadio IA o IB de grado I, en quienes la cirugía sola es prácticamente curativa y la supervivencia a los 5 años es de un 90%. Por el contrario, para pacientes con grado II (pobre diferenciación celular), estadio IC o II o histología de células claras, el riesgo de recaída es de un 20-30% y estaría indicado un tratamiento de quimioterapia tras la cirugía. Actualmente, el esquema de tratamiento estándar en los estadios iniciales del cáncer de ovario se basa en la asociación de un derivado de platino (cisplatino o carboplatino) con paclitaxel (o docetaxel), durante 3-6 ciclos.
El objetivo de la cirugía en estadios avanzados (IIIb-IIIc o IV) del cáncer de ovario es conseguir en primer lugar la reducción máxima de la masa tumoral, eliminándola macroscópicamente por completo o dejando una mínima porción de la misma. Si existe enfermedad voluminosa o el estado físico de la paciente es malo, se opta por la quimioterapia neoadyuvante, consistente en la administración de quimioterapia previamente a la cirugía, con el objetivo de poder realizar después, en pacientes respondedoras, una cirugía citorreductora de máximo esfuerzo; en pacientes con mal estado general, permite reducir las complicaciones y la dificultad de la cirugía. La quimioterapia neoadyuvante (basada en platino) no debe aplicarse más allá de 3-4 ciclos, puesto que cada ciclo adicional restaría supervivencia. Por ello, se reserva para pacientes con mal estado general o en quienes se prevea que la obtención de una citorredución óptima no va a ser posible por la extensión de la enfermedad.
En líneas generales, el esquema estándar actual de tratamiento quimioterapéutico en primera línea es la aplicación de 6 u 8 ciclos de paclitaxel más carboplatino2 (en ocasiones combinados con bevacizumab en caso de enfermedad avanzada en estadios IIIb/c-IV), tras los que se pueden alcanzar medianas de supervivencia libre de progresión de hasta 20 meses (más comúnmente 16-18) y medianas de supervivencia global de hasta 57 meses en pacientes en las que se ha conseguido una citorredución óptima. Sin embargo, hay que recordar que más de un 75% de los casos de cáncer de ovario se diagnostica en estadios avanzados (III-IV) y, a pesar de la mejora en los tratamientos y las altas tasas de respuesta (del 70-80%), la mayoría de las pacientes recaerán en los 2 primeros años: las tasas de recaída para el cáncer de ovario epitelial son de un 62-70%, pudiendo aumentar hasta el 85% en pacientes diagnosticadas con enfermedad más avanzada, convirtiéndose en una enfermedad incurable en el 50-60% de los casos.
La justificación para el uso de la quimioterapia intraperitoneal la constituye el hecho de que el carcinoma de ovario se disemina fundamentalmente por siembra peritoneal a toda la cavidad abdominal. Con ella, se consigue una mayor concentración de los fármacos en la cavidad abdominal, aunque la penetración en el tejido tumoral puede ser limitada, por lo que se reserva para pacientes en las que se haya conseguido una citorredución óptima y presenten enfermedad residual menor de 1 cm. Los principales inconvenientes son la posible compartimentación de la cavidad abdominal, su toxicidad y las potenciales complicaciones asociadas al catéter intraperitoneal, necesario para su administración.
En los casos de enfermedad en recaída tras un primer tratamiento (quirúrgico más quimioterapéutico), el objetivo de la quimioterapia es aumentar la calidad de vida, controlar los síntomas y aumentar la supervivencia libre de enfermedad (SLP) y supervivencia global (SG). En este sentido, el factor más importante a considerar para la decisión sobre el tratamiento es el tiempo transcurrido desde la última administración de platino (intervalo libre de platino), que condicionará la respuesta a ulteriores tratamientos: cuanto mayor sea el intervalo, mayor será la sensibilidad a un nuevo esquema basado en platino. Así, se han establecido varias categorías: enfermedad refractaria a platino (progresión durante o en las 4 últimas semanas tras la finalización del platino), resistente (progresión en menos de 6 meses), parcialmente sensible (recaída entre 6 y 12 meses) y sensible (recaída tras más de 12 meses de la última dosis de platino).
Por lo general, el beneficio de la segunda cirugía citorreductora en caso de recurrencia de la enfermedad es controvertido y en la práctica clínica la decisión, por ahora, se basa en la respuesta potencial al tratamiento adyuvante, dependiente del intervalo de tiempo hasta progresión tras quimioterapia basada en platino, el tratamiento previo y la respuesta obtenida, la toxicidad residual, las comorbilidades y preferencias de la paciente.
Como se ha indicado, tanto en pacientes sensibles como resistentes a platino que han recaído, otra opción de tratamiento –bien en monoterapia o en combinación con quimioterapia– es el bevacizumab (Avastin®), un anticuerpo monoclonal antiangiogénico que bloquea todas las isoformas del VEGF (factor de crecimiento endotelial vascular), que se sobreexpresa frecuentemente en el carcinoma de ovario y se asocia con progresión de la enfermedad, formación de ascitis y peor pronóstico. Se han registrado tasas de respuesta de hasta un 17% en monoterapia en pacientes politratadas y resultados aún mejores en combinación con quimioterapia (paclitaxel y carboplatino) en líneas avanzadas. Por ejemplo, bevacizumab en combinación con gemcitabina más carboplatino ha demostrado un incremento de la mediana de la SLP de 4 meses en comparación con gemcitabina más carboplatino solos en pacientes con cáncer de ovario recurrente sensible a platino (Aghajanian et al., 2012).
Otra diana farmacológica interesante en esta indicación son las denominadas PARP (poli ADP-ribosa polimerasas), una familia de enzimas implicada en la reparación de la cadena simple del ADN, tanto en células normales como neoplásicas. En las células normales existe un mecanismo alternativo para reparar el ADN que requiere la intervención de dos proteínas, BRCA1 y BRCA2, facilitando la vía de la recombinación homóloga. En el carcinoma de ovario hereditario, los genes BRCA1 y BRCA2 están mutados; sin embargo, en el carcinoma de ovario esporádico, se postula que puede existir también algún tipo de disfunción de esta vía, aunque no se encuentre la mutación.
Por ello, la utilización de inhibidores de PARP –como los ya disponibles olaparib (Lynparza®) y niraparib (Zejula®)– hace a las células más quimiosensibles, pues todos sus sistemas de reparación del ADN estarían bloqueados. Así, la Sociedad Española de Oncología Médica recomienda, en pacientes con cáncer de ovario epitelial seroso, de trompas de Falopio o peritoneal primario, de alto grado, sensibles a platino, el tratamiento estándar con una combinación de platino y la posible adición de bevacizumab en la primera recaída (si los pacientes no han sido tratados con bevacizumab en primera línea); en pacientes con mutación BRCA que tienen respuesta a la quimioterapia basada en platino, se debe considerar terapia de mantenimiento con olaparib (Santaballa et al., 2016) o niraparib. Ambos inhibidores de PARP se usan en monoterapia en pacientes que están en respuesta (completa o parcial) a un retratamiento con platino, independientemente del estado mutacional de BRCA.
Olaparib ha demostrado –en ensayos clínicos– la capacidad de prolongar la mediana de la SLP del tratamiento de mantenimiento en 6,9 meses (11,2 meses en el grupo de olaparib frente a 4,3 meses en el grupo de placebo) para pacientes con la mutación de BRCA. Posteriormente, se han publicado datos de SG para olaparib tras más de 5 años de seguimiento (Ledermann et al. 2016), que muestran un efecto beneficioso del tratamiento en el brazo de olaparib en comparación con el placebo, con una reducción del riesgo de muerte de un 38% (mediana de SG de 34,9 meses en el brazo de olaparib vs. 30,2 meses en el brazo de placebo).
Por su parte, niraparib ha demostrado la capacidad de prolongar la mediana de SLP en tratamiento de mantenimiento en 15,5 meses en pacientes con BRCA mutante (21,0 meses en el grupo de niraparib vs. 5,5 meses en el grupo placebo) y de 5,4 meses (9,3 vs. 3,9) en pacientes con BRCA no mutante, siendo la estimación de la tasa de SLP a los 12 meses de tratamiento del 27% (vs. 7% con placebo). Con datos aún inmaduros de SG, la eficacia de niraparib fue consistente en todos los subgrupos analizados, hubo aumentos notables en el tiempo hasta el primer tratamiento posterior de 12,6 y 4,6 meses y en el intervalo libre de quimioterapia de 13,4 y 4,1 meses a favor de niraparib en las cohortes con BRCA mutante y no mutante, respectivamente. La calidad de vida reportada por las pacientes no se vio perjudicada por el tratamiento con niraparib.
El rucaparib es un nuevo agente antineoplásico que actúa como inhibidor de las isoenzimas 1, 2 y 3 de las poli-ADP-ribosa polimerasas (PARP) –PARP1, PARP2 y PARP3– implicadas en los mecanismos de reparación del ADN, tanto en células normales como neoplásicas. El medicamento ha sido autorizado en monoterapia para el tratamiento de pacientes adultas con cáncer de ovario epitelial, de trompas de Falopio o peritoneal primario, de alto grado, sensible a platino, recidivado o en progresión, que presentan mutación de BRCA (germinal y/o somática), que han recibido dos o más líneas de quimioterapia previa basadas en platino y que no pueden tolerar más quimioterapia a base de platino. También se ha autorizado en monoterapia como tratamiento de mantenimiento de pacientes adultas con cáncer de ovario epitelial, de las trompas de Falopio o peritoneal primario, de alto grado, recidivado, sensible a platino que están en respuesta (completa o parcial) a quimioterapia con platino, con independencia del estado mutacional de BRCA.
Las PARP –Poly (ADP-ribose) Polymerases, en inglés– constituyen una amplia familia de proteínas implicadas en un gran número de procesos celulares, pero fundamentalmente en los procesos de reparación del ADN y de muerte celular programada (apoptosis). La importancia de este mecanismo de reparación del ADN en el tratamiento de determinadas formas de cáncer viene determinada por el hecho de que las células sanas (no malignas) disponen de otros mecanismos alternativos a dichos mecanismos reparadores, pero algunos tipos tumorales presentan tales mecanismos alternativos3 deteriorados o inactivados, lo que impide que los daños del ADN puedan ser subsanados. Ello conduce al deterioro y a la muerte celular al activarse los mecanismos de apoptosis.
La recombinación homóloga, por ejemplo, es un tipo de recombinación genética que las células emplean ampliamente para reparar de forma precisa las roturas dañinas que ocurren en el ADN, conocidas como roturas de doble cadena. El sistema de reparación por recombinación homóloga requiere que los genes BRCA sean funcionales. La presencia de mutaciones en estos genes conlleva que la reparación del ADN dependa de vías adicionales de reparación en las que están implicadas las enzimas PARP, las cuales, si son inhibidas, no pueden llevar a cabo la reparación del ADN.
Los resultados de estudios bioquímicos in vitro han confirmado que rucaparib ejerce una inhibición potente y selectiva de la actividad enzimática de las PARP (con valores de IC50 de 0,8, 0,5 y 28 nM para PARP1, PARP2 y PARP3, respectivamente); se han descrito constantes de inhibición de PARP-1 y PARP-2 por rucaparib de 1,4 y de 0,17 nM, respectivamente. Los análisis cristalográficos demostraron que rucaparib se une al sitio catalítico activo de PARP1, donde se une normalmente el NAD+.
La inhibición de las PARP por rucaparib conduce a un aumento de citotoxicidad mediado por una mayor formación de complejos PARP-ADN, que provocan un daño dosis-dependiente del ADN y activan la apoptosis y muerte celular. Esto se ha demostrado por métodos de inmunohistoquímica en líneas celulares tumorales de ovario y de mama con mutación/deficiencia de los genes supresores BRCA1/2 a través de un mecanismo conocido como letalidad sintética, en el que se necesita la pérdida de dos vías de reparación del ADN para la muerte celular. Además, en estudios in vivo en ratones a los cuales se les habían trasplantado tumores de xenoinjerto derivados de pacientes con cáncer de ovario epitelial seroso de alto grado, rucaparib ha demostrado una capacidad de inhibir significativamente el crecimiento tumoral tanto en tumores con o sin mutaciones/deficiencias de BRCA1/2 (EMA, 2018).
El fármaco se presenta en el medicamento como la sal camsilato de rucaparib, cuyo nombre químico es el de ácido 8-fluoro-2-{4-[(metilamino)metil]fenil}-1,3,4,5-tetrahidro-6H-azepino[5,4,3-cd]indol-6-ona ((1S,4R)-7,7-dimetil-2-oxobiciclo[2.2.1]hept-1-il)metanosulfónico, que se corresponde con la fórmula C19H19FN3O · C10H16O4S y un peso molecular relativo de 555,67 g/mol. Rucaparib es el tercer miembro comercializado de la serie de inhibidores de las PARP, tras olaparib y niraparib; inicialmente también se investigó al iniparib, pero su investigación fue detenida por la compañía titular en 2013 debido a sus resultados clínicos insatisfactorios.
En términos estructurales, guarda cierto paralelismo químico (menor del que podría preverse por el similar mecanismo de acción) con olaparib o niraparib, los cuales presentan a su vez cierta similitud con la serie de los inhibidores de tirosina cinasas (“inib”), particularmente con gefitinib y otros como vandetanib, afatinib, lapatinib
o erlotinib.
Todos ellos tienen un núcleo con heterociclos nitrogenados, en los inib de tipo quinazolínico, en el caso del olaparib es ftalazínico, en el de niraparib de indazol y en el de rucaparib, un sistema tricíclico (un indol con un azepano heptaciclíclico adherido). A ese núcleo, olaparib lleva ligado un resto bencílico (que es de fenilamina en los inhibidores de tirosina cinasas) con la presencia de un átomo de flúor, niraparib presenta una piperidina unida a un anillo bencénico, y rucaparib, un resto de bencilmetilamina (Figura 2). El citado paralelismo estructural, al menos en una parte de la molécula, sugiere que el mecanismo de bloqueo inhibitorio de las enzimas diana es similar, aunque las diferencias en el resto de la molécula sean el determinante de la especificidad del sustrato que, en el caso de los “inib” son diversas tirosina cinasas, mientras que en el de los “parib” son las PARP.
En su forma de camsilato, rucaparib se presenta como un polvo blanco o amarillo pálido, no higroscópico y cristalino. Es ligeramente soluble en medio acuoso a pH entre 3 y 7, viéndose su solubilidad significativamente reducida a pH por encima de 9,5. Se han descrito dos formas poliméricas enantiotrópicas, si bien rucaparib por sí mismo no presenta centros quirales (es el contra-ión ácido canforsulfónico el que contiene 2 estereocentros).
La eficacia y seguridad clínicas de rucaparib por vía oral han sido adecuadamente contrastadas en la indicación y dosis autorizadas mediante dos ensayos clínicos de fase 2 (Estudio 10 y ARIEL2) no aleatorizados, abiertos, multicéntricos y de un solo brazo (no controlados), que evaluaron la farmacodinamia del fármaco en un total de 106 pacientes adultas (≥ 18 años) con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario avanzado con BRCA mutante que había progresado después de 2 o más líneas de quimioterapia previas, ninguna de ellas con inhibidores de PARP.
Ambos estudios incluyeron una mayoría de pacientes (75%) sensibles a platino en el tratamiento anterior (solo 6,6% fueron refractarias a platino y 19,8% resistentes a platino) y que presentaban un buen estado funcional (puntuación ECOG de 0 o 1). En términos histológicos, la práctica totalidad de pacientes presentaban tumores serosos de alto grado (91,5%), endometrioides (2,8%) y mixtos (4,7%); un 83% de las pacientes presentaban una mutación de BRCA de línea germinal y un 17% una mutación somática. Por otro lado, un criterio de exclusión fue la hospitalización debida a obstrucción intestinal –que pudiera interferir con la absorción del fármaco– en los 3 meses anteriores al inicio del tratamiento; también se excluyeron pacientes embarazadas, lactantes, que habían recibido tratamiento dentro de los 14 días previos (quimioterapia, radioterapia, inmunoterapia o vacunas), pacientes con metástasis cerebrales o enfermedad cardiaca clínicamente significativa.
Las características demográficas y de la enfermedad fueron similares en las poblaciones de pacientes de ambos estudios (Tabla 1). Se realizó un análisis conjunto de los datos de eficacia del tratamiento en todas las pacientes que recibieron al menos una dosis. La variable principal de eficacia de ambos estudios fue la tasa de respuesta objetiva (TRO) según evaluación del investigador y definida mediante el empleo de los criterios RECIST v1.1; la TRO también fue evaluada por un comité de revisión independiente. Como variables secundarias se evaluaron la duración de la respuesta (DR), la supervivencia libre de progresión (SLP) y el perfil de seguridad de rucaparib. En la Tabla 1 se recogen también los principales resultados de eficacia en el global de las pacientes.
Cabe destacar que el resultado de TRO confirmado por el investigador principal (54,7%) fue ligeramente superior a la TRO confirmada por el comité de revisión independiente (44,3%, con un 12,3 y 32,1% de respuestas completa y parciales, respectivamente), si bien los resultados de DR (mediana de 294 días vs. 288 días) y SLP (mediana de 294 días vs. 289 días) arrojados por la revisión independiente fueron mayores.
