Resumen
NC1 es un novedoso medicamento de terapia avanzada cuyo principio activo son células mesenquimales troncales adultas obtenidas de la médula ósea del propio paciente (autólogas), expandidas ex vivo (in vitro) y resuspendidas previamente a su administración en plasma del mismo paciente. Estas células pluripotenciales, inyectadas –por punción lumbar– en el espacio subaracnoideo en el lugar de la lesión medular traumática, podrían posiblemente sufrir el proceso de transdiferenciación a células nerviosas (al entrar en contacto con factores tróficos solubles liberados localmente por células gliales del tejido hospedador), así como desencadenar fenómenos de neurogénesis endógena y regeneración tisular en la región lesionada. En base a ello, la AEMPS ha otorgado al Hospital Universitario Puerta de Hierro la primera autorización de uso para la fabricación y administración del medicamento, con indicación en el tratamiento de pacientes adultos (≤65 años) con secuelas de lesión medular traumática crónica, que presenten lesiones incompletas a nivel dorsal o lumbar; se excluyen pacientes con lesiones completas, excepto aquellas localizadas quísticas con cavidad centro-medular que no se extienda más de 1-3 segmentos medulares.
A pesar de que los datos clínicos tienen limitaciones (proceden de ensayos con un número reducido de pacientes y con diseño abierto y no controlado), el perfil beneficio-riesgo es muy prometedor, con una eficacia dependiente de la dosis pero independiente de factores como la edad del paciente, el nivel, la gravedad o el tiempo de cronificación de la lesión. Tras periodos de seguimiento de 10-12 meses, todos los pacientes mostraban una mejoría estadísticamente significativa de la sensibilidad y de las capacidades motoras (con indicios electromiográficos de reinervación muscular) por debajo de la lesión, según puntuaciones de la escala ASIA; dicho beneficio se observaba desde los primeros meses tras la administración del medicamento. Además, una gran mayoría de pacientes mejoraron el grado de discapacidad funcional (según escala IANR-SCIFRS) y el control de los esfínteres y la función vesical; también hubo mejorías notables en la función sexual, el dolor neuropático y la espasticidad muscular de algunos pacientes. Tales resultados se acompañan de una buena tolerabilidad: la mayoría de eventos adversos notificados fueron de gravedad leve-moderada y aquellos posiblemente relacionados con el tratamiento (32-40%), ninguno grave, se asocian al procedimiento de punción lumbar (cefalea, dolor postoperatorio, dolor ciático, hipertermia transitoria o infiltración subcutánea en la herida quirúrgica). No obstante, los seguimientos máximos de 1 año postratamiento no permiten descartar el riesgo de carcinogénesis a largo plazo, que deberá ser esclarecido con seguimientos más largos.
Así, el fármaco inaugura una prometedora vía de tratamiento en su indicación, tratándose del primer medicamento de terapia avanzada (terapia celular) que demuestra un beneficio clínico en la reversión –al menos parcial– de las secuelas de una lesión medular traumática, con importantes mejorías en aspectos tan trascendentes para estos pacientes como la sensibilidad en las extremidades y el control de los esfínteres. Supone una innovación terapéutica disruptiva que mejorará su calidad de vida, en situaciones que hasta ahora no tenían ningún tratamiento eficaz. La mejora del conocimiento científico con la experiencia de uso en la práctica real podría abrir la puerta a la esperanza de alcanzar la curación de la discapacidad que generan las lesiones medulares.
Introducción
Al amparo de lo estipulado en el Real Decreto 477/2014, de 13 de junio, por el que se regula la autorización de medicamentos de terapia avanzada de fabricación no industrial, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) autorizó el pasado 29 de enero de 2019 el medicamento “NC1 – Suspensión celular en plasma autólogo 100-300×106 células 1 jeringa precargada”, concediéndole un código nacional (CN 724688), como tienen el resto de medicamentos.
Desarrollado por el Hospital Universitario Puerta de Hierro de Majadahonda (Madrid), se trata del primer medicamento de terapia avanzada de fabricación no industrial –entre la comunidad biomédica, también se les conoce coloquialmente como medicamentos académicos o terapias avanzadas académicas– que recibe una autorización de uso (AU) por parte de la AEMPS, la cual presenta ciertas características diferentes a las autorizaciones de comercialización comúnmente concedidas a los medicamentos de fabricación industrial, pues habilita a la producción y administración del medicamento exclusivamente a ese hospital (se trata de un medicamento calificado como de Uso Hospitalario por la AEMPS), el único centro que podrá llevarlas a cabo.
