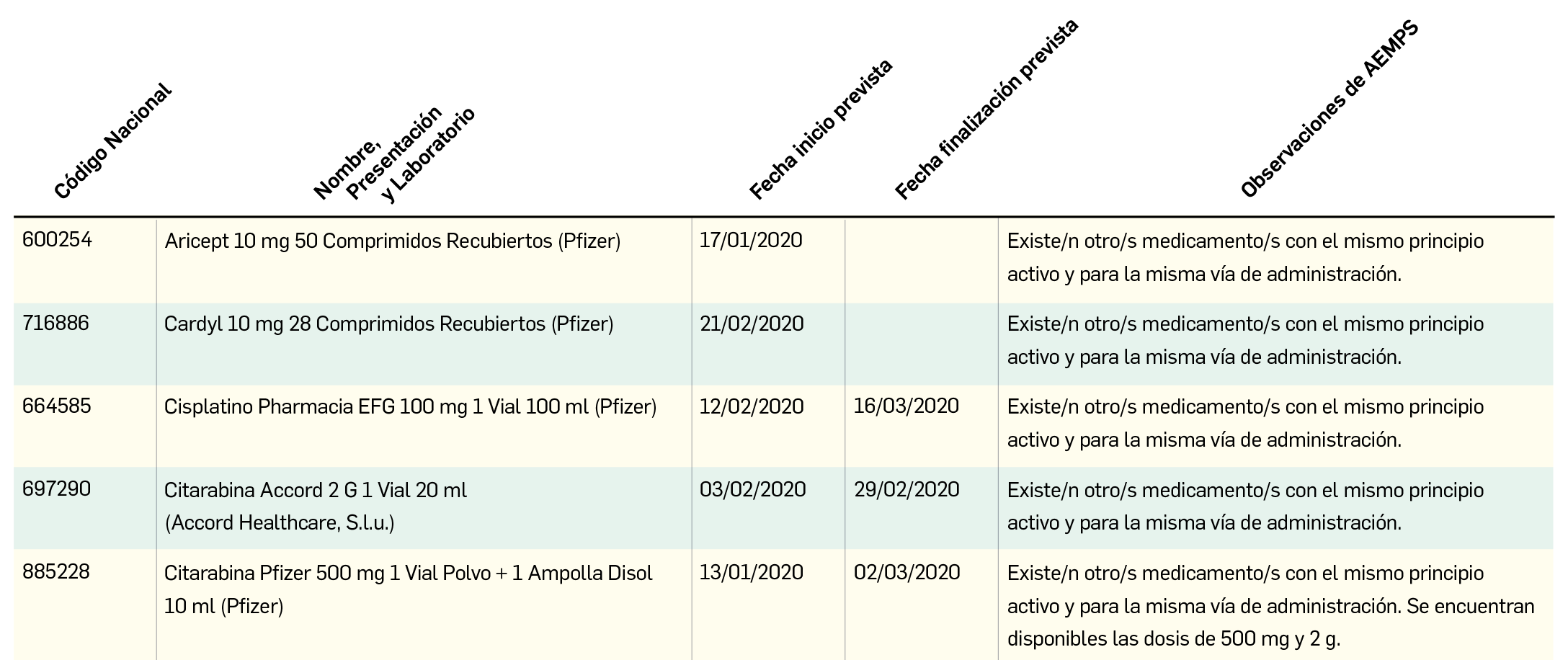
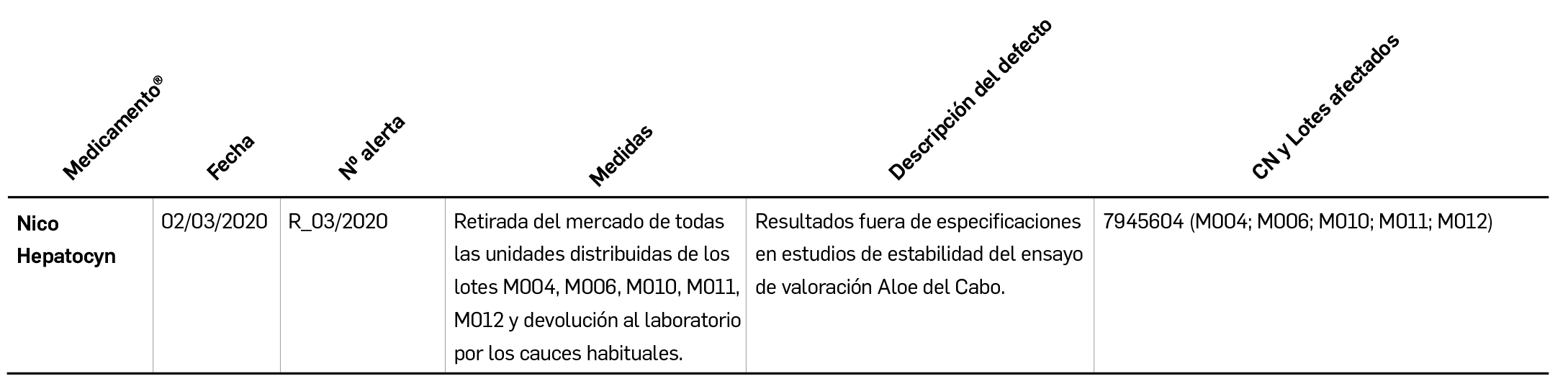
Los resultados del análisis de supervivencia global del estudio ALCYONE confirman los datos ya divulgados para la supervivencia libre de progresión: con una mediana de 5 años de seguimiento, la asociación de daratumumab al régimen quimioterapéutico VMP (bortezomib, melfalán, prednisona) reduce en un 40% el riesgo de muerte en pacientes no candidatos a trasplante de células madre en comparación con el la quimioterapia sola. La tasa de SG a los 36 meses fue 78% en los pacientes tratados con D-VMP frente al 67,9% con VMP.