El análisis por subgrupos según el evaluador principal reveló que las 79 pacientes que eran sensibles –al menos parcialmente– al platino (55 mostraban un intervalo libre de progresión tras la última dosis de platino de ≥6-12 meses y 24 pacientes de > 12 meses) tenían TRO notablemente superiores (61,8% para las pacientes parcialmente sensibles y 70,8% en pacientes sensibles) a la de las pacientes resistentes (35,0%) y refractarias (0%) a platino. Algo similar ocurría con la DR, mayor en las pacientes platino-sensibles (mediana de 270 y 383 días en pacientes parcial y completamente sensibles, respectivamente, vs. 196 días en pacientes resistentes). La TRO fue similar en las pacientes con cáncer de ovario con BRCA mutante de línea germinal o somática y en las pacientes con una mutación en el gen BRCA1 o en el BRCA2.
La eficacia y seguridad clínicas de rucaparib como tratamiento de mantenimiento del cáncer de ovario recidivante han sido demostradas en un amplio ensayo pivotal aleatorizado de fase 3 (ARIEL3), multicéntrico y multinacional (96 centros en 11 países), con diseño doble ciego y controlado con placebo. Dicho estudio evaluó los efectos de la monoterapia con el nuevo fármaco en pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario de alto grado sensible a platino, recurrente, pre-tratadas con al menos 2 esquemas de quimioterapia basados en platino y que se encontrasen en respuesta al último de ellos (administrado en las 8 semanas previas a la aleatorización).
Un total de 564 pacientes se asignaron al azar (2:1) a recibir por vía oral rucaparib 600 mg/12 h (N= 375) o placebo (N= 189) en ciclos de 28 días, hasta progresión, toxicidad inaceptable/muerte o pérdida de seguimiento. Todas las pacientes incluidas debían de ser adultas (≥ 18 años), tener una histología del tumor epitelial serosa, endometrioide o mixta, ser sensibles al penúltimo tratamiento con platino –tras un mínimo de 4 ciclos– antes de la inclusión, presentar niveles normales de CA-125 y un buen estado funcional general. En cambio, se excluyeron pacientes con antecedentes de otros tumores, con metástasis cerebrales, que habían recibido tratamiento previo con otro inhibidor de PARP o que habían sido sometidas a citorreducción; de igual modo, se excluyeron aquellas pacientes embarazadas o lactantes, con ciertas infecciones virales crónicas o cualquier trastorno gastrointestinal que pudiera interferir en la absorción del fármaco.
En líneas generales, las características basales demográficas y médicas de las pacientes estuvieron bien balanceadas entre los dos brazos del estudio. La media de edad de las pacientes fue de 61 años (rango 36-85), la mayoría de ellas eran de raza blanca (80% caucásicas) y presentaban tumor ovárico (84%) e histología serosa (95%), con puntuación en la escala ECOG de 0 (74%). El 28% de las pacientes incluidas habían recibido ≥3 líneas previas de quimioterapia con platino (el 23% habían sido tratadas con bevacizumab), el 32% mostró una respuesta completa al tratamiento más reciente y el intervalo libre de progresión tras el penúltimo tratamiento con platino fue de 6 a 12 meses en 39% de las pacientes y de > 12 meses en 61%.
Los datos de eficacia se analizaron por intención de tratar (ITT) mediante comparaciones múltiples ordenadas, para lo cual se definieron 3 cohortes de pacientes: i) con mutaciones en BRCA1/2 (tBRCA); ii) con deficiencia de la recombinación homóloga o HDR (todas las pacientes con mutaciones en BRCA1/2 y pacientes sin mutaciones en BRCA pero con pérdida de heterocigosidad en el tumor alta); y iii) la población ITT. Como variable primaria de eficacia se determinó la SLP según criterios RECIST v1.1 y evaluación por el investigador. Entre las secundarias, destaca la SLP determinada por comité radiológico independiente, la SG, la seguridad y los síntomas físicos relacionados con la enfermedad, medidos por la escala validada FOSI18.
Con una mediana de duración del tratamiento con rucaparib de 8,3 meses y con placebo de 5,5 meses, los principales resultados obtenidos en el estudio pivotal ARIEL3 se reflejan en la Tabla 2. Rucaparib demostró una mejora estadísticamente significativa de la SLP (de 5,4 meses) en comparación con placebo en las tres cohortes analizadas. Además, un análisis exploratorio sugiere que rucaparib también aporta un beneficio significativo frente a placebo en pacientes con BRCA no mutado, obteniéndose un aumento de 4,3 meses en la mediana de SLP –evaluada por el investigador– en pacientes con pérdida de heterocigosidad en el tumor alta (LOH+) (9,7 vs. 5,4 meses con placebo; HR: 0,44) y un aumento de 1,3 meses en pacientes LOH- (6,7 vs. 5,4 meses con placebo; HR: 0,58); tal beneficio fue confirmado por los resultados obtenidos en la evaluación por el comité radiológico independiente, que apuntaban a un beneficio aún mayor (aumento de 5,5 y 2,9 meses en pacientes LOH+ y LOH-, respectivamente).
En cuanto a otras variables secundarias, un análisis exploratorio de la TRO en las pacientes con enfermedad medible al inicio indicaba que ésta era mayor en pacientes tratadas con rucaparib (18%, 7% de respuestas completas) respecto al grupo placebo. Sin embargo, el tiempo hasta el empeoramiento de los síntomas según la escala FOSI18 fue inferior para rucaparib respecto a placebo en la población ITT y la cohorte HDR, si bien no se observaron diferencias estadísticamente significativas en la cohorte tBRCA. En el momento del análisis de SLP, los datos de SG no tenían la madurez suficiente (22% de los eventos) como para extraer conclusiones firmes, pero apuntan a que no hay diferencias notables entre las pacientes tratadas con rucaparib y con placebo en ninguna de las tres cohortes.
Por último, el perfil de seguridad de rucaparib en las dos indicaciones propuestas se basa en los datos de 937 pacientes que participaron en los ensayos clínicos con rucaparib en monoterapia, con medianas de administración del fármaco de unos 6-8 meses, siendo mayor de 12 meses en el 25-37% de las pacientes. La totalidad de las pacientes sufrió algún evento adverso; los más frecuentemente relacionados con el fármaco en sus dos indicaciones fueron las siguientes: náuseas (69-71% vs. 27% con placebo), fatiga (63-69% vs. 31%), aumento de los niveles de alanina-aminotransferasa y aspartato-aminotransferasa (41%), anemia (34-40% vs. 4%), disgeusia (31-37% vs. 75) y vómitos (31%).
La incidencia de eventos adversos graves (grado ≥3) relacionados con el tratamiento en el grupo de pacientes tratadas con rucaparib fue del 9,4-9,8% (vs. 1,6% con placebo), siendo los más frecuentes la anemia (en torno al 4%), la neutropenia febril (>1%) y la fatiga. Si bien la toxicidad de rucaparib pudo manejarse adecuadamente con interrupciones o ajustes posológicos en más de la mitad de pacientes (54-65%), un 8-16% de pacientes abandonaron el tratamiento, fundamentalmente como consecuencia de la fatiga, las náuseas y la anemia. Se han registrado casos de síndrome mielodisplásico/leucemia mieloide aguda en pacientes tratadas con rucaparib, algunos de ellos (2) con desenlace fatal.
El rucaparib es un nuevo antineoplásico que actúa como inhibidor potente y selectivo de las isoenzimas 1, 2 y 3 de la familia de las poli-ADP-ribosa polimerasas (PARP) –PARP1, PARP2 y PARP3– implicadas en los mecanismos de reparación del ADN, tanto en células normales como tumorales: mediante la inhibición de dichas enzimas, la citotoxicidad de rucaparib –que ha demostrado ser independiente de la presencia de mutaciones en los genes BRCA– se debe a un aumento de la formación de complejos PARP-ADN, conducente a un daño del ADN y a la activación de la apoptosis y la muerte celular.
En base a ese mecanismo, el medicamento ha sido autorizado en el tratamiento de mantenimiento, en monoterapia, de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, en recidiva, sensible al platino, que responde completa o parcialmente a la quimioterapia con platino. También está indicado en monoterapia para el tratamiento de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, con mutación BRCA (germinal y/o somática), sensible al platino, en recaída o progresión, que hayan sido tratadas con dos o más líneas previas de quimioterapia con platino y que no son capaces de tolerar más quimioterapia a base de platino.
La evidencia de la eficacia y seguridad de rucaparib en la primera indicación (mantenimiento) deriva de un amplio ensayo de fase 3, aleatorizado, doble ciego y controlado por placebo. Se acepta que este estudio no incluya como comparador activo otro inhibidor de PARP (olaparib o niraparib) porque el ensayo pivotal fue previo a su autorización, y la población de estudio (N= 564) se considera representativa de la población de pacientes en la que se empleará el fármaco en la práctica clínica real.
Rucaparib alcanzó en ese ensayo pivotal el objetivo principal de prolongar significativamente la SLP –evaluada por el investigador– frente a placebo, aumentando así el intervalo libre de quimioterapia basada en platino en un escenario clínico en el que la recurrencia, el tratamiento quimioterápico y su toxicidad acumulativa son inevitables. Esa eficacia se verificó en las tres cohortes evaluadas, prolongando la SLP 5,4 meses en la población global –y hasta 11,2 meses en pacientes con BRCA mutado– en comparación con la ausencia de tratamiento activo: las medianas fueron de 10,8, 13,6 y 16,6 meses en la población ITT, en la cohorte con deficiencia de recombinación homóloga y en pacientes con BRCA mutado, respectivamente, frente a los 5,4 meses en el grupo placebo. Tal beneficio fue aún más pronunciado en la evaluación radiológica por un comité independiente, alcanzando las pacientes tratadas con rucaparib y mutación de BRCA una prolongación de SLP de más de 20 meses (medianas de 26,8 vs. 5,4 meses con placebo). Todo ello se tradujo en reducciones notables del riesgo de progresión de la enfermedad, del 64% para el global de las pacientes. Los análisis de subgrupos confirman la eficacia de rucaparib también en pacientes sin mutación BRCA, aunque el beneficio parece inferior. No obstante, la ausencia de datos maduros impide confirmar un beneficio en SG con el fármaco.
El posicionamiento de rucaparib frente al resto de alternativas terapéuticas actualmente aprobadas como mantenimiento en la misma indicación, los inhibidores de PARP olaparib y niraparib, se ve dificultado por la ausencia de comparaciones directas. Una comparación indirecta ajustada, con un rigor cualitativo y cuantitativo intrínseco limitado, no ha podido detectar diferencias estadísticamente significativas entre las tres opciones en términos de SLP, si bien los resultados disponibles para olaparib proceden de un ensayo fase 2 (Staropoli et al., 2018). Tampoco se dispone de comparaciones directas o indirectas con bevacizumab, la otra alternativa de mantenimiento para casos en recaída (tras tratamiento con el fármaco en combinación con carboplatino y gemcitabina o paclitaxel); bevacizumab en monoterapia de mantenimiento reveló datos de SLP tumoral 4 meses mayores que con placebo sin afectar a la SG, lo cual puede sugerir una eficacia similar a la obtenida con los paribs.
En cuanto a su indicación de tratamiento de pacientes en recaída, pre-tratadas (con ≥2 líneas de platino) y que no pueden recibir nuevo tratamiento a base de platino, los resultados de eficacia proceden de dos estudios abiertos de fase 2 y sin comparador, por lo que no son confirmatorios y su principal limitación radica en la ausencia de un brazo control. En el análisis conjunto de las pacientes tratadas con rucaparib en ambos estudios (N= 106), mayoritariamente sensibles a platino y que presentaban mutación BRCA, la tasa de respuesta objetiva evaluada por el investigador fue del 54,7%, con una mediana de duración de la respuesta de unos 9,7 meses; la eficacia fue mayor en el subgrupo de pacientes sensibles a platino (TRO de 64,6%), el único incluido en la indicación autorizada.
Conviene recordar, a este respecto, que, aunque la quimioterapia con platino es la base del tratamiento del cáncer de ovario, para aquellas pacientes que recaen y no son candidatas a más ciclos con platino, no existe un tratamiento estándar y las opciones son limitadas. Se suele recurrir a esquemas de quimioterapia como doxorubicina liposomal pegilada (DLP), topotecán o la combinación de trabectedina con DLP, que se han relacionado con TRO de 19-29% en el total de la población; los escasos datos en pacientes BRCA mutadas apuntan a TRO de 63% para la combinación de trabectedina-DLP y del 29% con DLP. Se desconoce por ahora el beneficio real en términos de SLP y SG de rucaparib frente a las alternativas de tratamiento disponibles –resulta difícil posicionarlo entre ellas–, que podrá ser dilucidado cuando se divulguen los resultados de un estudio de fase 3 en marcha (ARIEL4). No obstante, parece que el nuevo fármaco podría ofrecer una mejor tolerabilidad que la quimioterapia, con menor incidencia de toxicidad hematológica (sobre todo, neutropenia) y otros efectos secundarios relevantes; además, se administra por vía oral, lo que podría suponer una ventaja adicional para una mejor adherencia terapéutica.
A grandes rasgos, rucaparib muestra una tolerabilidad similar a olaparib y niraparib: tiene un perfil de seguridad importante, pero clínicamente manejable –con ajustes posológicos– y la mayoría de reacciones adversas son leves-moderadas. Destacan por su frecuencia los eventos adversos de naturaleza gastrointestinal y hematológica: náuseas, vómitos, fatiga/astenia, anemia, neutropenia y mielotoxicidad; los eventos graves más comunes relacionados con rucaparib (<4%) fueron la anemia, la neutropenia febril y la fatiga. Se notificó una frecuencia ligeramente superior respecto a otros inhibidores de PARP de aumento de transaminasas y fotosensibilidad. Con los tres fármacos se han descrito casos de síndrome mielodisplásico y/o leucemia mieloide aguda, y se hace necesario considerar este riesgo y conocer, en futuros estudios, más datos sobre los posibles efectos a medio y largo plazo del tratamiento con rucaparib.
En resumen, rucaparib ha demostrado un beneficio relevante frente a placebo en el mantenimiento pos-quimioterapia a base de platino (permitirá que las pacientes se beneficien en mayor medida de un nuevo régimen posterior basado en platino), y parece que viene a representar una alternativa terapéutica más a olaparib y niraparib entre dos intervalos de quimioterapia en pacientes con buen estado funcional, pues los datos clínicos disponibles no permiten establecer una superioridad en términos supervivencia (aún hay incertidumbres, sobre todo en pacientes con BRCA no mutado) o diferencias importantes en el balance beneficio/riesgo, ni avalan el uso secuencial de estos inhibidores de PARP. En tratamiento de pacientes pre-tratadas en recaída, los datos de rucaparib son limitados y requieren confirmación, pero sugieren que puede dar lugar a tasas de respuesta similares a otras alternativas disponibles con un mejor perfil de seguridad y de adherencia terapéutica. En definitiva, sin incorporar ninguna novedad mecanística respecto a otros inhibidores de PARP usados frente al cáncer de ovario, tampoco parece que vaya a aportar una innovación clínica notable en su tratamiento.
Dupilumab es un nuevo anticuerpo monoclonal de administración subcutánea dirigido frente a la subunidad α del receptor de la IL-4 (IL-4Rα), que impide la señalización mediada por la unión de esa citocina a su receptor tipo I (IL-4Rα/γc) y también por IL-4 e IL-13 a través del receptor tipo II (IL4Rα/IL-13Rα). Puesto que IL-4 e IL-13 son los principales mediadores de la inflamación tipo 2, dupilumab ejerce interesantes efectos antiinflamatorios. En base a ello, el medicamento ha sido autorizado para el tratamiento de la dermatitis atópica (DA) moderada-grave en pacientes adultos y adolescentes de ≥12 años candidatos a tratamiento sistémico, y también como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 (caracterizada por eosinófilos elevados en sangre y/o FeNO elevado), en adultos y adolescentes de ≥12 años que no están adecuadamente controlados con corticosteroides inhalados en dosis altas en combinación con otro medicamento.
El fármaco ha demostrado superioridad frente a placebo en el tratamiento de la DA tanto en pacientes que inician tratamiento sistémico tras fracaso al tratamiento tópico como en pacientes pre-tratados con ciclosporina: en monoterapia, el fármaco induce aumentos significativos en la proporción de pacientes respondedores con aclaramiento de la piel (+27-28% según escala IGA y +37-44% que alcanzan EASI-50), que también se verifican en combinación con un corticoide o inhibidor de calcineurina tópicos (+24-28% en IGA y +40-48% con EASI-50); un estudio en pacientes adolescentes confirmó resultados similares. En el tratamiento adicional del asma no controlada, evidenció una eficacia notable en pacientes con biomarcadores inflamatorios de tipo 2 elevados. Así, en presencia de altos niveles de eosinófilos en sangre y de FeNO en aire exhalado, reduce el riesgo de exacerbaciones en un 66-67% y mejora la funcionalidad pulmonar (duplicando el valor de VEF1 pre-broncodilatador) en comparación con placebo. Además, los resultados de un estudio específico demuestran que dupilumab permite reducir/suprimir la dosis de corticoides orales –y, con ello, su toxicidad– en un 70% de pacientes (vs. 42% con placebo). En ambas patologías, el efecto es de inicio rápido (2 semanas) y duradero en periodos de hasta 1 año, aportando beneficio en síntomas y calidad de vida reportados por los pacientes.
