Queridos lectores,
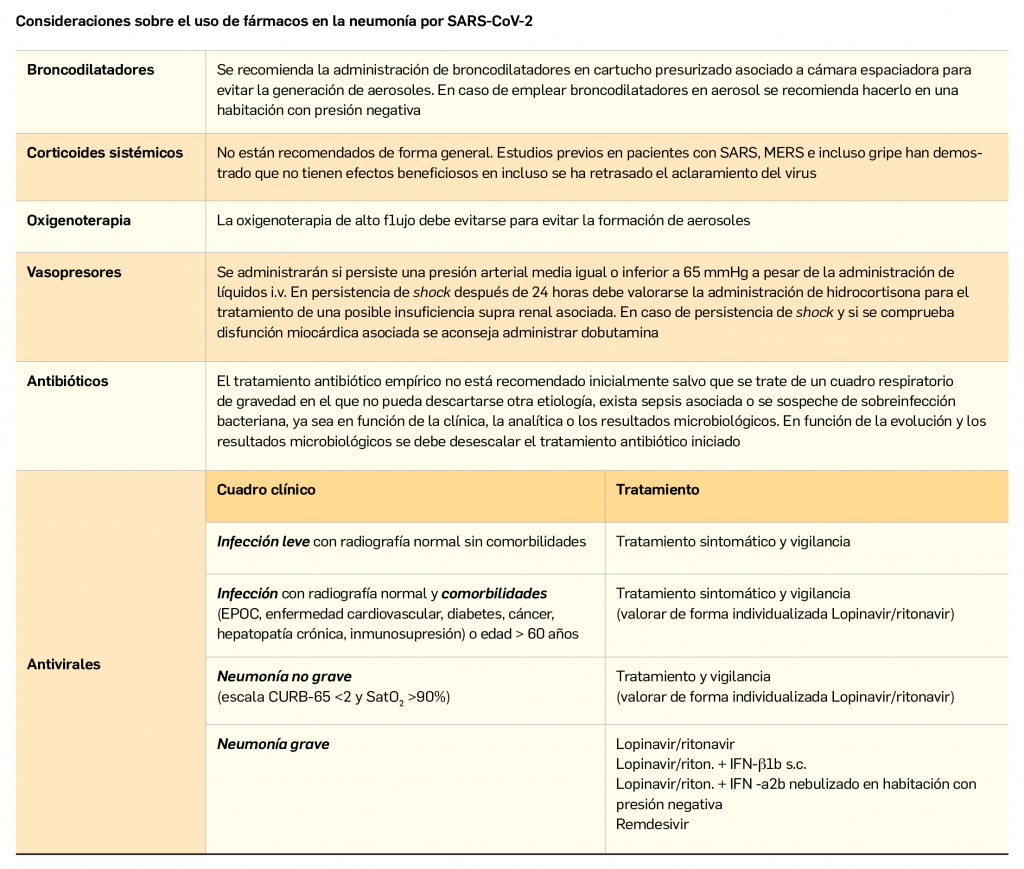
Os presentamos el segundo número de Panorama Actual del Medicamento del año 2020, un año que, por el momento, tiene marcada su actualidad en el ámbito sanitario por el brote epidémico del nuevo coronavirus, bautizado como SARS-CoV-2. Ante la aparición de numerosos casos de infección respiratoria por dicho virus –llamado COVID-19– en España, los Farmacéuticos nos ofrecemos a seguir colaborando con las autoridades sanitarias y el resto de profesionales sanitarios para difundir información fidedigna y actualizada, trasmitiendo a los pacientes y a la población general las medidas higiénicas y preventivas que contribuyan a reducir la propagación del virus. De igual modo, contribuiremos a transmitir un mensaje de calma y aseguraremos la prestación farmacéutica en todo momento.La ausencia actual de medicamentos o vacunas para combatir o prevenir la infección no debe hacernos desconfiar de los investigadores y laboratorios farmacéuticos, muchos de los cuales ya están centrando sus esfuerzos en encontrar y testar una vacuna eficaz y segura frente al SARS-CoV-2. Como ya se consiguió con el virus de la gripe estacional y otros virus, no podemos sino mantener la esperanza de que, en plazos razonables (la OMS habló de 1,5-2 años), estaremos divulgando en PAM la autorización –al menos condicional– de nuevas herramientas terapéuticas frente a esta nueva infección. Por el momento, se presenta en este número de PAM un artículo sobre las generalidades del COVID-19.
En cuanto a otros contenidos de PAM 431, destaca la revisión monográfica de apertura que versa sobre el estado actual del uso terapéutico del cannabis y los cannabinoides, un tema que en los últimos años viene generando cierto debate, no solo entre los profesionales vinculados a la sanidad sino también en la sociedad. Habida cuenta de que la EMA (European Medicines Agency) ha autorizado hace escasamente 3 meses un nuevo medicamento a base de cannabidiol, este artículo pretende, además de revisar la farmacología, mostrar la situación legal de los productos derivados del cannabis tanto en España como en otros países de Europa. Tratamos en este número también temas de salud ocular tan interesantes como las enfermedades de la córnea o el glaucoma.