No obstante, conviene recordar que el procedimiento para la solicitud de una AU no es diferente a la presentación de cualquier otro dossier para la autorización de un medicamento, debiendo acreditar, con las especificidades propias de las terapias avanzadas, los elementos pre-clínicos, clínicos y de calidad a fin de que se pueda evaluar el balance beneficio/riesgo positivo. Las AU contienen generalmente elementos de condicionalidad (se conceden por 3 años y son revisables), pues obligan y comprometen a su titular a continuar con la investigación clínica –y presentar un informe anual1 – para mantenerla, igual que ocurre con el resto de terapias avanzadas a nivel de la UE, por lo que podría considerarse como un proceso similar a una autorización condicional por procedimiento centralizado. La exención hospitalaria se ha planteado como una posible herramienta que facilite la investigación y el desarrollo de terapias avanzadas por parte de organizaciones sin ánimo de lucro (instituciones académicas y hospitales), como paso previo a la solicitud de autorización de comercialización, siempre y cuando aseguren la demostración de un perfil adecuado de seguridad y eficacia clínicas.
De forma posterior a su autorización, el medicamento NC1 fue incluido por primera vez en el mes de octubre de 2019 en el Nomenclátor oficial de la prestación farmacéutica del Sistema Nacional de Salud, que mensualmente actualiza el Ministerio de Sanidad.
Lesiones medulares traumáticas
La médula espinal es la parte del sistema nervioso central –compuesta de tejido blando y rodeada de estructuras óseas específicas (vértebras)– que se extiende caudalmente desde la base del cerebro por la parte central de la espalda, a través de la cual transcurren una serie de grupos de nervios llamadas vías. Éstas permiten emitir señales desde el cerebro para controlar el movimiento de los músculos del resto del cuerpo (vías motoras o eferentes) y recibir sensaciones muy diversas en el cerebro desde las distintas partes del cuerpo (vías sensoriales o aferentes).
Normalmente se define como lesión de la médula espinal aquella que provoca disfunción neurológica, con o sin alteración de la estructura ósea (columna vertebral) que la protege. Implica daño en alguna parte de la médula espinal o en los nervios del extremo caudal del conducto vertebral –comúnmente conocidos como “cola de caballo” –, que provoca afectaciones de la fuerza motriz, la sensibilidad y otras funciones corporales por debajo del sitio de la lesión.
A grandes rasgos, las manifestaciones clínicas que puede provocar cualquier lesión de la médula espinal incluyen, entre otros, los siguientes signos y síntomas: pérdida de movimiento y pérdida o alteración de la sensibilidad (como el tacto o la capacidad de sentir calor y frío) sobre todo en extremidades, pérdida del control intestinal o de la vejiga, espasmos o reflejos exagerados, cambios en la función sexual (bien en la sensibilidad sexual o bien en la fertilidad), dolor o sensación de ardor intenso –en la espalda– a causa del daño en las fibras nerviosas de la médula espinal, y hasta dificultad para respirar, toser o eliminar las secreciones de los pulmones.
El nivel de control de las extremidades después de una lesión medular depende del lugar de la lesión (esto es, el llamado nivel neurológico de la lesión) y de la gravedad de la misma (según la integridad de la médula). A modo de ejemplo, una lesión medular a nivel del tórax o en la parte inferior de la espalda (lumbar) puede afectar al torso, las piernas y, en los casos más graves, a la función sexual y al control del intestino y de la vejiga; por su parte, una lesión a nivel del cuello (cervical) afecta las mismas áreas, pero también al movimiento de brazos y, posiblemente, la capacidad para respirar.
En función de la gravedad, se pueden clasificar las lesiones medulares en: a) completas, si toda la función sensorial y la capacidad motora están ausentes por debajo del lugar de la lesión; o b) incompletas, si el paciente conserva cierto grado –variable– de actividad motora y/o sensorial por debajo de la zona afectada. Además, según el tipo de parálisis sensorial y motora que producen, las lesiones medulares pueden denominarse: a) tetraplejia (también denominada cuadriplejía), que implica que brazos, manos, tronco, piernas y órganos pélvicos se encuentran afectados; o b) paraplejia, cuando la parálisis afecta a la totalidad o parte del tronco, las piernas y los órganos pélvicos, pero no a las extremidades superiores.