A grandes rasgos, dupilumab muestra una buena tolerabilidad a corto-medio plazo, con un perfil toxicológico aceptable y manejable. En línea con la seguridad de otras proteínas terapéuticas, entre las reacciones adversas más frecuentes destacan reacciones en el lugar de la inyección (eritema, edema y prurito) e infecciones (conjuntivitis, blefaritis, nasofaringitis, infecciones respiratorias del tracto superior, sinusitis y herpes oral), en su mayoría leves-moderadas. El riesgo de desarrollo de reacciones alérgicas e inmunogenicidad parece bajo, si bien se requieren aún datos de seguridad a largo plazo que permitan esclarecer los potenciales riesgos de desarrollo de neoplasias malignas.
Dupilumab inaugura una nueva vía farmacológica en sus dos indicaciones, patologías de curso crónico-recurrente que afectan a población pediátrica y adulta y que en ciertos casos pueden resultar incapacitantes y tener un elevado impacto socio-laboral. Aporta, además, un beneficio clínico superior a placebo en casos graves-moderados, siendo quizás más relevante su indicación en dermatitis atópica, pues supone el primer avance terapéutico desde hace mucho tiempo (primer tratamiento biológico autorizado) y será una alternativa adecuada en pacientes sin respuesta o no candidatos a tratamiento con ciclosporina.
La dermatitis atópica (DA) es una enfermedad inflamatoria de la piel de carácter crónico o crónicamente recurrente, que se caracteriza fundamentalmente por la presencia de lesiones eccematosas, sequedad de la piel (xerosis) y prurito intenso. Suele cursar con brotes de duración e intensidad variable, entre los que se intercalan periodos de remisión, aunque, en algunos casos, los síntomas pueden ser continuos. Bien es sabido que el sistema inmunitario es determinante en la aparición de muchas dermatitis1, y la dermatitis atópica, a diferencia de las dermatitis de contacto, se ha asociado con reacciones de hipersensibilidad tipo I o inmediatas, pues frecuentemente se observa un incremento notable de inmunoglobulinas IgE (como también ocurre en otros trastornos como el asma o la rinitis alérgica) (Montalvo-Calvo et al., 2019).
La DA es la expresión cutánea de un estado denominado atópico o atopia, un término que se define genéricamente como la existencia de hipersensibilidad frente a proteínas heterólogas. La reacción de hipersensibilidad de tipo I se produce tras la exposición a un antígeno exógeno (polen, polvo, etc.), provocando la liberación inmediata de una amplia variedad de sustancias activas a partir de los mastocitos y células cebadas del organismo. Estas sustancias activas, tales como la histamina, prostaglandinas, factor activador de las plaquetas, leucotrienos, factor quimiotáctico de eosinófilos, etc., junto con el aumento de la síntesis de IgE, conforman una situación típica de alergia. La eosinofilia es otro rasgo característico de este tipo de las reacciones tipo I.
La etiopatogenia subyacente justifica la notable relación existente entre los pacientes que padecen DA con los antecedentes personales o familiares de crisis asmáticas, rinitis, reacciones cutáneas desproporcionadas tras la picadura de insectos, urticaria masiva, etc. Así, epidemiológicamente, la DA se caracteriza por un historial familiar en el 70% de los pacientes de asma bronquial, rinitis alérgica, fiebre del heno o dermatitis.
Se trata de una de las enfermedades de la piel más comunes, que puede aparecer en cualquier época de la vida. Se ha estimado que un 15-20% de los niños que acuden al dermatólogo en los países desarrollados padecen este tipo de afección, frente a menos del 1% en los países en vías de desarrollo; la prevalencia en adultos a nivel mundial se cree que oscila entre el 1-3%. Por tanto, se trata de una patología que va muy ligada al nivel de desarrollo de los países, hasta el punto de que su prevalencia parece haberse triplicado en el último medio siglo en los países desarrollados. En España, se estima que la DA grave tiene una prevalencia del 0,08% de la población.
Puede afectar a las personas de cualquier raza, es más frecuente en el sexo femenino (1,5:1) y se presenta más a menudo en las clases socioeconómicas altas, así como en las grandes ciudades, posiblemente por una mayor exposición a estímulos capaces de desencadenar el cuadro. La probabilidad de padecer la enfermedad es del 80% cuando ambos progenitores la sufrieron, del 55% cuando solo la padeció uno de ellos y del 60% cuando un progenitor presentaba dermatitis y el otro atopia respiratoria.
En cuanto a las manifestaciones, el prurito el síntoma predominante en todos los casos (puede dar lugar a lesiones secundarias debido al rascado). La inflamación epidérmica propia de las dermatitis provoca una serie de lesiones que, progresivamente, según se agrava o se hace crónico, pasan sucesivamente por las fases2 de eritema, edema, vesiculación, exudación, costra, descamación y liquenificación.
No obstante, la presentación y el curso clínico de la DA varían con la edad y suelen ser heterogéneos. Una gran mayoría de pacientes (85%) desarrollan por primera vez la enfermedad antes de los 5 años, la cual se resuelve espontáneamente durante la infancia (en torno a la mitad de los casos). Es bastante frecuente que la DA debute en lactantes a los 3-6 meses de edad con aparición de placas inflamatorias y supuración, o placas con descamación en cara, cuello, superficies de extensión y en la ingle. También es relativamente frecuente su desaparición espontánea entre los 3 y los 5 años de edad, pero puede mantenerse como una condición crónica durante la edad adulta hasta para un 40% de pacientes. Cuando debuta en adultos, la DA suele presentarse como un cuadro eccematoso de aparición alrededor de los 20 años.
Según se ha sugerido, se estima que un 61% de los pacientes adultos con DA padecen al menos un tipo de comorbilidad atópica, como asma o rinoconjuntivitis alérgica (las tres patologías constituyen la clásica triada de la atopia). También se ha descrito un mayor riesgo de comorbilidades no atópicas, como otros trastornos cutáneos, ciertas enfermedades cardiovasculares, trastornos inmunitarios sistémicos o cáncer. Además, la pérdida de la barrera protectora de la piel y la disregulación del sistema inmunitario aumentan el riesgo de infecciones cutáneas, las cuales pueden ser potencialmente graves. Por todo ello, y teniendo en cuenta las manifestaciones clínicas producen alteraciones importantes del sueño, secuelas psicológicas y sociales, la DA –sobre todo, en sus formas moderadas y graves– representa un importante problema sociosanitario y tiene un impacto importante en la calidad de vida de los pacientes.
De forma general, la severidad de la DA se determina mediante el empleo de distintas escalas de valoración validadas que consideran la extensión de las áreas de piel afectadas, la gravedad de las lesiones y los síntomas subjetivos del paciente. Las más aceptadas son: SCORAD (Scoring Atopic Dermatitis) y EASI (Eczema Area and Severity Index), que no incluyen síntomas subjetivos; según estas escalas se puede definir la DA moderada a partir de una puntuación SCORAD> 14 o un EASI 7,1-21 y DA grave como SCORAD >40 o EASI 21,1-50 (muy grave >50,1-72). Otras escalas ampliamente empleadas son la IGA (Investigator global assessment), escala del 0 al 5 (dermatitis aclarada, mínima, ligera, moderada, intensa y grave) que tiene en cuenta el eritema, la formación de pápulas y de exudados, y la escala NRS (Numerical Rating Scale), escala numérica que mide la intensidad del prurito, siendo 10 la mayor intensidad (AEMPS, 2020).
Volviendo sobre la etiopatogenia de la DA, cabe destacar que no solo intervienen mecanismos inmunitarios, sino también genéticos, ambientales y otros desconocidos actualmente, característicos de la propia piel del individuo atópico (por ejemplo, se ha postulado el posible papel de anomalías en la sudoración y un aumento de la pérdida transepidérmica de agua), así como factores externos como la dieta, la existencia de aero-alérgenos, infecciones por microorganismos (estafilococos u hongos como Malassezia furfur) o incluso otros factores como sequedad ambiental, disminución de la temperatura, tejidos irritantes, etc., que podrían actuar como estímulos desencadenantes o agravantes de un episodio de DA.
En una elevada proporción de los pacientes con DA –en torno al 80%– se detecta la producción excesiva de IgE y la disminución de la inmunidad mediada por células. De hecho, existe alteración en las subpoblaciones de linfocitos T y de las células de Langerhans (células presentadoras de antígenos existentes en dermis y epidermis): se produce un predominio de células T que contribuye a incrementar la presencia de reacciones inflamatorias, con un descenso asociado de linfocitos CD8+ y un aumento del cociente CD4+/CD8+; además, se ha demostrado que las células de Langerhans de los pacientes tienen en su superficie una alta expresión del receptor para IgE.
En las lesiones agudas de DA se observa un predominio de linfocitos T facilitadores (Th) de tipo Th2 (subpoblación de CD4+), mientras que en las lesiones liquenificadas y crónicas lo que predominan son las células Th1. Los linfocitos Th2 liberan diversos factores (interleucinas IL-4, IL-5, IL-6, IL-10) activadores de las células B; la producción aumentada de IL-4 estimula la producción de IgE e inhibe la de interferón (IFN) gamma, con lo que se inhibe la diferenciación de células T hacia Th1. Por su parte, las células Th1 median en la producción de IL-2, IFN-γ y factor de necrosis tumoral (TNF), activando los macrófagos y favoreciendo la reacción de hipersensibilidad retardada.
Por tanto, son las células Th2 las responsables de la producción aumentada de IL-4 e IL-13, estimuladoras de la producción de IgE, IL-5 (responsable de la activación de los eosinófilos) e IL-10 (responsable de la disminución de la inmunidad celular). Estos hallazgos son importantes ya que la IL-4 y IL-13 inducen la expresión de moléculas de adhesión involucradas en la migración de células inflamatorias en las zonas de inflamación tisular. Además, se ha descrito que la IL-4 podría inhibir la producción de IFN-γ e inhibir la diferenciación de las células T hacia Th1 (Figura 1).
Este conjunto de alteraciones inmunitarias condicionan, en consecuencia, la aparición frecuente de infecciones cutáneas recidivantes, tanto de origen bacteriano como vírico o fúngico. De ellas, quizás la más frecuente sea la colonización por Staphylococcus aureus, presente en el 90% de las lesiones cutáneas.
En base a todo lo anterior, los objetivos fundamentales del tratamiento de la dermatitis atópica consisten en limitar los síntomas (sobre todo, el prurito), prevenir las exacerbaciones, evitar las infecciones dérmicas y minimizar los riesgos del tratamiento.
Para ello, previamente a la farmacoterapia, en esta patología es fundamental la adopción de medidas de higiene a fin de prevenir todas aquellas circunstancias que desencadenen prurito. Entre otras medidas, se pueden citar las siguientes: el baño –con agua templada y jabón de pH ácido– es preferible a la ducha, se deben administrar cremas emolientes, se debe evitar la temperatura elevada y una baja humedad ambiental (la calefacción por corrientes de aire caliente puede ser perjudicial), así como el uso de ropas de abrigo excesivas y el contacto con ciertos tejidos (lana, plásticos y gomas); por el contrario, el uso de guantes y la exposición solar suelen resultar beneficiosas. En cuadros de carácter crónico, es recomendable el empleo frecuente de soluciones oleosas que limpien y humedezcan la piel.
El tratamiento farmacológico estándar para combatir los síntomas de la DA se basa en la administración de agentes antiinflamatorios tópicos, sobre todo, corticosteroides (beclometasona, betametasona, fluocinolona, metilprednisolona, etc.) e inhibidores de la calcineurina (tacrolimus o pimecrolimus) en forma de cremas; sin embargo, las preparaciones tópicas producen resultados pobres (sobre todo en casos moderados-graves), posiblemente debido a su escasa penetración cutánea. Junto a estos, suelen administrarse emolientes e hidratantes de la piel, tales como vaselina blanca, vaselina hidrófila, óxido de zinc, etc. La utilización de corticosteroides sistémicos en ciclos cortos se reserva para los casos agudos o de gran extensión corporal. Además, en caso de infección cutánea sobreañadida se usan antibióticos por vía sistémica (nunca tópica).
No es aconsejable la utilización por vía tópica de antihistamínicos, neomicina, sulfamidas ni perfumes. El tratamiento del picor asociado a la dermatitis se realiza generalmente mediante la utilización de antihistamínicos por vía oral: suele preferirse a los antiguos antihistamínicos (por ejemplo, dexclorfeniramina), que producen más somnolencia que los modernos, ya que aparentemente su eficacia se debe más a su efecto sedante que en su actividad antipruriginosa propiamente dicha. Cuando en las alteraciones de la epidermis predomina el edema y la exudación, es útil el empleo tópico de sulfato de cobre o permanganato potásico, por sus propiedades astringentes y desinfectantes.
Los pacientes con enfermedad grave o resistente a los tratamientos tópicos pueden requerir de fototerapia (UVA) o pueden ser candidatos para la administración de tratamiento sistémico con inmunosupresores convencionales: el único autorizado en la UE, y considerado de elección, es la ciclosporina, aunque también se emplean –con menor evidencia que respalde su uso– corticoides, azatioprina, metotrexato y micofenolato de mofetilo. En ciertos casos graves, según criterio del dermatólogo, se puede recurrir a los fármacos biológicos (anticuerpos monoclonales), aunque conviene recordar que, hasta ahora, cuando se han empleado en DA ha sido un uso off label (fuera de sus indicaciones en ficha técnica). En todo caso, la decisión de iniciar el tratamiento sistémico en los pacientes con DA grave se debe de basar en la evaluación de la severidad y la calidad de vida y, al mismo tiempo, en la consideración del estado general de salud de forma individualizada para cada paciente, valorando factores como preferencias del paciente, comorbilidades y coste del tratamiento.
A día de hoy, el tratamiento de las formas graves de DA es un reto terapéutico y sigue siendo una necesidad médica no resuelta, sobre la cual no hay consenso en las principales guías clínicas europeas y estadounidenses. Algunos estudios apuntan a que más de la mitad de los pacientes adultos en diversos países de Europa (Reino Unido, Alemania, Francia) tienen un control clínico pobre, con altas tasas de enfermedad no controlada en las formas moderadas (56-75%) y graves (82-85%), que son sugerentes de la limitada eficacia del tratamiento hasta ahora disponible (incluyendo el inmunosupresor) y que se traducen en un mayor impacto en la calidad de vida y deterioro de las capacidades de la vida diaria.
De forma general, el asma se define como una enfermedad inflamatoria crónica de las vías respiratorias, en cuya patogenia intervienen diversas células y mediadores de la inflamación, condicionada en parte por factores genéticos y que cursa con hiperreactividad bronquial y una obstrucción variable al flujo aéreo, total o parcialmente reversible, ya sea por la acción de fármacos o espontáneamente (Cuéllar, 2016).
Se trata de un problema de salud de elevada prevalencia y, aunque las formas graves de la enfermedad solo suponen el 10% de todos los casos, tiene importantes implicaciones en la esperanza y calidad de vida de las personas afectadas, generando un importante consumo de recursos sanitarios y notables costes sociales. Según la OMS, el asma es la séptima enfermedad más prevalente en el mundo, con cerca de 235 millones de afectados. La cifra de afectados en Europa ronda los 29 millones. Además, supone la quinta causa de muerte en países desarrollados.
En España, la prevalencia de síntomas asmáticos se ha mantenido constante en los niños de 13-14 años, mientras que ha sufrido un aumento significativo en el grupo de 6-7 años. En adultos, la prevalencia es inferior a la de países anglosajones y centroeuropeos. Se estima que en España sufren asma aproximadamente el 5% de los adultos y el 10% de niños. El Estudio Europeo del Asma (GEEEA, 1996) constató en nuestro país unas tasas variables que van desde el 4,7% en Albacete hasta el 1% en Huelva, alcanzando en algunas zonas cifras cercanas al 14%; y lo que es más preocupante: un 52% de las personas con asma no habían sido diagnosticadas y hasta un 26% de éstas, a pesar de padecer síntomas frecuentes, no seguía ningún tratamiento.
El asma en adultos suele clasificarse, según el grado de gravedad, en cuatro categorías: intermitente, persistente leve, persistente moderada y persistente grave. En los niños se definen dos patrones principales: asma episódica (ocasional o frecuente) y asma persistente (este último siempre considerado como grave). Las manifestaciones clínicas características del asma son:
En realidad, el asma no es una enfermedad única, sino más bien un síndrome que comparte manifestaciones clínicas similares en apariencia pero ligadas a diversos mecanismos celulares y biomoleculares. Ha pasado de ser considerada una reacción de hipersensibilidad de tipo I (la relevancia se daba al episodio de broncoespasmo desencadenado por la liberación de mediadores tras la desgranulación del mastocito al producirse la reacción alérgeno-IgE específica) a ser interpretada en la actualidad como un proceso inflamatorio crónico de las vías aéreas puesto en marcha por una serie de factores desencadenantes, entre los destacan determinadas actividades profesionales, ejercicio físico, infecciones, fármacos (betabloqueantes y AINE, en particular el ácido acetilsalicílico), reflujo gastroesofágico y ciertos factores emocionales.