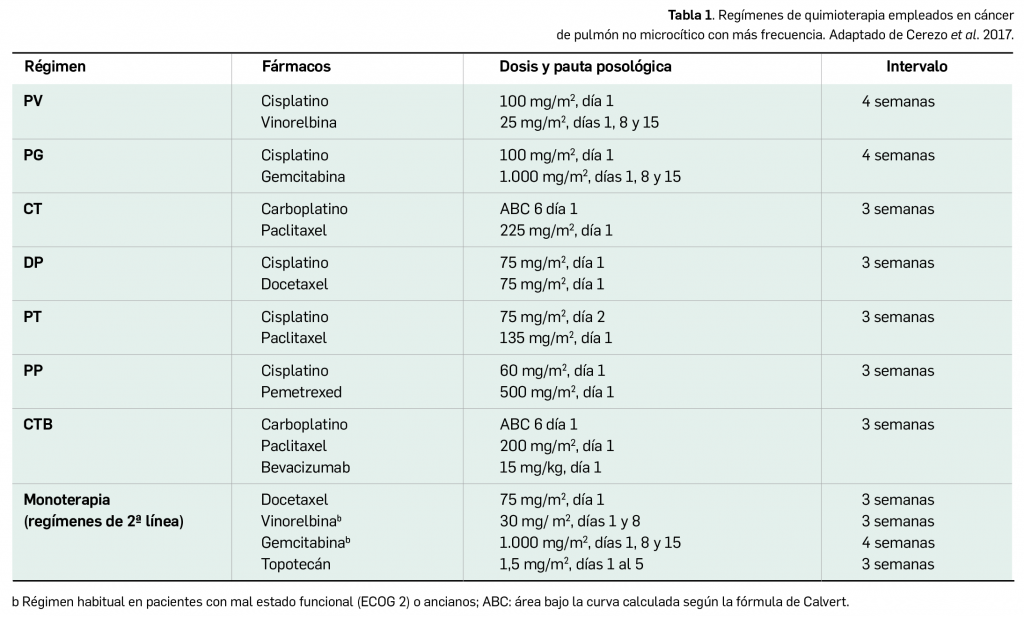
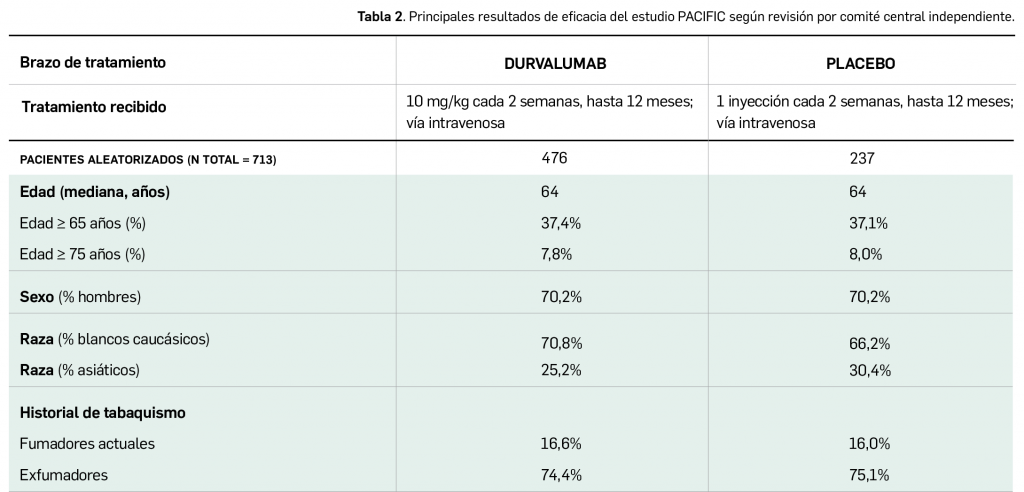
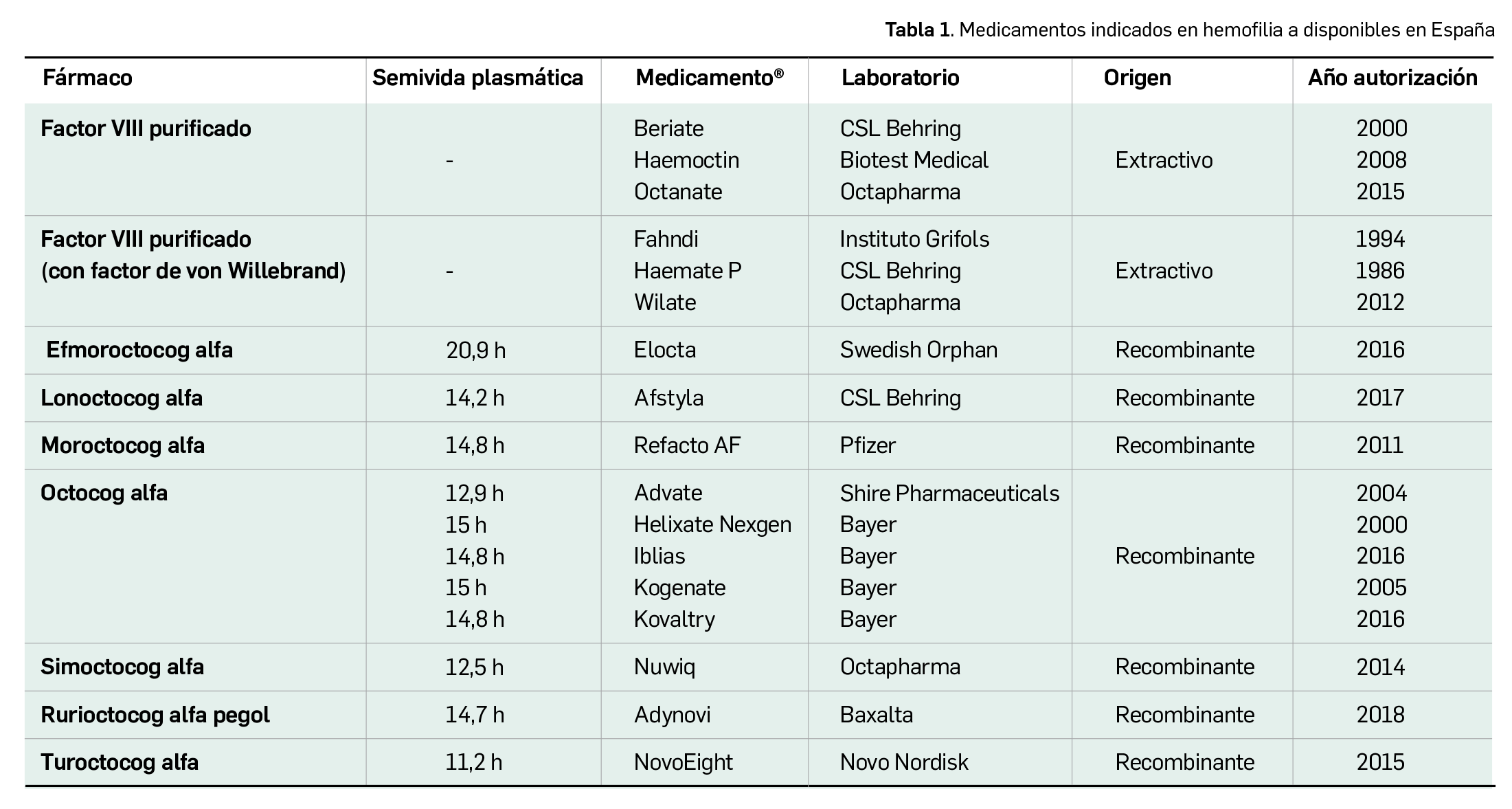
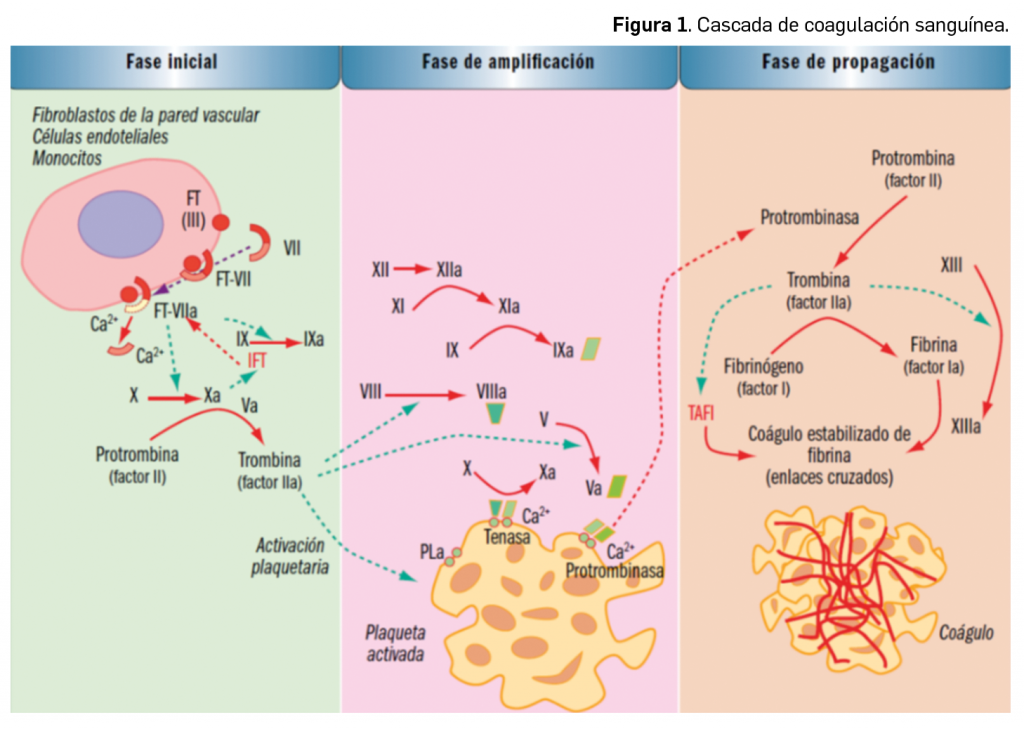
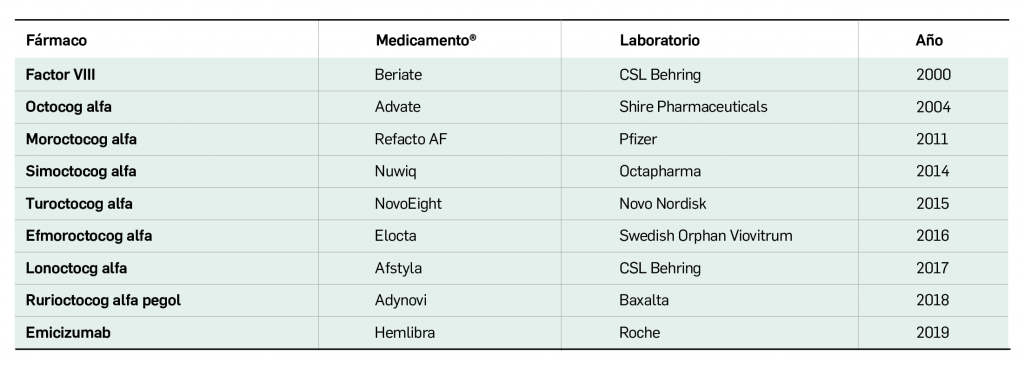
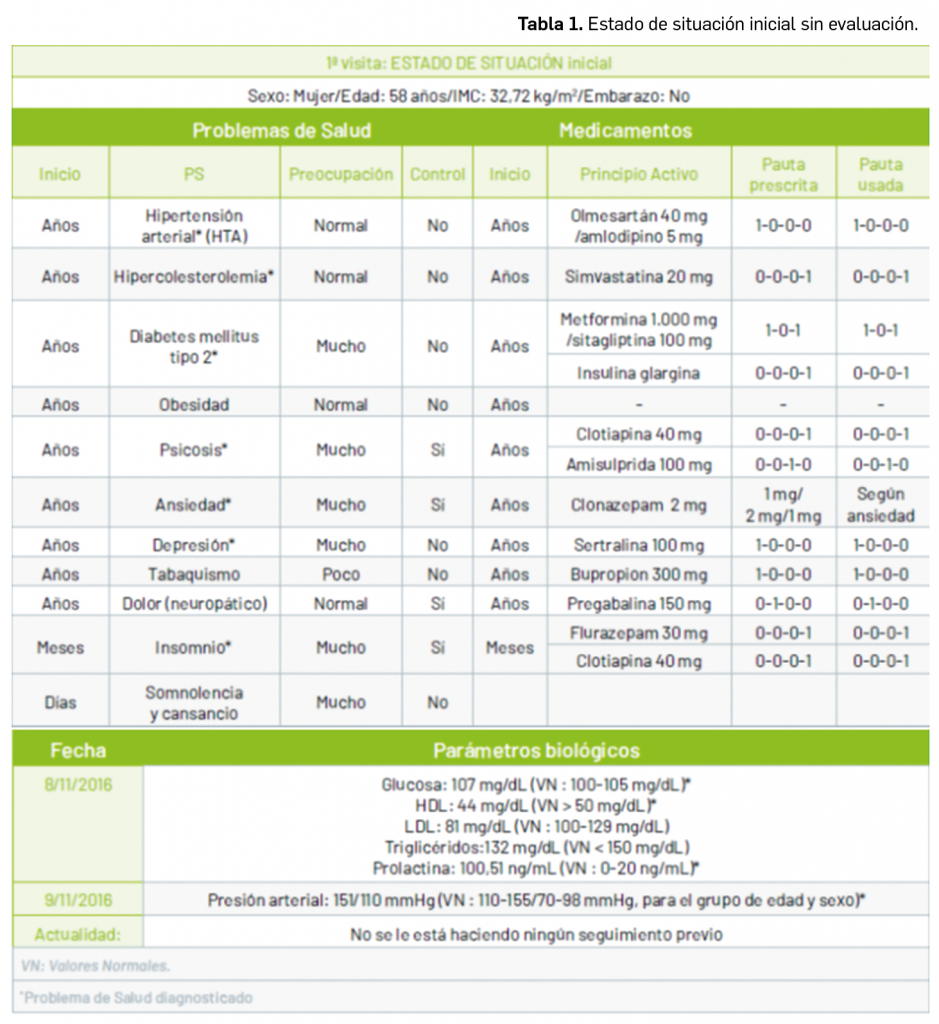