A día de hoy, el tratamiento estándar de primera línea en pacientes recién diagnosticados de mieloma múltiple (MM) –enfermedad frente a la cual ha habido avances terapéuticos importantes en los últimos años (alcanzando casi la duplicación de la supervivencia de los pacientes)– que no son candidatos a trasplante autólogo de progenitores hematopoyéticos consiste en combinaciones de agentes quimioterapéuticos. De hecho, la asociación de un inhibidor de proteasoma (bortezomib), un agente alquilante (como mefalán) y un corticosteroide (dexametasona o prednisona) es una de las más ampliamente empleadas. La más reciente aparición de fármacos biológicos específicos planteó la posibilidad de combinarlos con los regímenes de quimioterapia en dicha indicación. Tal es el caso de daratumumab (Darzalex®), un anticuerpo monoclonal humano IgG1κ dirigido frente a CD38, proteína que se expresa con un nivel alto en la superficie de las células tumorales de MM.
En este sentido, el análisis primario de un ensayo clínico aleatorizado de fase 3 –ALCYONE–, multinacional y multicéntrico (con importante participación de hospitales españoles), abierto y controlado, demostró que la supervivencia libre de progresión (SLP) con la combinación de daratumumab, bortezomib, melfalán y prednisona (D-VMP) fue significativamente mayor que con la asociación de estos tres últimos fármacos sin daratumumab (VMP) –empleada como comparador activo– en pacientes no candidatos a trasplante, bien por edad (>65 años) o bien por comorbilidades.
Recientemente se han presentado los resultados del análisis intermedio de supervivencia global (SG) de este estudio, en el que 706 pacientes fueron asignados (1:1) a recibir ambos tratamientos durante 9 ciclos1, tras una seguimiento de más de 3 años (mediana de 5 años). En la población por intención de tratar, la asociación de daratumumab a la quimioterapia demostró un beneficio significativo en la SG respecto al grupo control, con una reducción del riesgo de muerte del 40% (HR: 0,60; IC95% 0,46-0,80; p= 0,0003); se estimó una tasa de SG a los 36 meses del 78% en los pacientes tratados con D-VMP vs. 67,9% con VMP. Con ese nuevo periodo de seguimiento, en que casi el doble de pacientes en el grupo de tratamiento con daratumumab tuvieron respuesta completa respecto al comparador, la SLP se mantenía notablemente más alta en el grupo D-VMP, suponiendo una reducción del riesgo de progresión de la enfermedad de casi el 60% (HR: 0,42; IC95% 0,34-0,51; p< 0,0001). Los resultados revelan, además, eventos de seguridad esperables para la monoterapia de mantenimiento con daratumumab, siendo los eventos adversos más frecuentes las infecciones respiratorias del tracto respiratorio superior (19%) –mayoritariamente bronquitis (15%) e infecciones víricas (12%)–, la tos (12%) y la diarrea (10%).
En definitiva, parece evidente que el régimen D-VMP prolonga la supervivencia global en pacientes con mieloma múltiple recién diagnosticado no candidatos a trasplante de células madre (que normalmente tienen un peor pronóstico), manteniendo un menor riesgo de progresión de la enfermedad y sin aparición de nuevos problemas de seguridad. Si se considera que se trata del segundo tumor hematológico más frecuente tras los linfomas, estos resultados apuntan a que puede establecerse un nuevo estándar de tratamiento en el que daratumumab juega un papel importante.