Sin embargo, con mucho, el factor más frecuentemente desencadenante del asma es la alergia. Los alérgenos más comúnmente involucrados en su patogénesis son proteínas de los reinos animal y vegetal, como los procedentes de los ácaros (Dermatophagoides pteronyssinus y D. farinae) –los más comunes en nuestro medio–, los de pólenes (gramíneas), los de árboles (Corylus avellana, Olea sp.), de hongos (Alternaria sp., Aspergillus sp.) o de animales (epitelios y fluidos de gato, perro, etc.). El asma alérgico comienza con la sensibilización: en individuos con predisposición genética y ante determinadas condiciones ambientales (infección viral, humo de tabaco, etc.), se produce la interacción del antígeno con las células dendríticas y la posterior activación de linfocitos T, que generan citocinas inductoras de la diferenciación y activación de eosinófilos (IL-5), de la expresión de receptores de la IgE en mastocitos y esosinófilos (IL-4), de la expresión en el epitelio de receptores que atraen a los eosinófilos (IL-4) y de la producción y liberación de IgE por células B.
Una reexposición al alérgeno produce un episodio agudo o ataque de asma, que generalmente consiste en 2 fases: i) la fase inmediata se caracteriza por la aparición de un espasmo en el músculo liso bronquial como consecuencia de la interacción del antígeno con el mastocito que había expresado y fijado la IgE a sus receptores en la sensibilización, liberando principalmente histamina y leucotrienos C4 y D4 (LTC4 y LTD4), responsables del broncoespasmo; también se liberan otros mediadores (prostaglandina D2, neurocinina A, LTB4) que provocan una migración de células inflamatorias –eosinófilos y monocitos– hacia esa zona anatómica; ii) la fase tardía o respuesta diferida ocurre en un tiempo variable desde la exposición inicial al antígeno (6-8 horas), suele ser nocturna, y es claramente la progresión de una reacción inflamatoria iniciada en la primera fase que conlleva un acúmulo local de eosinófilos; se piensa que los gránulos de los eosinófilos infiltrantes liberan mediadores citotóxicos que afectan al epitelio respiratorio ciliado (Figura 2).
En el ataque de asma se produce un desacoplamiento en la relación ventilación-perfusión con un aumento del gradiente alveolo-arterial de oxígeno, lo cual implica la disminución de la presión parcial de O2 en sangre y, si la ventilación alveolar no aumenta lo suficiente (el trabajo respiratorio no compensa el aumento de la resistencia y la carga elástica pulmonar ni la menor eficiencia contráctil del diafragma), un aumento de la presión parcial de CO2. En suma, la hipoxemia suele ser moderada y responde favorablemente al aumento de la concentración de oxígeno inspirado.
Son diversos los tipos de células inflamatorias implicadas en el asma, destacando entre ellas:
Por la relación existente entre el asma y la atopia, se ha descrito que hasta el 35% de las personas con asma grave sufren dermatitis atópica y hasta la mitad de los afectados de DA padecen asma. Ambas patologías se engloban dentro de las llamadas enfermedades mediadas por inflamación tipo 2. Durante muchos años el asma fue considerada como una patología mediada por una respuesta inmunitaria adaptativa, pero cada vez hay más evidencias de que la inmunidad innata también tiene un rol importante en las distintas vías que subyacen en su compleja patobiología.
Las respuestas inflamatorias tipo 2 en el asma –predominantes en la mayoría de casos– son iniciadas por citocinas como la IL-25 o IL-33, llamadas alarminas, secretadas tras la exposición a contaminantes, infecciones o alérgenos. Esas citocinas derivadas del epitelio activan a las células presentadoras de antígenos (dendríticas y NK) para inducir la respuesta inmunitaria adaptativa Th2. Una vez activas, las células Th2 migran hacia el epitelio y la mucosa subepitelial de la vía aérea, donde secretan las citocinas tipo 2, IL-5 e IL-13, que ejercen un papel crucial en patogénesis y el desarrollo de muchas características de la enfermedad. Además, las alarminas también activan directamente a las células linfoides innatas pulmonares tipo 2 (CLI-2) para que secreten una gran cantidad de IL-5 e IL-13. Una vez estimuladas, esas CLI-2 pulmonares producen pequeñas cantidades de IL-4, aunque no está claro si esto representa una interacción entre los mecanismos inmunitarios innatos y adaptativos.
En consecuencia, las citocinas tipo 2 promueven el reclutamiento de células efectoras (mastocitos, basófilos y eosinófilos) y median la producción de IgE por las células B, un aumento de los niveles de óxido nítrico (FeNO) en la fracción exhalada y un incremento de los niveles séricos de periostina. La inflamación eosinofílica en las vías aéreas y la síntesis de IgE son, pues, eventos centrales en el asma tipo 2 y en la patogénesis de las exacerbaciones (Robinson et al., 2017).
El objetivo principal del tratamiento del asma es lograr y mantener el control de la enfermedad lo antes posible, además de prevenir las exacerbaciones y la obstrucción crónica al flujo aéreo, para reducir al máximo su mortalidad.
Los ataques o crisis de asma son episodios agudos o subagudos caracterizados por un aumento progresivo de uno o más de los síntomas típicos (disnea, tos, sibilancias y opresión torácica) acompañados de una disminución del flujo espiratorio (VEF1). Según la rapidez de instauración, existen dos tipos: las crisis de instauración lenta (normalmente en días o semanas, por una infección respiratoria o mal control de la enfermedad) y las de instauración rápida (en menos de 3 horas, por un broncoespasmo causado por inhalación de alérgenos, fármacos, alimentos o estrés emocional), que deben identificarse por tener causas, patogenia y pronóstico diferentes. La intensidad de las exacerbaciones es variable, cursando en ocasiones con síntomas leves e indetectables por el paciente y en otras con episodios muy graves que ponen en peligro su vida.
El objetivo inmediato del tratamiento de una crisis es preservar la vida del paciente revirtiendo la obstrucción al flujo aéreo y la hipoxemia (si está presente) de la forma más rápida posible, y posteriormente instaurar o revisar el plan terapéutico para prevenir nuevas crisis. El tratamiento en la exacerbación leve debe incluir la administración de broncodilatadores agonistas β2-adrenérgicos de acción corta (SABA), glucocorticoides orales y oxígeno (si es necesario).
Los SABA inhalados son los fármacos broncodilatadores más eficaces y rápidos en el tratamiento de la crisis asmática. Si en las primeras 2 horas del tratamiento se constata una evolución favorable (desaparición de síntomas, VEF1 > 80% del teórico o del mejor valor personal del paciente) y ésta se mantiene durante 3-4 horas, no son necesarios más tratamientos. No obstante, en pacientes no controlados con combinaciones inhaladas de corticosteroides y SABA administradas a demanda, puede recurrirse a combinaciones similares con agonistas β2 de acción prolongada (LABA), como salmeterol, formoterol, vilanterol, etc.
En el tratamiento de mantenimiento a largo plazo del asma se distinguen varios escalones terapéuticos de complejidad creciente, que deben individualizarse en base a la respuesta y el nivel de control que manifieste el paciente, siendo necesaria una evaluación clínica periódica para determinar si se cumplen los objetivos clínicos. En todos los escalones de tratamiento, los corticosteroides inhalados (CSI) son de elección para el control a largo plazo del asma, a dosis crecientes según la gravedad del cuadro asmático, y empleándose junto con un LABA en los casos más graves. Además, a estos puede asociarse tiotropio (anticolinérgico) y/o un antagonista de los receptores de leucotrienos (montelukast) y/o administrar corticosteroides orales (CSO) si la gravedad de la patología así lo recomienda (GEMA, 2018). En todos los escalones se pueden administrar a demanda agonistas β2-adrenérgicos de acción corta.
El uso de glucocorticoides sistémicos (CSO) acelera la resolución de las exacerbaciones. Excepto en crisis muy leves, deben administrarse siempre, especialmente si: a) no se consigue una reversión de la obstrucción de las vías respiratorias con SABA; b) el paciente estaba tomando ya CSO; c) el paciente no controlado ha probado sin éxito otras opciones terapéuticas; d) existen antecedentes de exacerbaciones previas que requirieron CSO. Aunque son moderadamente eficaces para el control de la mayoría de los casos de enfermedad moderada-grave, es importante señalar que el uso prolongado de corticoides sistémicos suele asociarse con efectos adversos en diversos órganos, en ocasiones severos, por lo que existe una necesidad de desarrollar tratamientos que puedan emplearse con anterioridad en pacientes cuya patología no es tan grave como para requerir corticoterapia sistémica, previniendo los riesgos de ésta.
En esa línea, los anticuerpos monoclonales han abierto recientemente un importante capítulo en el tratamiento de mantenimiento del asma grave. Básicamente, los fármacos biológicos ya autorizados contemplan las siguientes vías inmunofarmacológicas:
Cabe recordar que el asma grave no controlada se define como aquella que persiste mal controlada3 a pesar de recibir tratamiento con una combinación de CSI a dosis elevadas junto con un LABA en el último año, o bien CSO durante al menos 6 meses del mismo periodo. En nuestro medio, la prevalencia de pacientes con asma grave no controlada es aproximadamente del 4% del total de la población asmática (algunos autores hablan de falta de control en hasta el 45% de pacientes con asma grave). Entre ellos, el asma eosinofílica –caracterizada por la presencia de altos niveles de eosinófilos en biopsias bronquiales y esputo a pesar de dosis altas de corticosteroides– representa aproximadamente el 25% de los casos (y el 40-60% de todos los casos de asma).
Finalmente, la inmunoterapia con alérgenos específicos es un tratamiento de mantenimiento indicado en el asma alérgica bien controlada, siempre que se haya demostrado una sensibilización mediada por la inmunoglobulina E frente a aeroalérgenos comunes, y se utilicen extractos bien estandarizados.
Dupilumab es un anticuerpo monoclonal completamente humano dirigido de forma específica frente a la subunidad α del receptor de la interleucina 4 (IL-4Rα). Impide, por tanto, la señalización molecular mediada por la IL-4 a través de su unión al receptor tipo I (IL-4Rα/γc) y también la señalización de IL-4 e IL-13 a través del receptor de Tipo II (IL-4Rα/IL-13Rα). Teniendo en cuenta que IL-4 e IL-13 son los principales mediadores de la inflamación tipo 2, mediada por linfocitos Th2 y mayoritaria en las patologías relacionadas con la atopia, dupilumab bloquea esta ruta de señalización y ejerce efectos antiinflamatorios (Shirley, 2017).
En base a ello, el medicamento ha sido autorizado para el tratamiento de la dermatitis atópica de moderada a grave en pacientes adultos y adolescentes a partir de 12 años que son candidatos a tratamiento sistémico, y también como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 (caracterizada por eosinófilos elevados en sangre y/o FeNO elevado), en adultos y adolescentes a partir de 12 años que no están adecuadamente controlados con corticosteroides inhalados en dosis altas en combinación con otro medicamento.
La IL-4 es una citocina reguladora que se une a su receptor IL-4Rα, ampliamente expresado sobre las células Th (contribuye a su diferenciación a Th2), eosinófilos, mastocitos, células B, epitelio bronquial, endotelio y células de la musculatura lisa de la vía aérea. Induce la producción de las llamadas citocinas de tipo 2 (IL-5, IL-9, IL-13, TARC4 y eotaxinas-3) por las células Th2, el cambio de isotipo de células B a células plasmáticas productoras de IgE y el reclutamiento de eosinófilos y otras células inflamatorias. Para activar la señalización bioquímica, el receptor IL-4Rα necesita formar un heterodímero con uno de los dos receptores de superficie adicionales (IL-2γc o IL-13Rα1): la heterodimerización con el complejo IL-2γc produce una señal reguladora (tipo 1) que modula la diferenciación Th0 a Th2 y Treg, mientras que la heterodimerización con el IL-13Rα1 induce la misma señal (reguladora tipo 2) que la se produce con la unión de la IL-13 a este receptor, fundamentalmente expresado en la musculatura lisa y células epiteliales de la vía aérea.
Se ha descrito que la IL-13 es una citocina efectora que ejerce acciones pleiotrópicas sobre la inflamación tipo 2, tales como el cambio de isotipo de las células B, el reclutamiento de eosinófilos, la hipersecreción mucosa, la hiperplasia de las células caliciformes, la fibrosis subepitelial y la hiperreactividad bronquial. Por tanto, la IL-4 y la IL-13 comparten muchas actividades que se superponen; la principal diferencia radica en que la IL-13 no se une a las células T y tiene funciones predominantemente estimuladoras de tejidos, mientras que la IL-4 es una citocina básicamentalmente reguladora que dirige la respuesta inmunitaria a Th2.
Ambas interleucinas juegan un rol multifactorial en la patogénesis de la dermatitis atópica y el asma, por lo que se comprende que la inhibición de esa ruta de señalización por dupilumab –al unirse al receptor IL-4Rα– permite un control de las manifestaciones clínicas. Dupilumab inhibe tanto las vías de la inflamación tipo 1 (mediadas por la formación del dímero IL-4Rα/γc) como de tipo 2 (mediada por el dímero IL-4Rα/IL-13Rα), siendo la acción sobre esta última la más importante en su efecto terapéutico.
De hecho, en los ensayos clínicos de DA, el tratamiento con dupilumab se asoció a descensos, respecto al nivel basal, de las concentraciones de biomarcadores de la inmunidad de tipo 2 (tales como TARC), de IgE total en suero y de IgE específica de alérgenos en suero; se observó también una disminución de la lactato deshidrogenasa (un biomarcador asociado a la actividad y gravedad de la DA). En ensayos clínicos de asma, el tratamiento con dupilumab redujo significativamente en comparación con placebo los niveles de FeNO y la concentración circulante de eotaxina-3, de IgE total, de IgE alérgeno-específica, de TARC y de periostina; la supresión máxima de estos marcadores se produjo a las 2 semanas de tratamiento (excepto la IgE, que disminuyó más lentamente), y el efecto se mantuvo a largo plazo (AEMPS, 2017).
El dupilumab es un anticuerpo monoclonal completamente humano de tipo IgG4, que se produce por técnicas de ingeniería genética (ADN recombinante) en células de ovario de hámster chino (CHO).
Entre sus especificidades moleculares, dupilumab –que tiene un peso molecular predicho de 146,9 kDa– contiene un sitio de N-glicosilación conservado en el residuo de asparagina en posición 302 de la región Fc de cada una de las subunidades de las cadenas pesadas. Dichas cadenas pesadas contienen, además, una mutación en el aminoácido en posición 233 (que pasa de serina a prolina), localizada en la región bisagra del dominio Fc.
La eficacia y la seguridad clínicas de dupilumab por vía subcutánea han sido adecuadamente contrastadas en pacientes adultos a la dosis autorizada mediante tres ensayos clínicos pivotales de fase 3 (confirmatorios de eficacia y seguridad), multicéntricos, doblemente ciegos, aleatorizados y controlados con placebo, de 16 a 52 semanas de duración. Estos estudios (SOLO1, SOLO2 y CHRONOS) tuvieron diseños similares e incluyeron un total de 2.119 pacientes de >18 años, con DA moderada grave –definida según puntuaciones IGA≥ 3, EASI≥ 16 y una afectación mínima de la superficie corporal (BSA)≥ 10%– y respuesta insuficiente a la farmacoterapia tópica previa.
Las características basales de los pacientes eran similares y estuvieron bien equilibradas en los distintos brazos experimentales. En los estudios SOLO (1 y 2), la media de edad de los pacientes fue de 38,3 años, el 58% eran hombres y el 68% de raza blanca; con respecto a la patología, el 52% tenían una puntuación de 3 en la escala IGA (severidad moderada) y el 48% de 4 puntos (grave), el 32% habían recibido tratamiento sistémico inmunosupresor, la puntuación media en la escala EASI fue de 33, la media en la NRS del prurito fue de 7,4 puntos por semana y, en la escala SCORAD, la puntuación media basal fue de 67,8. Por su parte, el estudio CHRONOS incluyó una población de pacientes con una edad media de 37,1 años, el 60% eran hombres y el 66% de raza blanca; además, la puntuación media basal de la escala IGA fue de 3 y 4 puntos, respectivamente, en el 53% y 47% de los pacientes, el 34% habían recibido tratamiento previo con inmunosupresores sistémicos, la puntuación media basal del EASI fue de 32,5 puntos, de 7,3 puntos cada semana en la escala NRS, y la puntuación media basal de la escala SCORAD fue de 66,4.
Los tres estudios evaluaron los mismos regímenes posológicos de dupilumab y placebo, pudiéndose administrar tratamiento de rescate (inmunosupresores sistémicos o esteroides tópicos de mayor potencia) a criterio del investigador según la gravedad de los síntomas de DA; en tal caso, los pacientes se consideraron no-respondedores. También emplearon las mismas variables co-primarias: proporción de pacientes que alcanzó un IGA de 0 o 1 (aclaramiento total o casi total de la piel) con una reducción de al menos 2 puntos y proporción de pacientes con una mejora de al menos el 75% en la EASI (EASI-75) desde el momento basal hasta la semana 16. Otras variables secundarias fueron la proporción de pacientes con EASI-50 y EASI-90 (mejoras ≥ 50% y ≥ 90% en la escala EASI), el porcentaje de cambio en la escala SCORAD o la proporción de pacientes con una reducción de ≥4 puntos en la escala NRS del prurito.