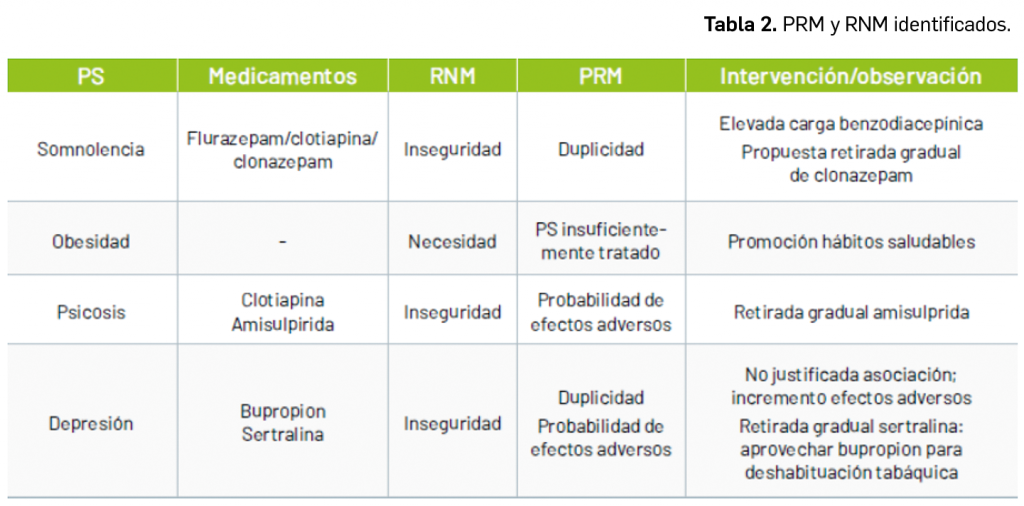
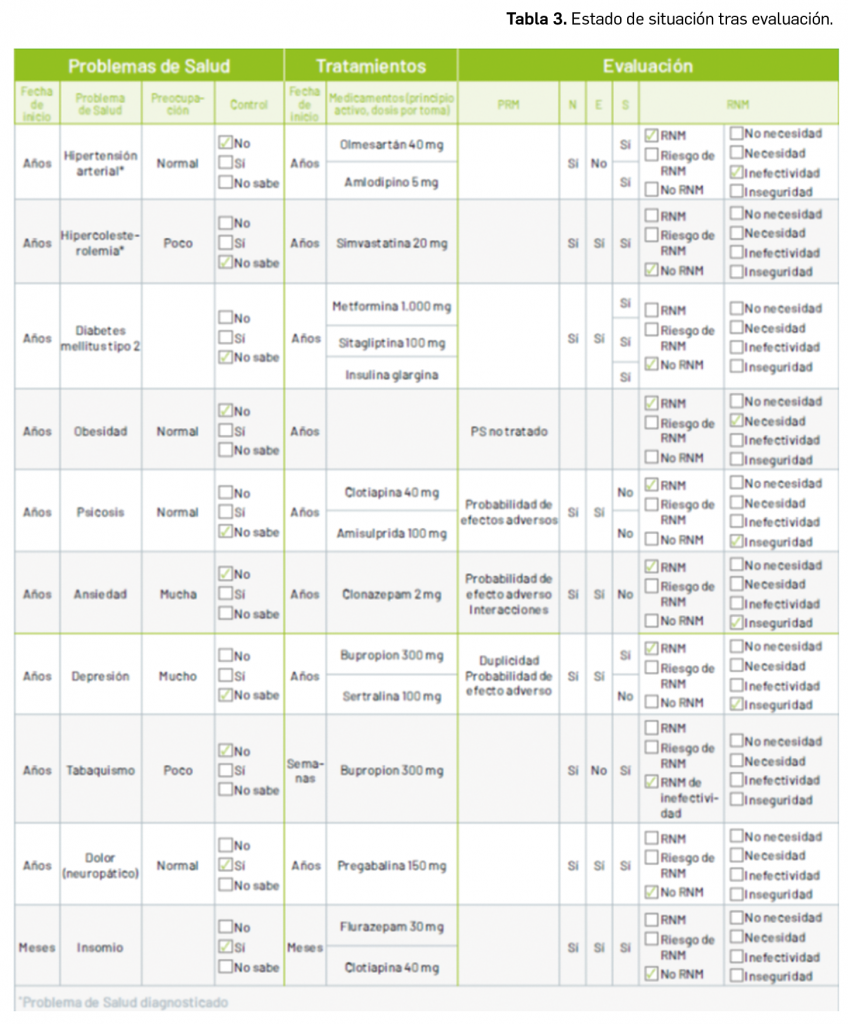
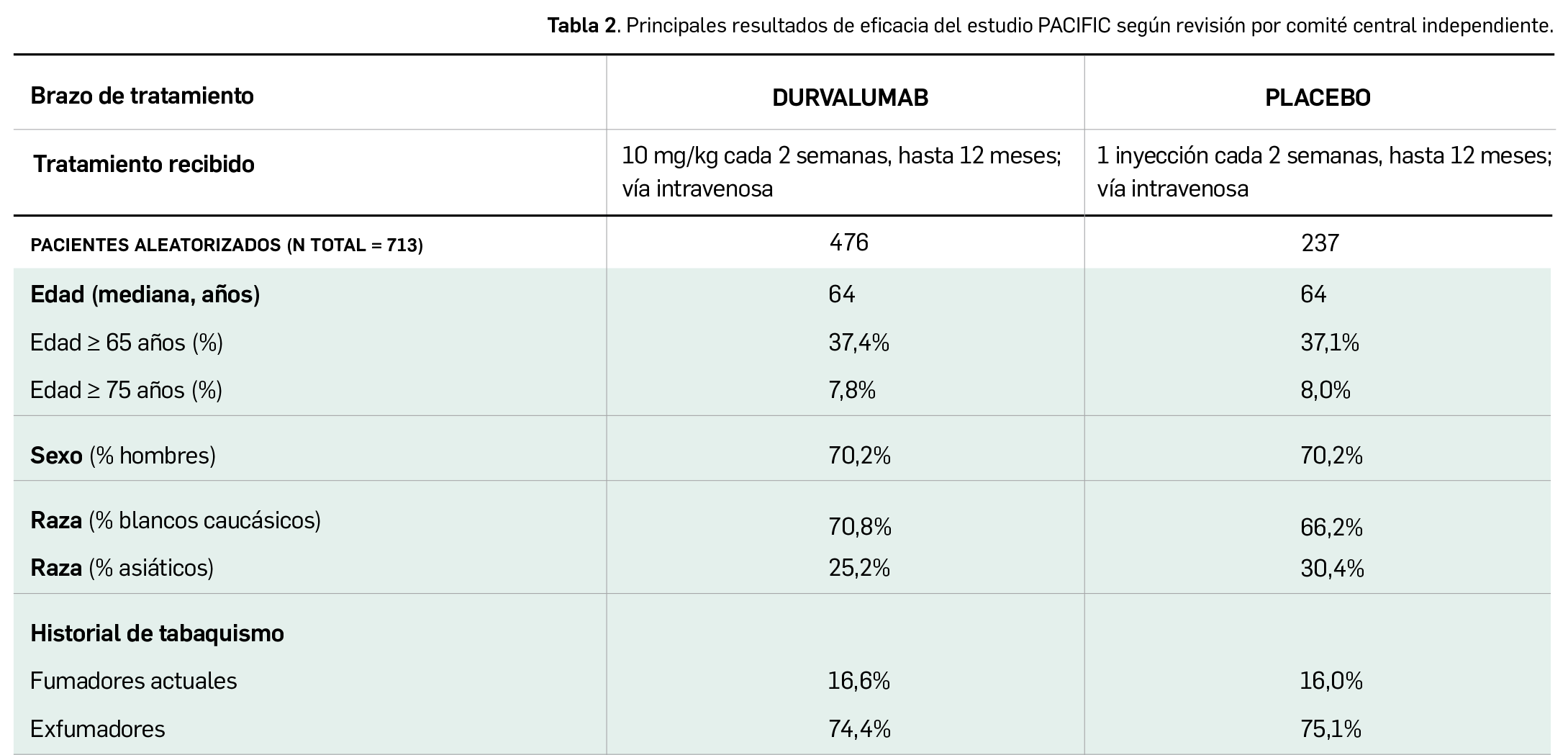
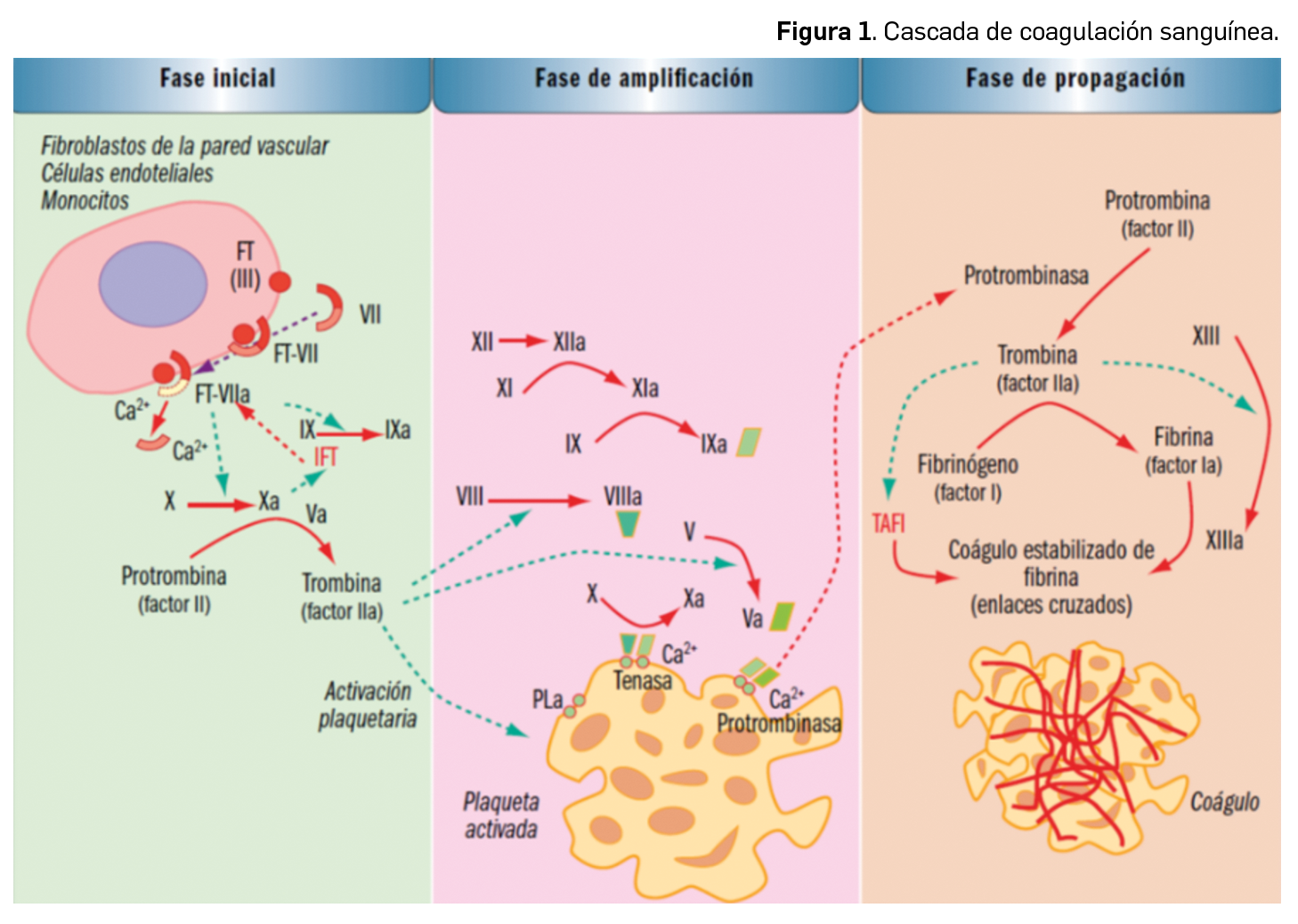
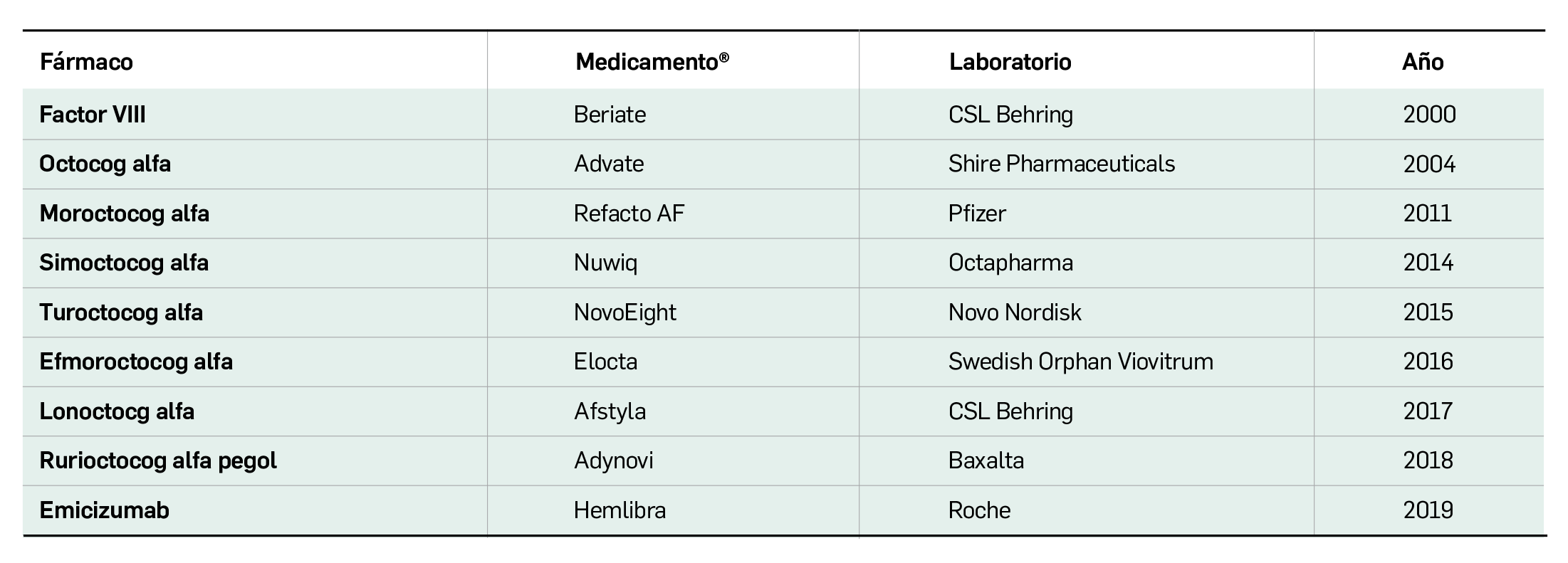
Por otra parte, continuando con la evaluación de medicamentos con