Los principales resultados de eficacia divulgados para cada uno de los tres ensayos clínicos con dupilumab en adultos, referentes al total de pacientes aleatorizados, se recogen en la Tabla 1. Se observa una clara superioridad frente a placebo en el tratamiento de la DA, sin diferencias significativas de eficacia entre los dos regímenes posológicos del fármaco (300 mg cada semana o cada dos semanas). La eficacia se verificó en todos los subgrupos de pacientes evaluados (peso, edad, sexo, raza y tratamiento de base, incluidos los inmunosupresores), siendo los resultados coherentes con los de la población global. Aunque no se muestran en la tabla, también se observaron mejorías significativas respecto a placebo en la calidad de vida (escala DLQI), síntomas de depresión y ansiedad (escala HADS) y síntomas referidos por el paciente (escala POEM).
Uno de los subgrupos de especial interés analizados en el estudio CHRONOS es el de pacientes con respuesta inadecuada o intolerancia a ciclosporina o en los que dicho fármaco no fuera aconsejable. En la semana 16, estos pacientes mostraron una mejoría de signos y síntomas de la DA consistentes con los de la población general, alcanzándose un EASI-75 en el 69,6% de los pacientes tratados con dupilumab 300 mg/2 semanas frente al 18,0% de pacientes tratadas con placebo; el cambio porcentual medio –respecto al valor basal– de la NRS del prurito fue de -51,4% con dupilumab 300 mg/2 semanas vs. -30,2% en el grupo placebo (EMA, 2017). Estos resultados están en línea y fueron corroborados por los obtenidos en el estudio de soporte CAFE: en una población de pacientes con similares características, el tratamiento con dupilumab 300 mg/2 semanas y 300 mg/semana permitió que, a la semana 16, un 62,6% y un 59,1% de pacientes, respectivamente, alcanzara un EASI-75, frente al 29,6% con placebo (de Bruin-Weller et al., 2018).
Adicionalmente a los datos del ensayo CHRONOS, la evaluación de la duración de la respuesta clínica fue objeto de un estudio de extensión (SOLO CONTINUE) en que se enrolaron aquellos pacientes clasificados como respondedores en los estudios SOLO1 y SOLO2, quienes fueron aleatorizados a recibir un tratamiento adicional con dupilumab en distintas pautas o placebo por 36 semanas más, hasta el total de 52 semanas. Los resultados (Worm et al., 2019) confirman que las pautas de 300 mg cada 1 o 2 semanas son óptimas para mantener la respuesta, mientras que las más espaciadas (una dosis mensual o bimensual) tenían menor eficacia. De los respondedores (EASI-75) en el momento basal, una proporción significativamente mayor –en comparación con placebo– de pacientes en los grupos de dupilumab seguía en respuesta EASI-75 al final del estudio (72% de pacientes en pautas de 1-2 semanas vs. 30% en el grupo placebo).
Por otra parte, la eficacia de dupilumab en adolescentes de entre 12 y 17 años ha sido caracterizada en un ensayo de fase 3 (AD-1526), multicéntrico, doblemente ciego y controlado por placebo, en el que se aleatorizaron 251 pacientes con DA de moderada a grave (mismos criterios de definición de gravedad que en adultos) y respuesta insuficiente a fármacos tópicos. La media de edad de los pacientes fue de 14,5 años, un 59% eran varones y un 63% de raza blanca; en cuanto al estado patológico al inicio, una amplia mayoría de pacientes tenía alguna comorbilidad alérgica (66% rinitis y 54% asma), el 42% habían recibido previamente inmunosupresores sistémicos, el 46% tenían puntuación de 3 en la escala IGA y el 54% de 4, y la valoración media basal de las escalas EASI, NRS y SCORAD fue, respectivamente, de 35,5, 7,6 y 70, 3 puntos.
Los resultados (Simpson et al., 2019) ponen de manifiesto que dupilumab (a las dosis de 200 y 300 mg cada 2 semanas, según peso) es significativamente superior en eficacia a placebo. Así, a la semana 16, un 24,4% de pacientes tratados con el fármaco alcanzó una puntuación de 0 o 1 en la escala IGA (con cambio de ≥ 2 puntos) y un 41,5% presentaba respuesta en términos de EASI-75, frente al 2,4% y 12,9% de pacientes en el grupo placebo, respectivamente (p< 0,0001). Se verificó también una reducción en la intensidad del picor, con un 36% de pacientes con una mejora de ≥ 4 puntos en la escala NRS (vs. 4,8% con placebo), y una reducción significativa en la escala de puntación global SCORAD (cambio medio de -51,6% vs. -17,6% con placebo). Estos resultados se acompañaron de notables mejoras en la percepción subjetiva de los pacientes, el impacto de la DA en el sueño y en la calidad de vida (según resultados de escalas POEM y DLQA), todo lo cual condujo a que una menor proporción de adolescentes tratados con dupilumab requiriera tratamiento corticoideo o inmunosupresor de rescate (20,7% vs. 58,8% en el grupo placebo).
Por último, el perfil toxicológico de dupilumab en el tratamiento de la DA parece bien definido en base a datos de los más de 1.900 pacientes tratados en los estudios de su desarrollo clínico, de los cuales 305 recibieron inyecciones del fármaco durante al menos 1 año; además, el fármaco muestra una tolerabilidad similar en adolescentes y en adultos. En general, la incidencia de eventos adversos relacionados con el fármaco es baja, de forma que la proporción de pacientes que interrumpió el tratamiento por problemas de seguridad osciló entre el 1,5% y el 1,8% con la dosis autorizada en monoterapia y combinación con corticoides tópicos, respectivamente (vs. 1,9% y 7,6% con placebo).
Las reacciones adversas más frecuentes –respecto a placebo– fueron: reacciones en el lugar de la inyección (9,6-10%) e infecciones, como conjuntivitis (3-6,4%), blefaritis, nasofaringitis, infecciones respiratorias del tracto superior, sinusitis y herpes oral (2,7-3,8%); también se describió una mayor incidencia de eosinofilia (1,7% vs. 0,4% con placebo). La mayoría de ellas fueron autolimitadas y de intensidad leve-moderada, notificándose infecciones graves solo en el 0,5-0,6% de pacientes tratados con dupilumab. Las reacciones de hipersensibilidad fueron muy raras (se reportaron casos de eccema herpetiforme en 0,2-<1% de pacientes). En términos de inmunogenicidad, la aparición de anticuerpos anti-fármaco (transitoria y con bajo título) se describió en menos del 10% de pacientes tratados, aunque su relevancia en eficacia/seguridad no ha sido determinada y se requieren datos a más largo plazo para aclarar este potencial riesgo.
La aprobación de la indicación en asma para dupilumab por vía subcutánea se apoyó fundamentalmente en los resultados de eficacia y seguridad clínica contrastados en dos ensayos clínicos pivotales de fase 3, estudios multicéntricos aleatorizados, de grupos paralelos, doblemente ciegos, y controlados con placebo, de 24 a 52 semanas de duración, que incluyeron un total de 2.112 pacientes adolescentes (>12 años) y adultos con asma grave, pero sin requerimientos específicos de biomarcadores de inflamación tipo 2 (eosinofilia > 150 células/μl o FeNO ≥ 20 ppb).
El primero de ellos, el estudio QUEST, comparó dupilumab frente a placebo en 107 pacientes adolescentes y 1.795 adultos (media de edad de 47,9 años) con asma persistente en tratamiento con un corticosteroide inhalado (CSI) a dosis media-alta y combinado con un segundo fármaco; se incluyeron algunos pacientes que necesitaban un tercer fármaco de control. Los pacientes eran mayoritariamente mujeres (63%), de raza blanca (83%), no fumadores (81%) y presentaban una media de duración del asma de 21 años, con 2,1 exacerbaciones de media en el año anterior. En el momento basal, mostraban una media de VEF1 pre-broncodilatador de 1,78 l (con un porcentaje previsto de VEF1 del 58,4%) y una puntuación media en las escalas ACQ-55 y AQLQ(S)6 de 2,76 y 4,29 puntos, respectivamente.
Los pacientes fueron asignados al azar (2:2:1:1) a recibir dupilumab 200 mg (N= 631) o 300 mg (N= 633) cada 2 semanas (tras una dosis inicial doble), o un placebo equivalente (N= 317 y N= 321), y se evaluaron dos variables co-primarias: la tasa anualizada de exacerbaciones graves (TAEG) durante las 52 semanas del período controlado con placebo y el cambio en la semana 12 del VEF1 pre-broncodilatador.
Los resultados publicados del estudio QUEST (Castro et al., 2018) tras el análisis por intención de tratar revelan una reducción significativa en la TAEG en comparación con placebo: tomó el valor de 0,46 eventos/año (IC95% 0,39-0,53) con la dosis de dupilumab 200 mg frente a 0,87 en el grupo placebo, y de 0,52 eventos/año (IC95% 0,45-0,61) con la dosis de 300 mg frente a 0,97 de su correspondiente placebo (p< 0,0001 en ambo casos); esos datos se traducían en una reducción del riesgo de crisis grave del 48% y del 46% con las respectivas dosis del fármaco. El número de exacerbaciones/año se redujo en todos los subgrupos de pacientes tratados con dupilumab, si bien la eficacia fue más pronunciada en los pacientes con niveles basales elevados de biomarcadores inflamatorios de tipo 2:
En relación a la funcionalidad pulmonar, se observaron aumentos clínicamente significativos (p< 0,0001) en el VEF1 pre-broncodilatador en la semana 12 en los pacientes tratados con dupilumab respecto a placebo, evidentes ya desde la semana 2 y que se mantuvieron hasta la semana 52. La diferencia media respecto al estado basal a la semana 12 fue de +0,32 l con dupilumab 200 mg (vs. 0,18 l con placebo) y de +0,34 l con la dosis de 300 mg (vs. 0,13 l con placebo). De nuevo, las mayores mejoras en el VEF1 se produjeron en los subgrupos de sujetos con niveles basales de ≥ 300 eosinófilos/µl sangre (0,43-0,47 l con dupilumab vs. 0,21-0,22 l con placebo; p< 0,0001) y los de FeNO ≥ 50 ppb (0,30-0,39 l con dupilumab vs. 0,19-0,23 l con placebo; p< 0,0001).
Además, de forma interesante, los resultados comunicados por los pacientes del control del asma y la calidad de vida a través de los cuestionarios ACQ-5 y AQLQ(S) muestran un aumento notable, rápido y duradero –desde la semana 2 y hasta la 52– de la proporción de pacientes que responden a dupilumab (70-76% con la escala ACQ-5 y 67-71% en la AQLQ(S)) en comparación con placebo (64-67% con la escala ACQ-5 y 53-58% en la AQLQ(S)).
Por otra parte, el estudio VENTURE evaluó la eficacia de dupilumab 300 mg/2 semanas sobre la necesidad de corticosteroides orales (CSO) a las 24 semanas de tratamiento en 210 pacientes asmáticos que requerían a diario ese tipo de fármacos complementariamente al uso de corticoides inhalados a dosis altas más otro fármaco de control. Las características demográficas y basales de los pacientes eran muy similares a las del estudio QUEST: media de edad de 51,3 años, 61% mujeres, 94% de raza blanca, 81% no fumadores, duración media del asma de 20 años, 2,1 exacerbaciones de media en el año anterior, valor basal medio de VEF1 pre-broncodilatador de 1,58 l (porcentaje previsto de VEF1 del 52,1%) y puntuación media en las escalas ACQ-5 y AQLQ(S) de 2,5 y 4,35 puntos, respectivamente.
Los resultados divulgados del análisis por intención de tratar (Rabe et al., 2018) demuestran que los pacientes tratados con dupilumab (N= 103) tenían, en comparación con aquellos que recibieron placebo, un cambio significativamente mayor (N= 107) en el porcentaje de reducción de la dosis de CSO respecto al estado basal –variable primaria– (70,1% vs. 41,9% con placebo; p< 0,0001). Así, el 80% de pacientes tratados con el fármaco consiguió una reducción a la mitad de la dosis de CSO (vs. 50% de pacientes en el grupo placebo; p< 0,0001), y hasta un 48% pudo suprimir por completo el uso de CSO manteniendo un buen control del asma (vs. 25% con placebo; p< 0,0001). La proporción de pacientes que consiguió reducir la dosis diaria hasta < 5 mg/día fue del 69% con dupilumab y del 33% con placebo (razón de probabilidades: 4,5; p< 0,0001).
La eficacia del fármaco fue consistente en los distintos subgrupos analizados, destacando un mayor beneficio en pacientes con ≥ 150 eosinófilos/µl, y sobre todo, ≥ 300 eosinófilos/µl (media del porcentaje de reducción de la dosis de CSO del 80% con dupilumab vs. 43% con placebo) y con ≥ 25 ppb de FeNO (73% vs. 43%); en estas subpoblaciones, el 79-84% de pacientes pudo reducir la dosis de CSO por debajo de 5 mg/día (vs. 34-40% con placebo). Además, dupilumab también demostró superioridad frente a placebo en su efecto sobre la tasa de exacerbaciones (0,65 vs. 1,60 eventos/año; reducción del 59% del riesgo) y sobre la mejora en el VEF1 desde el momento basal (diferencia media de +0,22 l vs. placebo); no obstante, estos efectos fueron similares independientemente de los niveles basales de los biomarcadores inflamatorios de tipo 2. Los resultados percibidos por los pacientes y sobre la calidad de vida confirmaron la tendencia de las mejoras comentada en el estudio QUEST.
La seguridad clínica del nuevo fármaco en el tratamiento del asma moderada-grave también ha sido bien definida en base a los datos procedentes de más de 1.500 pacientes tratados en los estudios controlados con placebo (> 2.600 pacientes en todo su desarrollo clínico), la mayoría de ellos durante al menos 1 año. La incidencia de eventos adversos relacionados con el fármaco fue relativamente baja y estos fueron en su mayoría leves-moderados y de carácter transitorio. Las reacciones adversas más frecuentes fueron reacciones en el lugar de inyección (incidencia en torno al 20% vs. 9% con placebo), como eritema/edema y prurito; el dolor de cabeza y la eosinofilia (~1%) también se notificaron en proporción ligeramente superior que con placebo. En cambio, a diferencia de lo que ocurría en pacientes con dermatitis atópica, la incidencia de conjuntivitis fue baja y similar entre dupilumab y placebo. Los casos de reacción anafiláctica también fueron muy raros (2,5-3%).
En un análisis conjunto de seguridad, se observó que la proporción de pacientes que interrumpió el tratamiento por eventos adversos fue baja, oscilando entre el 3,2-6% con dupilumab (vs. 4,3% con placebo); los eventos que se relacionaron con discontinuaciones fueron fundamentalmente las reacciones en el lugar de inyección (eritema 0,5-1,5%, edema 0,1-0,9%, prurito 0,3-0,9% y dolor 0,1%-0,6%). La frecuencia de aparición de anticuerpos anti-fármaco también fue baja (6% vs. 5% con placebo) y no se asoció con problemas concretos de seguridad ni de eficacia. Un estudio abierto de extensión, actualmente en marcha, aportará más información de seguridad a largo plazo, que permitirá esclarecer el riesgo de desarrollo de neoplasias malignas a largo plazo, ya que los datos disponibles hasta la fecha son limitados (EMA, 2019).
Dupilumab es un nuevo anticuerpo monoclonal humano de administración por vía subcutánea que se une específicamente a la subunidad α del receptor de la interleucina 4 (IL-4Rα). Impide así la señalización molecular mediada por la unión de esa citocina a su receptor tipo I (IL-4Rα/γc) y también la señalización de IL-4 e IL-13 a través del receptor tipo II (IL4Rα/IL-13Rα). Puesto que IL-4 e IL-13 son los principales mediadores de la inflamación tipo 2 –regulada por linfocitos Th2–, dupilumab ejerce efectos antiinflamatorios bloqueando esta ruta de señalización, que juega un papel fundamental en patologías relacionadas con la atopia. En base a ello, el medicamento ha sido autorizado para el tratamiento de la dermatitis atópica (DA) moderada-grave en pacientes adultos y adolescentes de ≥12 años candidatos a tratamiento sistémico, y también como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 (caracterizada por eosinófilos elevados en sangre y/o FeNO elevado), en adultos y adolescentes de ≥12 años que no están adecuadamente controlados con corticosteroides inhalados en dosis altas en combinación con otro medicamento.
Los datos conducentes a su autorización en DA derivan de tres amplios estudios pivotales de fase 3, doblemente ciegos y controlados por placebo, que incluyeron pacientes adultos con enfermedad moderada-grave y refractarios a tratamiento tópico; la población estudiada puede considerarse representativa de la población diana a la cual se dirige el fármaco. Dos de esos estudios evaluaron el tratamiento en monoterapia y demostraron la superioridad de dupilumab frente a placebo tras 4 meses de tratamiento, con diferencias significativas en la proporción de pacientes que mejoran clínicamente: un aumento del 27-28% de respondedores según la escala IGA y del 37-44% en la proporción de los que alcanzan EASI-50 (aumentos del 23-28% para EASI-90). Un tercer estudio probó que dupilumab también es significativamente superior a placebo en combinación con un corticoide o inhibidor de calcineurina tópicos, con una eficacia que se mantiene por periodos de tratamiento de hasta 1 año (aumento de respondedores del 24-28% según escala IGA y de 40-48% en el número de pacientes con EASI-50 y de 28-36% con EASI-90).
Los resultados de un estudio controlado en adolescentes (12-17 años) corroboran la eficacia de dupilumab en este grupo etario, con aumentos frente a placebo del 22% y el 28% en la proporción de pacientes respondedores según escala IGA y que alcanzan EASI-75, respectivamente. En general, los efectos en el aclaramiento de la piel con el fármaco se acompañaron de mejorías notables del prurito (del orden del 25-30%), así como de la calidad de vida y los síntomas referidos por el paciente. El beneficio que aporta el fármaco, que parece instaurarse de forma rápida (2 semanas) y se mantiene en el tiempo, se verificó en los distintos subgrupos analizados, independientemente de factores como edad, sexo, raza o tratamiento de base; de forma interesante, los datos clínicos ponen de manifiesto la consistencia de la eficacia en pacientes con respuesta inadecuada, intolerancia a ciclosporina o en los que dicho fármaco no es aconsejable: tras 4 meses de tratamiento, alcanzaron EASI-75 un 59-69% de los pacientes vs. 18-29% con placebo.
Así pues, dupilumab ha demostrado superioridad frente a placebo en el tratamiento de la DA tanto en pacientes que inician tratamiento sistémico de novo tras fracaso del tratamiento tópico como en pacientes pre-tratados con ciclosporina, el fármaco que la mayoría de guías clínicas establecen de elección para el inicio del tratamiento sistémico (por la amplia experiencia de uso) y el único con eficacia contrastada y autorización para ello en la UE. No obstante, no se dispone de comparación directa de dupilumab con este fármaco (de hecho, la principal limitación de los estudios comentados es la ausencia de comparador activo) y los datos no permiten establecer la eficacia relativa entre ellos, por lo cual, hasta no se demuestre superioridad clínica, parece que ciclosporina7 se mantiene como el tratamiento de elección a corto plazo –máximo de 1 año– en la DA moderada-grave; a largo plazo los datos son más limitados y controvertidos, por el riesgo de nefrotoxicidad irreversible.
Con respecto a su uso en asma grave no controlada como tratamiento adicional (a corticoides inhalados más otro fármaco), la aprobación por la EMA se basó en los resultados de dos estudios controlados y doble ciego de fase 3. Un amplio ensayo de grupos paralelos demostró la superioridad de dupilumab frente a placebo en tratamientos de 1 año de duración, con una notable reducción de la tasa de exacerbaciones graves y mejora de la funcionalidad pulmonar, que en la práctica real se puede traducir en menor incidencia de hospitalizaciones y visitas a urgencias por crisis agudas. Esa eficacia, de inicio precoz (2 semanas) y duradera, se confirmó en el conjunto de pacientes incluidos, pero fue más pronunciada en la población con niveles basales elevados de biomarcadores inflamatorios de tipo 2 (altos niveles de eosinófilos en