nuevos principios activos comercializados por primera vez en España, incluimos aquí la evaluación de dos nuevos fármacos recombinantes del factor VIII de la coagulación, los cuales, si bien no suponen una innovación terapéutica disruptiva, contribuyen a ampliar el abanico de posibilidades terapéuticas en el tratamiento y prevención de hemorragias en una enfermedad rara como la hemofilia A. Además, el pasado mes de febrero se incorporó al arsenal terapéutico un nuevo anticuerpo monoclonal anti-PD-L1, durvalumab (Imfinzi®), que emerge como un posible nuevo estándar de tratamiento en la fase de consolidación de la quimiorradioterapia con intención curativa en pacientes con cáncer de pulmón no microcítico avanzado y no operable.
Sin más preámbulo, confiamos en que disfruten y se enriquezcan con su lectura.
- Ana Isabel López-Casero Beltrán – Directora de Panorama Actual del Medicamento y Tesorera del Consejo General de Colegios Farmacéuticos de España.
- Carlos Fernández Moriano – Editor científico y coordinador de Panorama Actual del Medicamento.














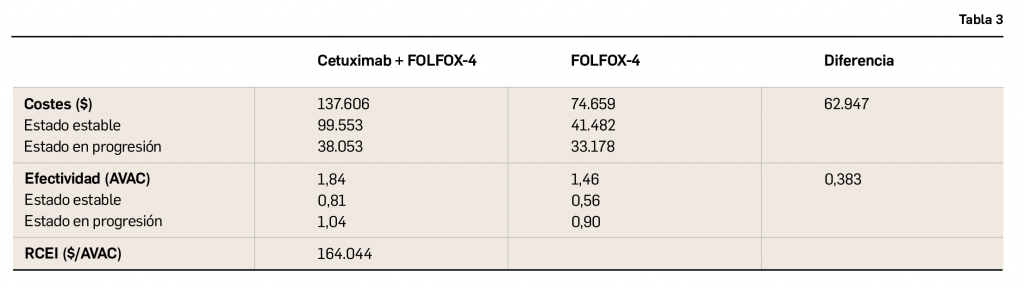
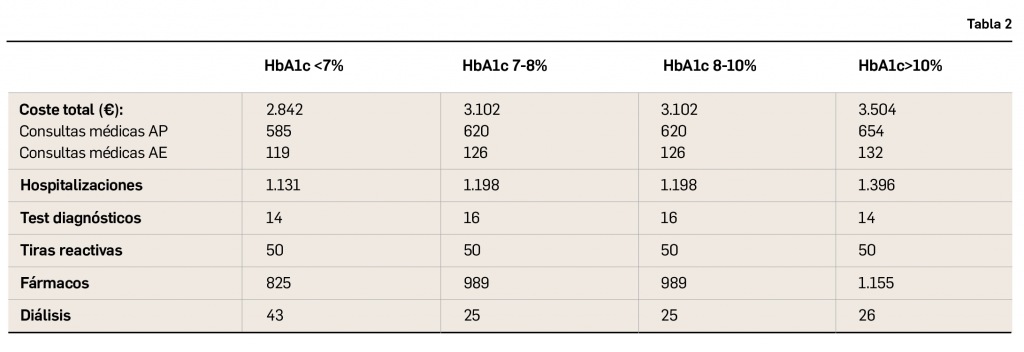
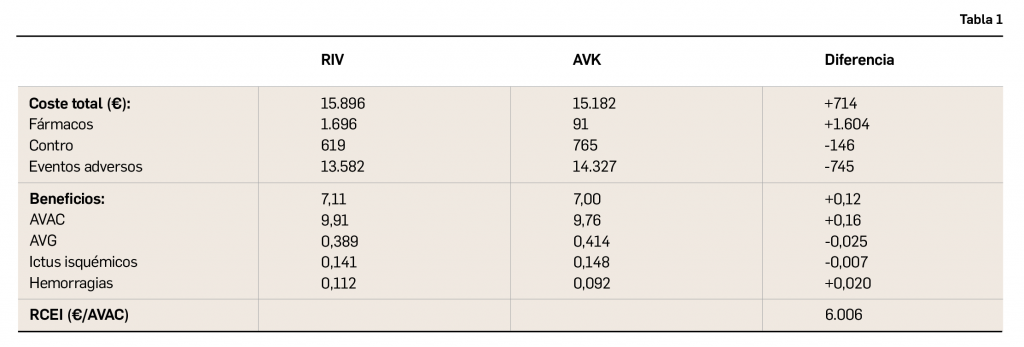
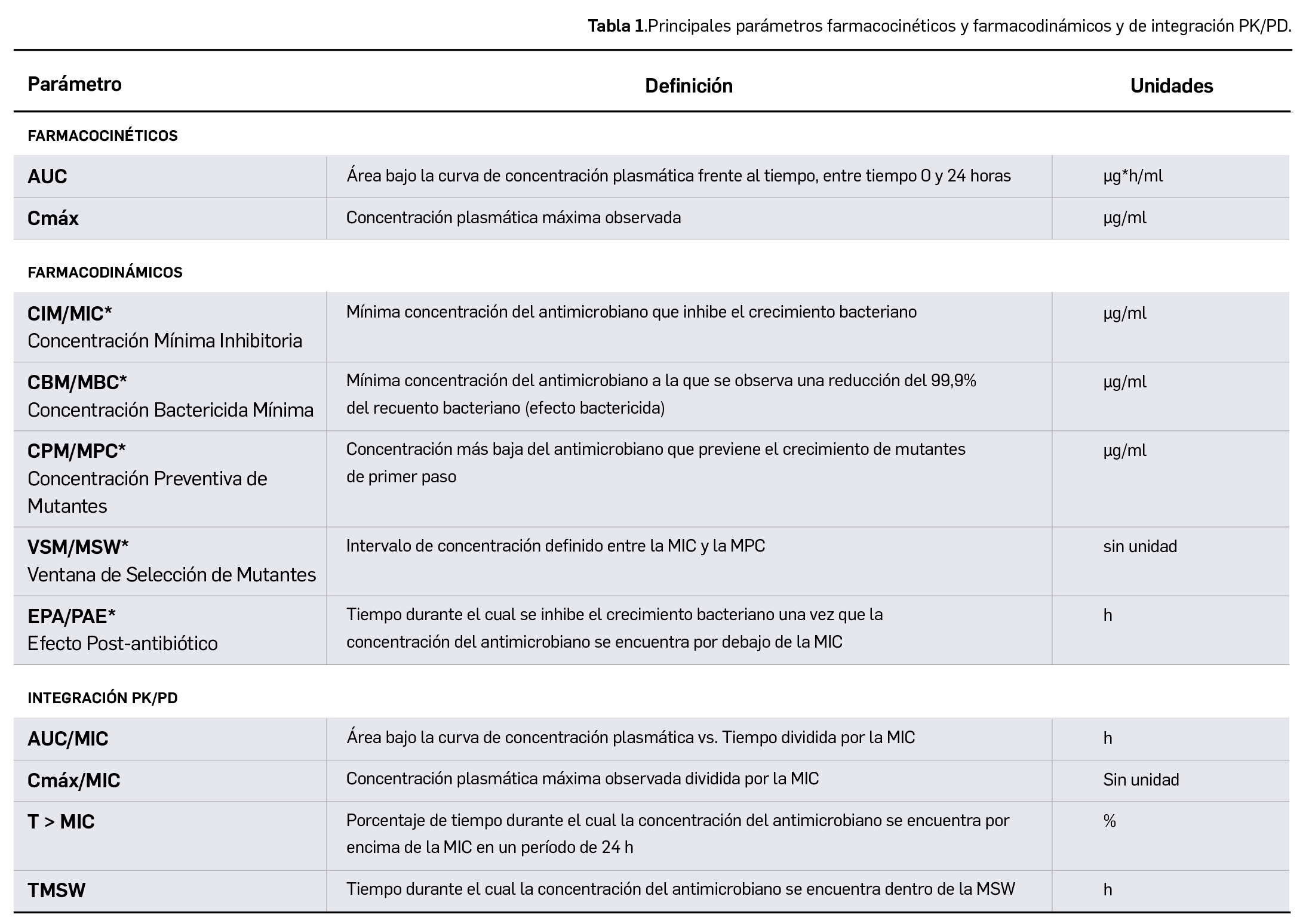
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}