sangre y de FeNO en el aire exhalado). Así, en pacientes con ≥ 300 eosinófilos/µl, dupilumab redujo el riesgo de exacerbaciones en un 66-67% (tasa de 0,37-0,40 vs. 1,08-1,24 eventos/año con placebo) y duplicó el valor de VEF1 pre-broncodilatador (0,43-0,47 l vs. 0,21-0,22 l con placebo). Valores similares se observaban en pacientes con FeNO ≥ 50 ppb: tasa de 0,33-0,39 exacerbaciones/año (vs. 1,06-1,27 con placebo) y VEF1 pre-broncodilatador de 0,30-0,39 l (vs. 0,19-0,23 l con placebo).
El segundo estudio reveló que en el 70% de los pacientes tratados con dupilumab el asma mejora hasta el punto de que pueden reducir la dosis de corticoides orales (CSO), frente al 42% de los tratados con placebo; de hecho, la mayoría de quienes recibieron el fármaco pudieron reducir a la mitad la dosis de CSO (80% vs. 50% con placebo) y casi la mitad pudieron suprimir por completo el uso de CSO (48% vs. 25% con placebo), manteniendo un buen control del asma. En esta indicación la eficacia del fármaco también fue consistente en los distintos subgrupos analizados, destacando un mayor beneficio en pacientes con ≥ 150 eosinófilos/µl y con ≥ 25 ppb de FeNO. De forma interesante, los resultados comunicados por los pacientes del control del asma y la calidad de vida en los dos estudios –a través de los cuestionarios ACQ-5 y AQLQ(S)– muestran un aumento notable de la proporción de pacientes respondedores a dupilumab en comparación con placebo.
Por tanto, el nuevo fármaco representará una alternativa terapéutica interesante a otros anticuerpos que se oponen a los efectos de la IL-5 (mepolizumab, reslizumab y benralizumab) o la IgE (omalizumab), autorizados para pacientes con alto recuento de eosinófilos o demostración de asma alérgica mediada por IgE, respectivamente, pero que no han demostrado eficacia en un amplio rango de pacientes asmáticos cuya enfermedad está mediada por una compleja red de inflamación celular. No se dispone de comparaciones directas e indirectas con estos fármacos, siendo difícil el posicionamiento de su eficacia relativa por el distinto perfil de los grupos de pacientes que se benefician clínicamente de ellos, a priori más amplio en el caso de dupilumab. En cualquier caso, su uso como tratamiento adicional se restringe a pacientes con mal control a pesar de dosis altas de CSI (más otro fármaco), pues según las guías actuales, en pacientes tratados con dosis medias se recomienda aumentar dicha dosis antes de iniciar fármacos biológicos; en ese sentido, no implica un cambio sustancial en la terapéutica estándar. No obstante, los CSI no son capaces de normalizar los niveles de biomarcadores de inflamación tipo 2, y un 30% de pacientes con asma grave requieren tratamiento adicional con corticoides orales: en ellos, dupilumab sí puede representar una ventaja sustancial, permitiendo reducir/suprimir su dosis y, con ello, su toxicidad.
En términos de seguridad, dupilumab presenta un perfil toxicológico bien definido, relativamente benigno a corto-medio plazo y clínicamente manejable, que determina unas tasas bajas de abandono del tratamiento (< 6%), y que es consistente entre las dos indicaciones y dosis autorizadas del fármaco, con independencia de la edad de los pacientes. En línea con la tolerabilidad de otros fármacos biológicos empleados en patologías con componente inflamatorio, destacan por su frecuencia reacciones adversas como reacciones en el lugar de la inyección (eritema, edema y prurito) e infecciones (conjuntivitis, blefaritis, nasofaringitis, infecciones respiratorias del tracto superior, sinusitis y herpes oral), que en su mayoría son leves-moderadas y autolimitadas. El riesgo de desarrollo de reacciones alérgicas e inmunogenicidad parece bajo (la aparición de anticuerpos anti-fármaco en menos del 10% de pacientes no se relacionó con problemas concretos de seguridad o eficacia), si bien los datos de uso a largo plazo del fármaco –periodos de tratamiento de >1 año– son limitados y se requieren futuros estudios para descartar riesgos potenciales de desarrollo de neoplasias malignas.
En definitiva, dupilumab inaugura una nueva vía farmacológica en el tratamiento de la dermatitis atópica y del asma (a la que previsiblemente se incorporarán otros prometedores fármacos ya en desarrollo clínico, como lebrikizumab), patologías de curso crónico-recurrente que afectan a población pediátrica y adulta y que en determinados casos pueden resultar incapacitantes y tener un elevado impacto socio-laboral. A la innovación mecanística se suma el beneficio clínico superior a placebo demostrado en casos graves-moderados, siendo quizás más relevante su indicación en dermatitis atópica, patología en la cual no se han incorporado avances terapéuticos desde hace mucho tiempo y representa el primer tratamiento biológico autorizado: emerge como una opción adecuada de tratamiento sistémico de novo en pacientes refractarios a medicación tópica o en aquellos pre-tratados pero en los que el uso de ciclosporina no se considere adecuado por falta de respuesta, contraindicación o intolerancia.
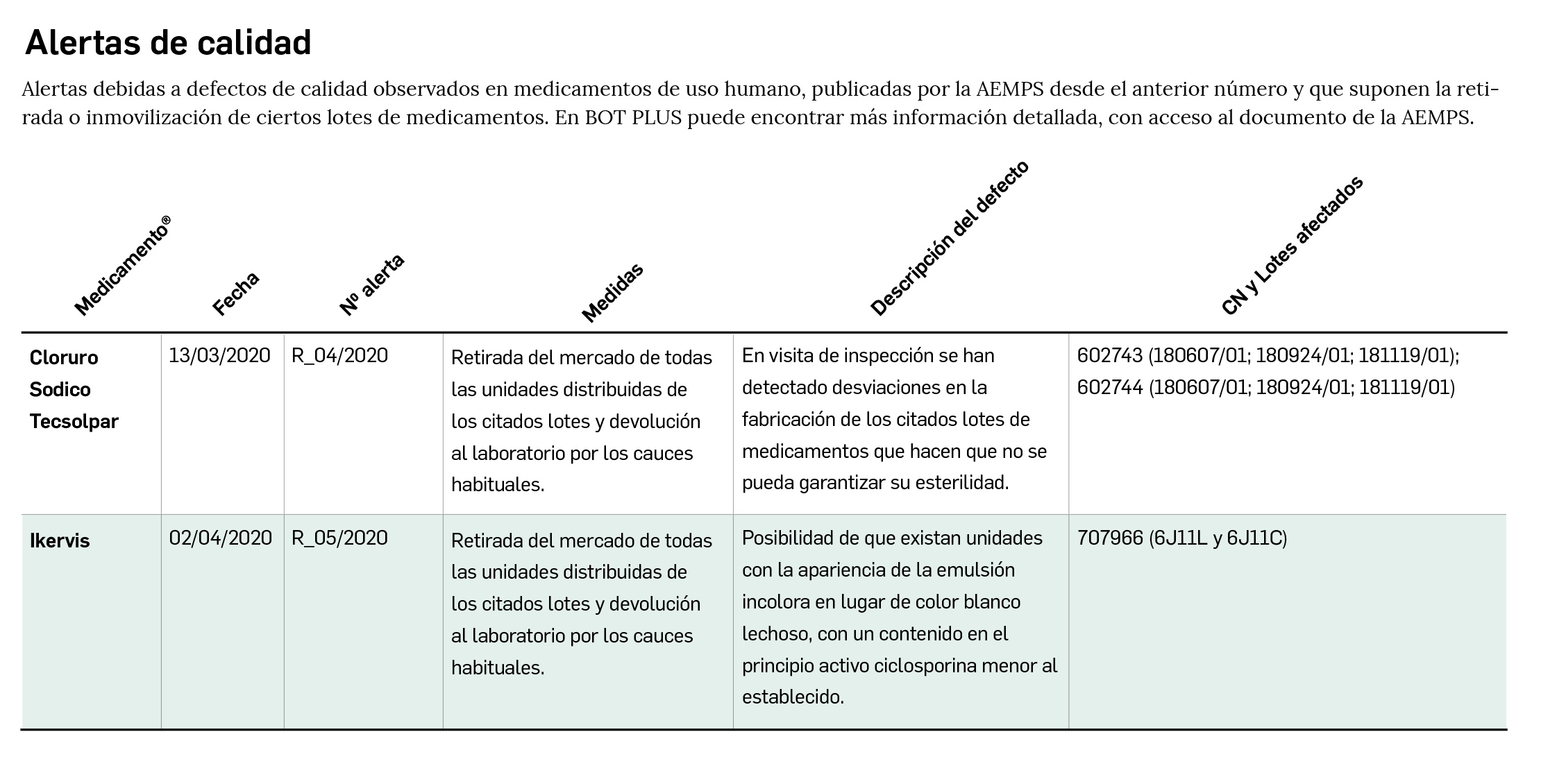
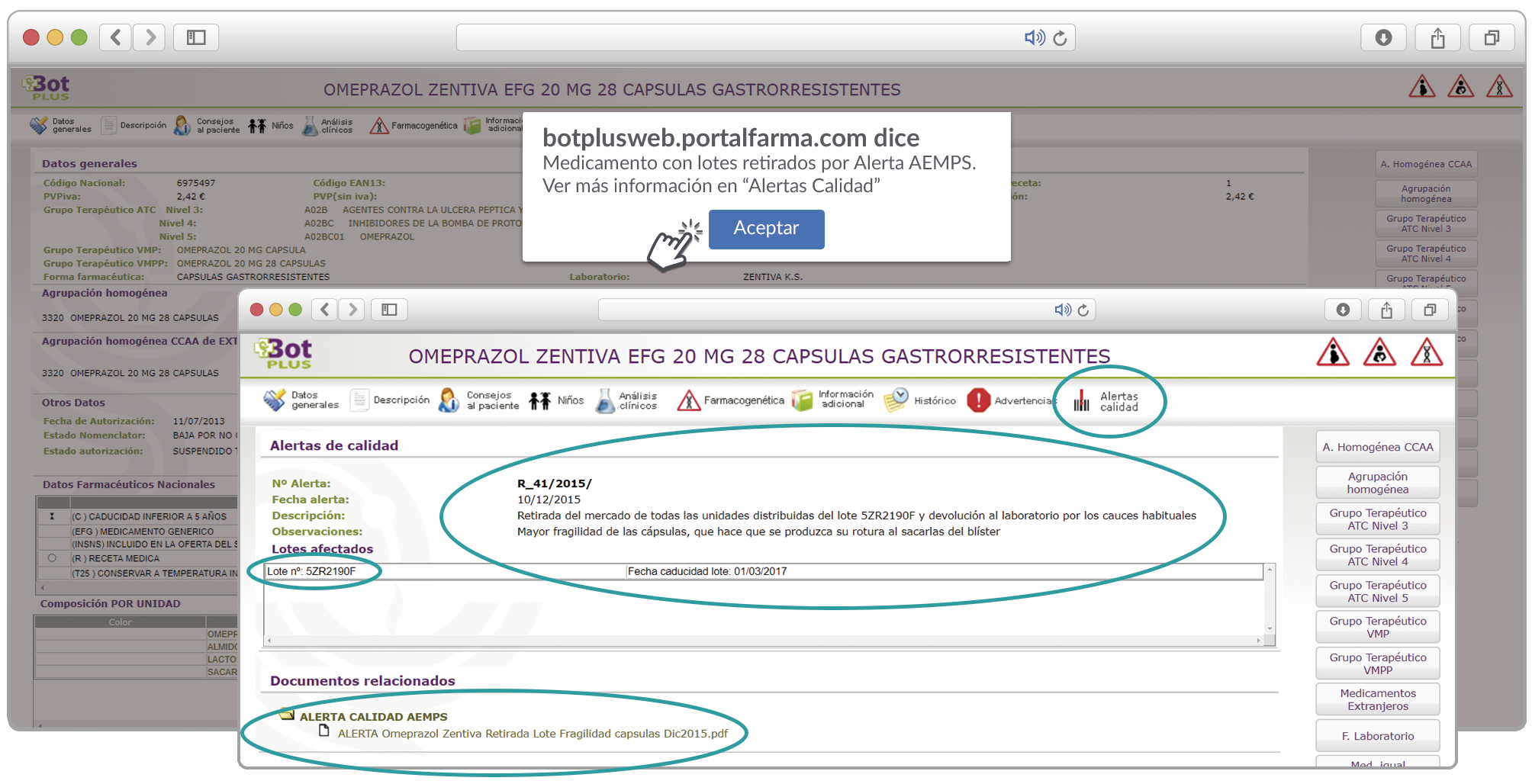
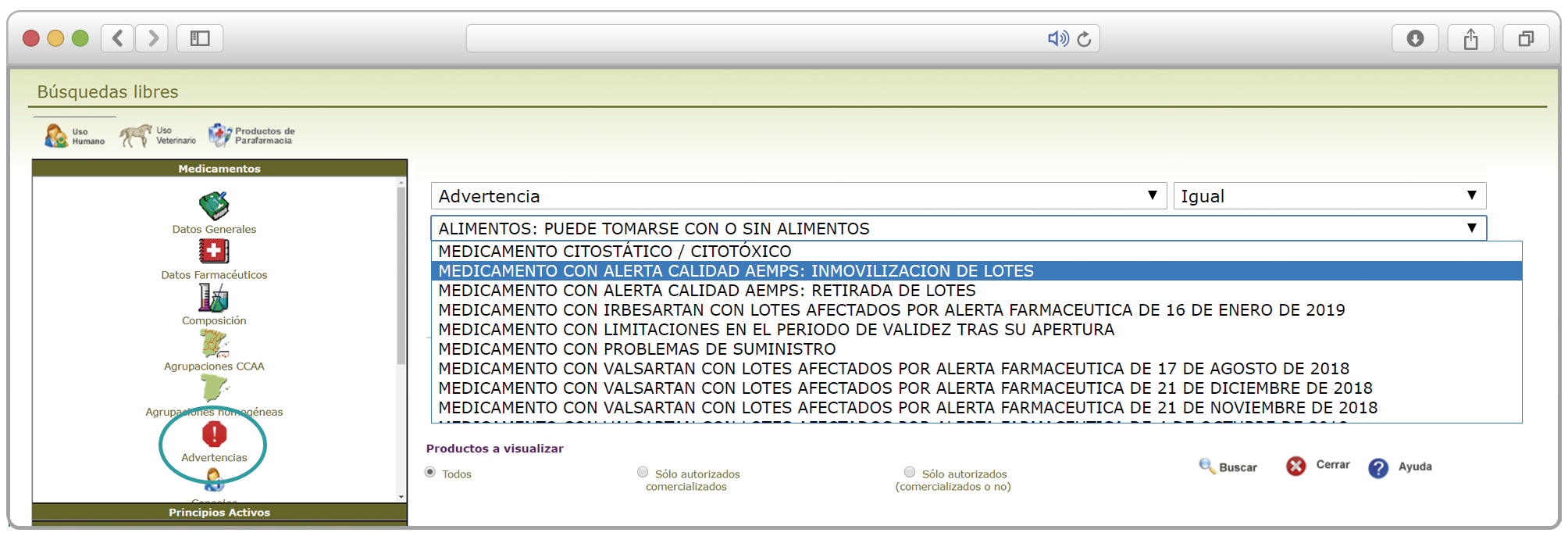
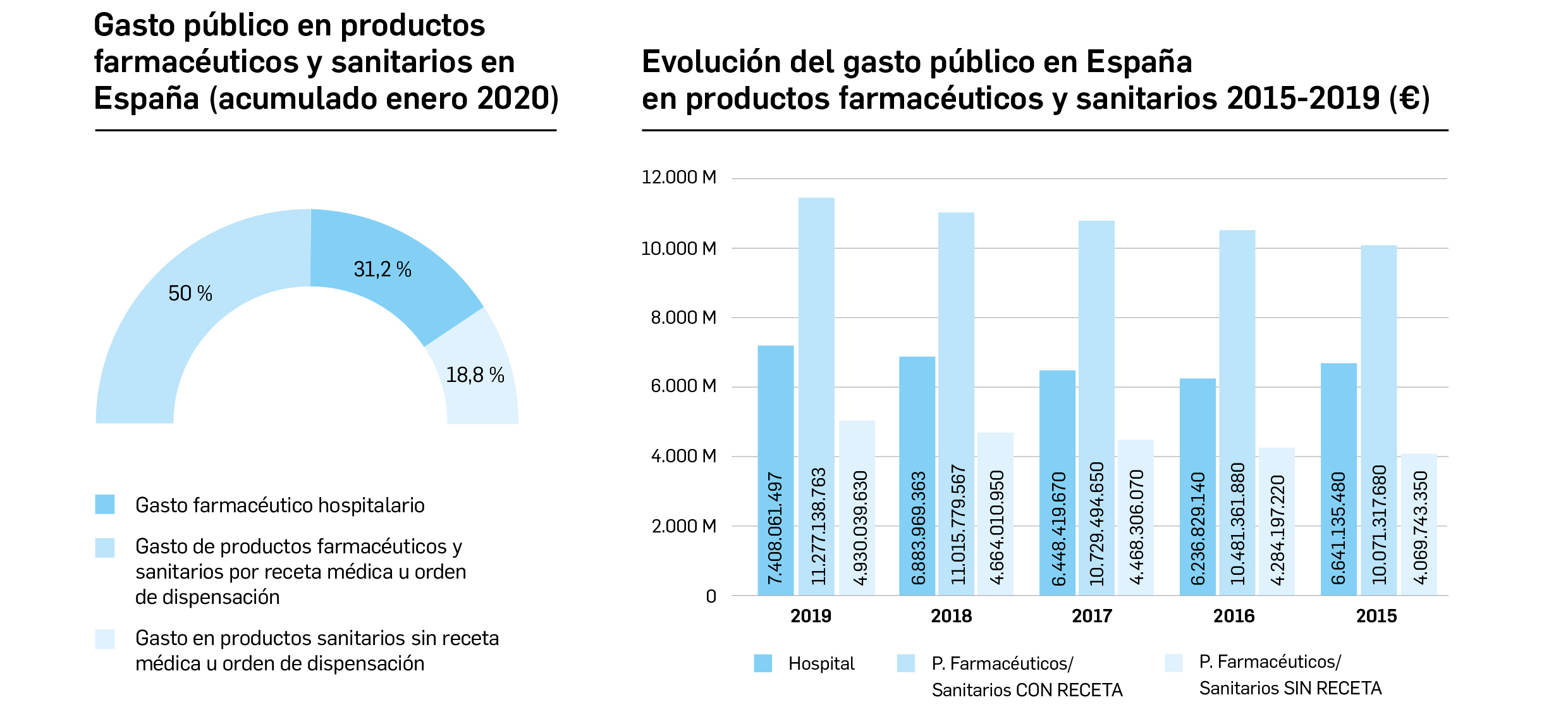
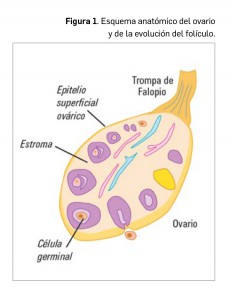
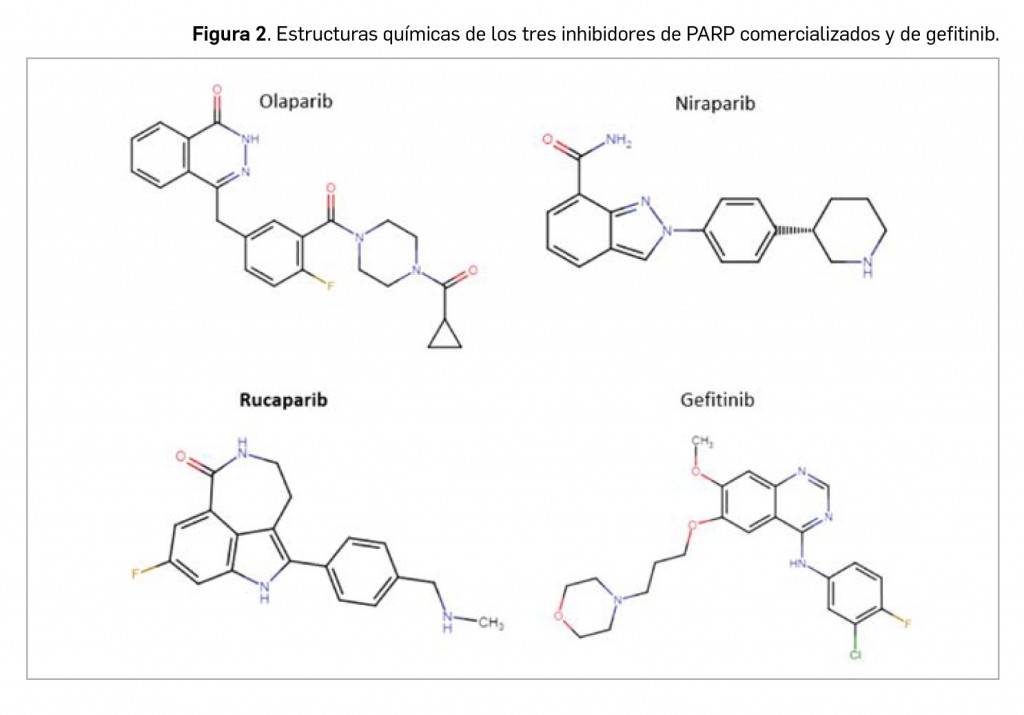
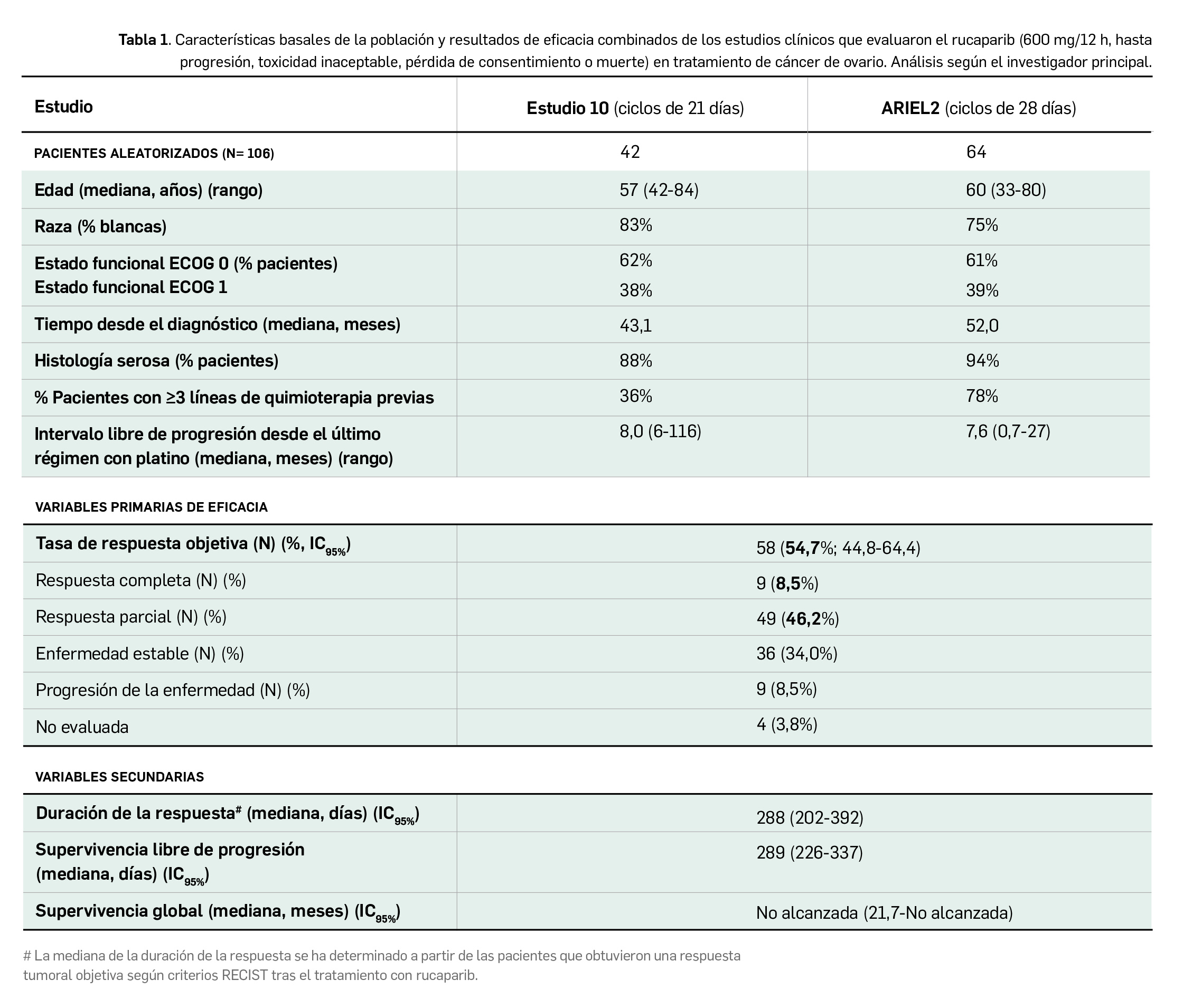
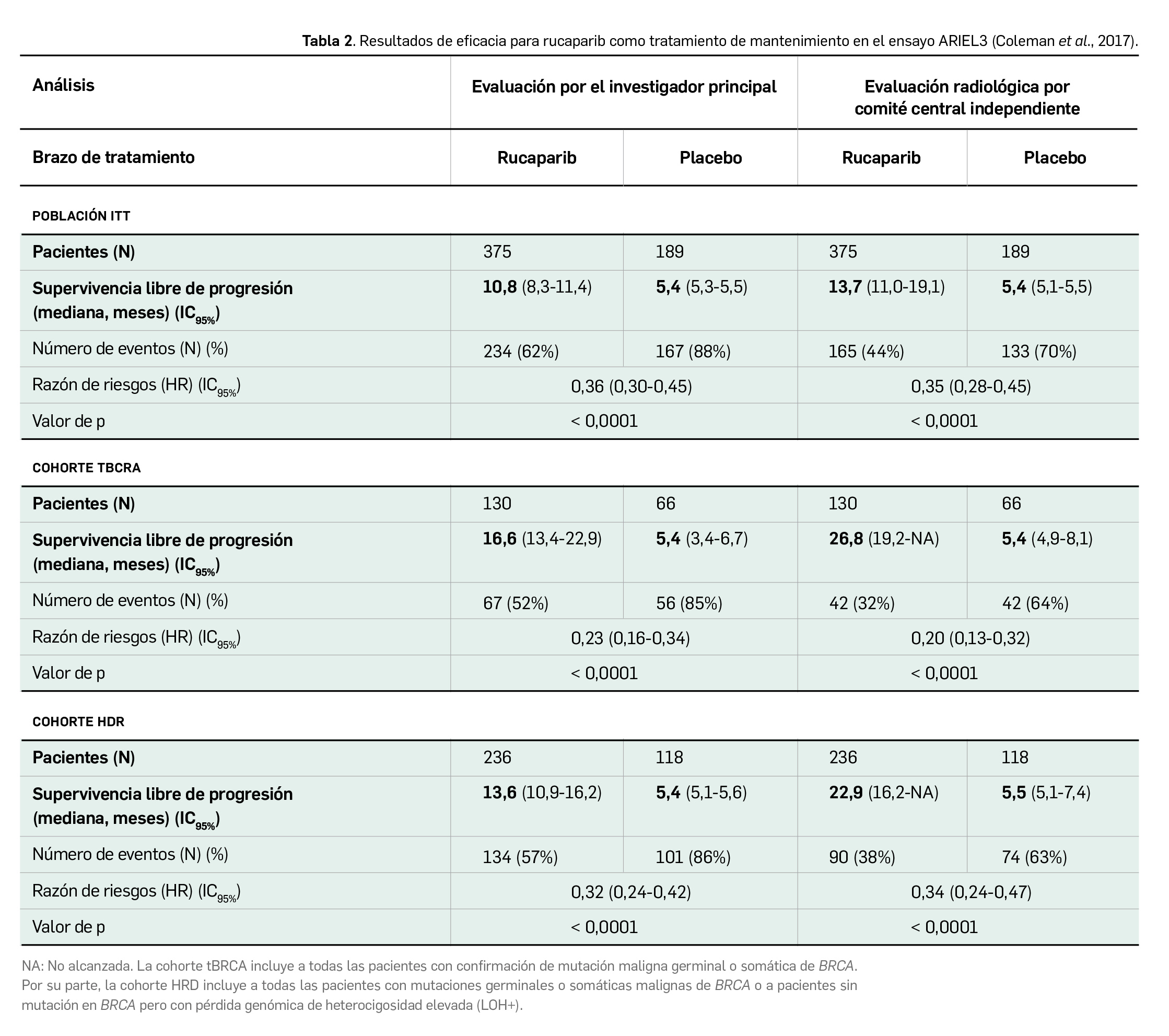


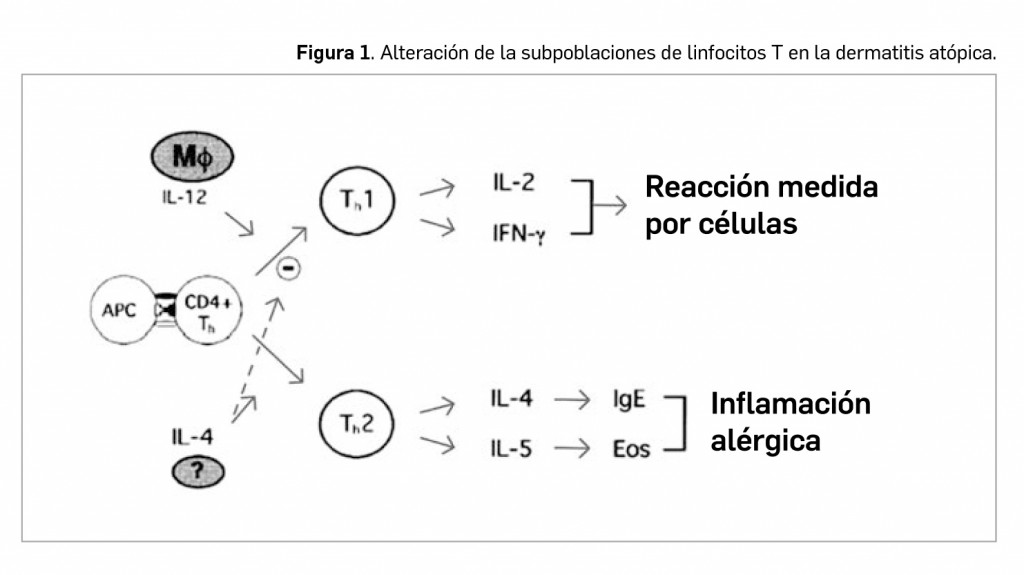
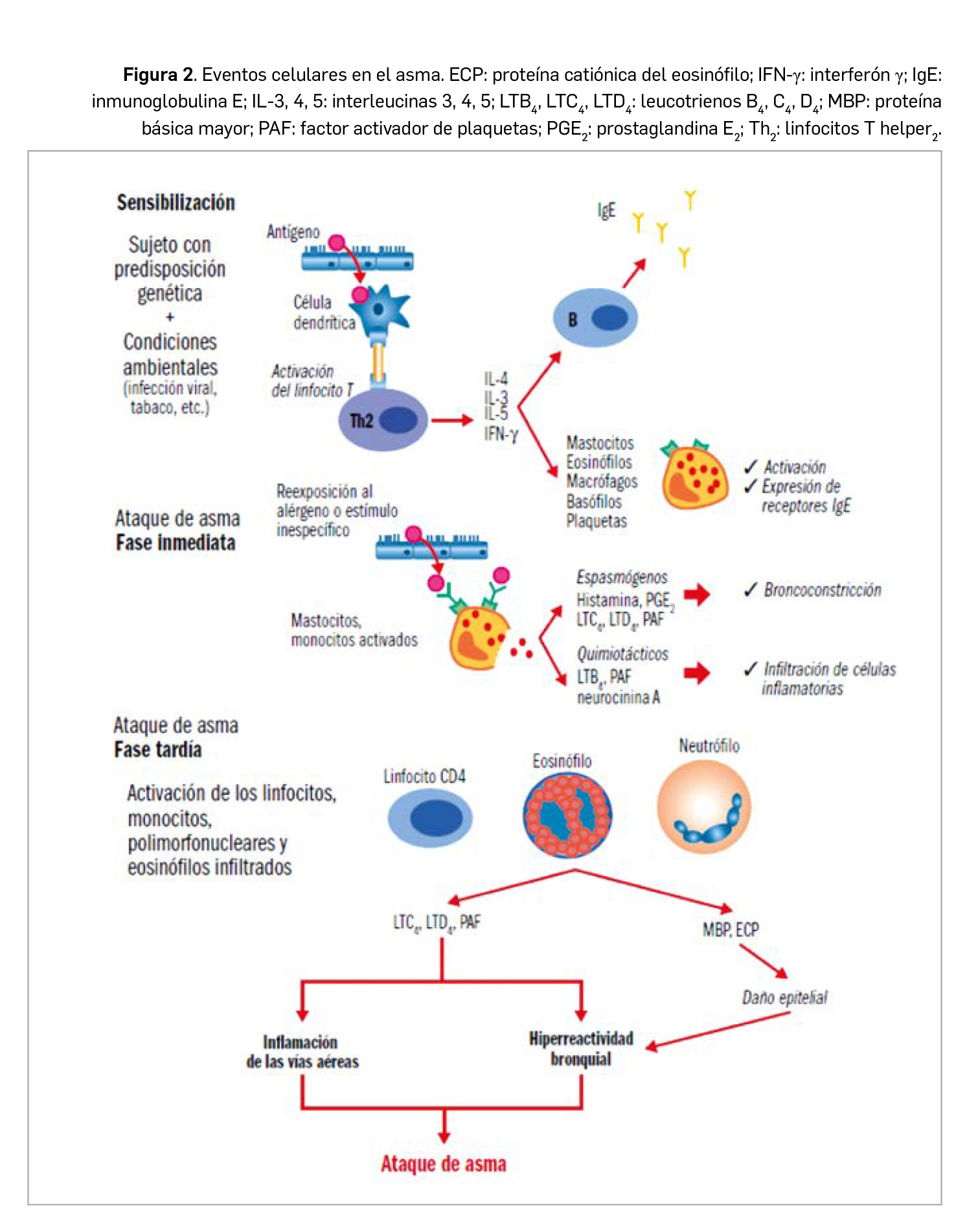
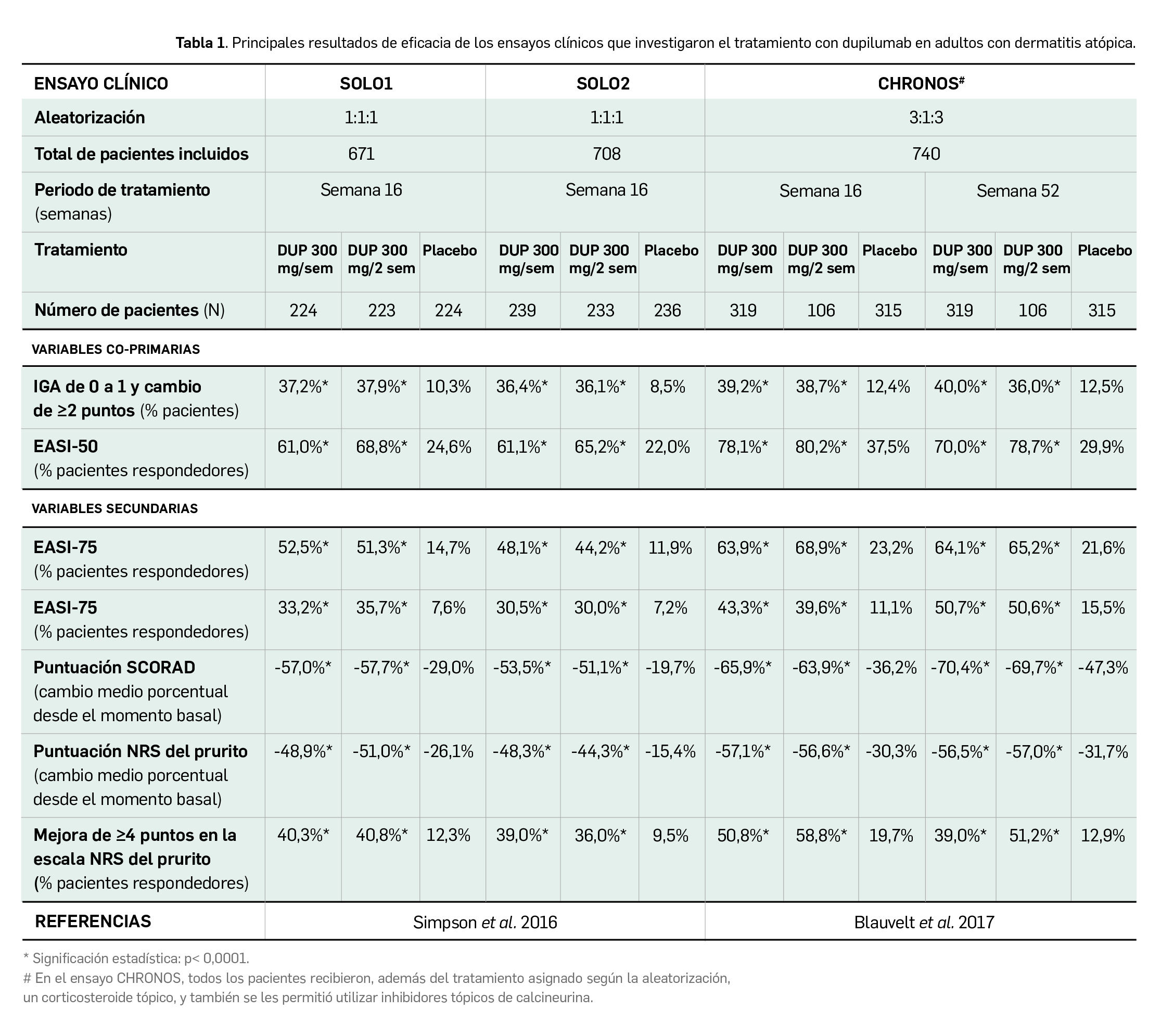

{kind=link}
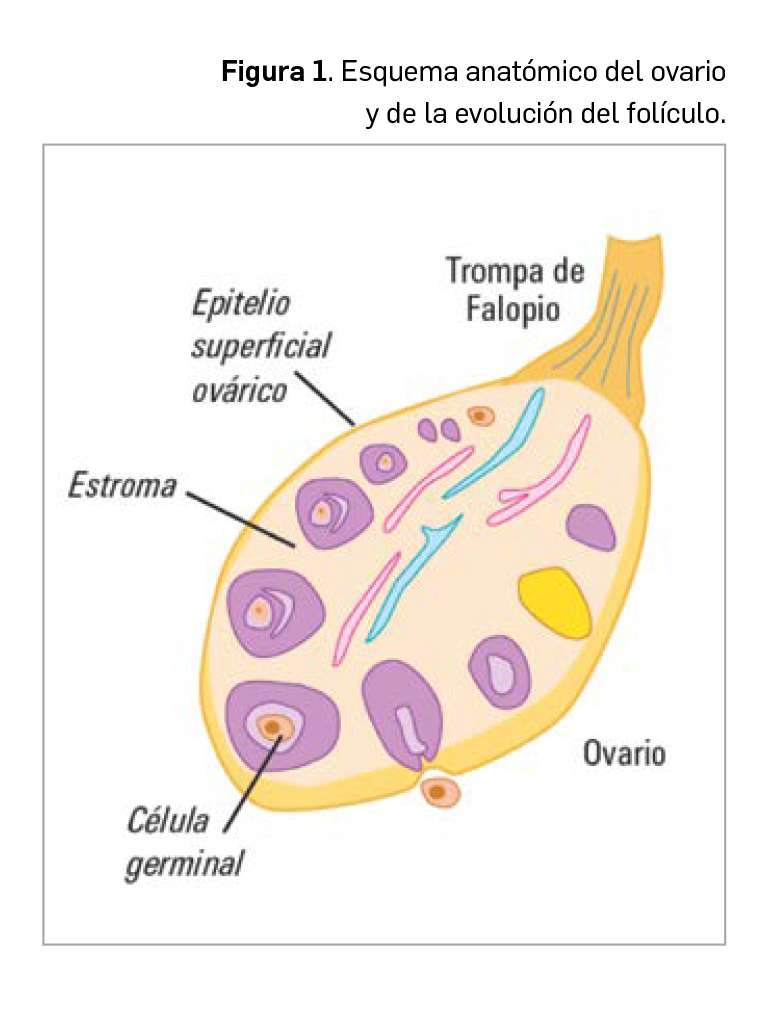{kind=link}
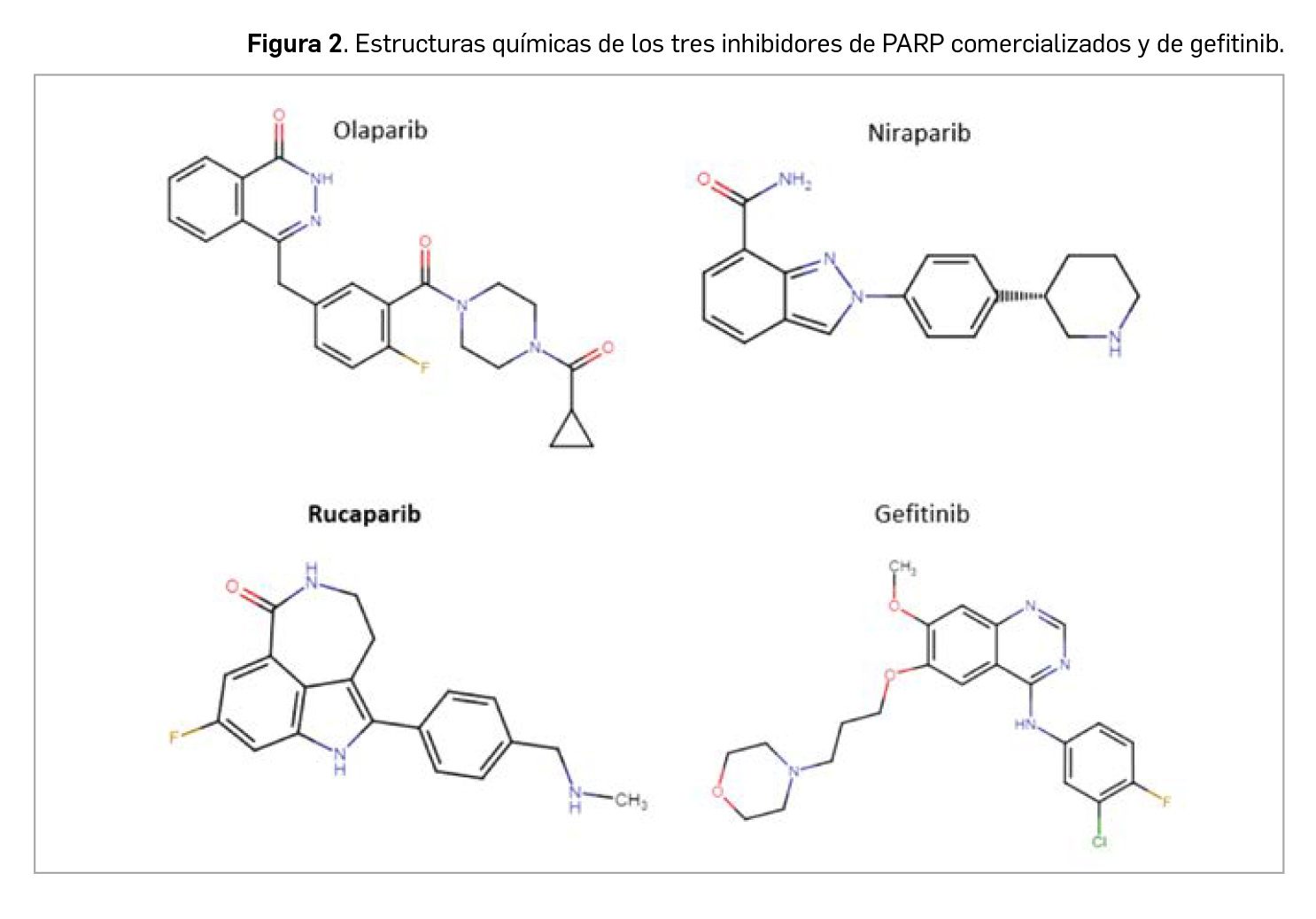{kind=link}
{kind=link}
{kind=link}
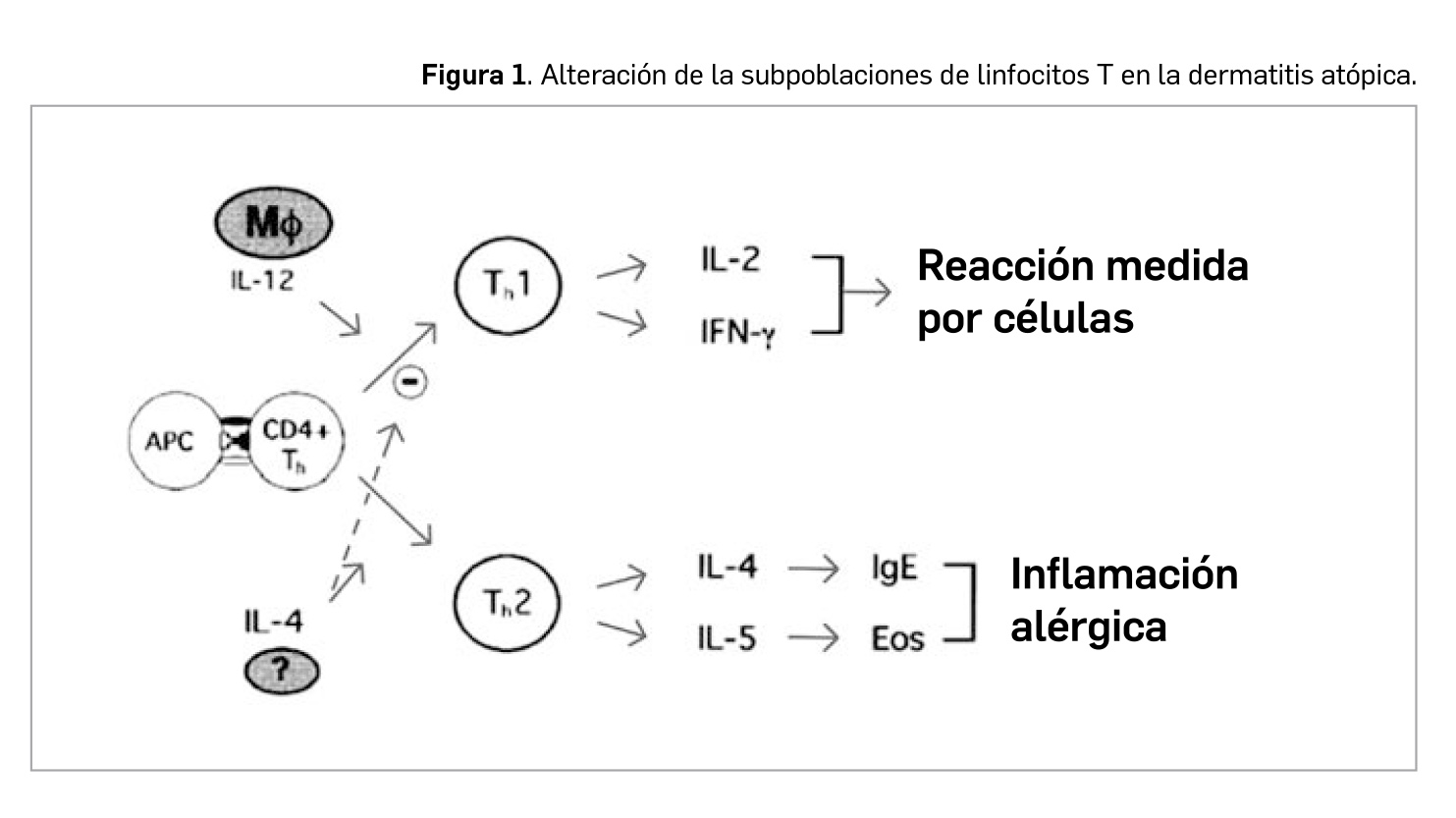{kind=link}
{kind=link}