En cuanto a la etiología, las lesiones medulares se deben a un daño que afecte a las vértebras, los ligamentos o los discos vertebrales de la columna, o incluso a la misma médula espinal. Las lesiones de origen traumático pueden deberse a un golpe o impacto repentino a la columna vertebral, tales como un accidente automovilístico (responsables de casi la mitad de casos notificados al año), una caída brusca (sobre todo en edades de >65 años, y que representan en torno al 15% de casos), una herida por violencia (con un arma blanca o de fuego, ~12%) o lesiones en la práctica deportiva u otras actividades recreativas (como deportes de impacto o buceo, ~10%). Cualquiera de esas situaciones puede provocar la fractura, dislocación, aplastamiento o compresión de una o más vértebras, con seccionamiento total o parcial de la médula espinal, y es común que se produzcan daños adicionales tras varios días o semanas desde el traumatismo, a consecuencia de posibles hemorragias, inflamación o edema en la zona.
Por otro lado, entre las lesiones no traumáticas de la médula espinal, se pueden citar aquellas que se desencadenan por infecciones, artritis, neoplasias, inflamación o degeneración de los discos de la columna vertebral (como ocurre en la osteoporosis). El consumo de alcohol se ha relacionado como factor de riesgo en hasta un 25% de casos de lesiones medulares.
Las cifras sobre la incidencia y prevalencia de lesiones de la médula espinal son generalmente imprecisas y variables dependiendo de la fuente, y también del país. La incidencia a nivel mundial se ha estimado en el rango de 11 a 53 casos por cada millón de habitantes y por año (Ackery et al. 2004). Según la Organización Mundial de la Salud (OMS), este rango oscila entre 40 y 80 casos nuevos al año por cada millón de habitantes: se calcula que entre 250.000 y 500.000 personas sufren cada año en todo el mundo lesiones medulares.
Existe cierto consenso sobre la idea de que la causa mayoritaria (hasta un 90% de esos casos) son causas traumáticas2 en su mayoría prevenibles, sobre todo accidentes de tráfico, seguidos de caídas, lesiones deportivas y heridas por violencia. No obstante, la proporción de lesiones medulares de origen no traumático parece ir en aumento (sobre todo, por enfermedades), con un perfil demográfico diferente y sin tantas complicaciones como las que caracterizan a las lesiones traumáticas. En cualquier caso, todos los pacientes afectados de lesiones medulares son entre 2 y 5 veces más propensos a morir de forma prematura en comparación con quienes no las padecen (ese riesgo de mortalidad es máximo en el primer año tras sufrir la lesión); además, las tasas de supervivencia más bajas corresponden a los países de ingresos bajos y medios3 . El pronóstico tras el diagnóstico de una lesión medular es complicado de establecer: a veces aparece cierta recuperación (en lesiones incompletas) entre 1 semana y 6 meses después, aunque algunas personas manifiestan pequeñas mejorías durante 1 o 2 años.
La mayoría de autores aluden a que en los hombres el riesgo de sufrir una lesión medular es mayor en adultos jóvenes (20 a 29 años) y ancianos (70 años o más), mientras que en mujeres, en cambio, el mayor riesgo se registra en la adolescencia (de 15 a 19 años) y a partir de los 60 años. Entre los principales factores de riesgo que predisponen a una lesión medular destaca el sexo masculino (la razón hombres:mujeres es de al menos 2:1, e incluso mayor), la edad (16-30 años y >65 son las que implican un mayor riesgo), la adopción de conductas de riesgo (conducir de forma temeraria, practicar actividades acuáticas en zonas poco profundas, etc.) y la presencia de trastornos óseos o articulares (artritis u osteoporosis).
Todas las lesiones de la médula espinal son devastadoras a cualquier edad, pero son especialmente trágicas y psicológicamente difíciles de asumir en pacientes de corta edad (niños, adolescentes y adultos jóvenes), y representan un problema médico y social de grandísima magnitud, ya que se asocian a menores tasas de escolarización y productividad laboral y suponen un importante coste económico tanto para quienes las padecen y sus familiares como para la sociedad en su conjunto.
Con respecto a la terapéutica, las lesiones medulares traumáticas suponen indudablemente una necesidad médica no cubierta, pues hasta ahora no existe ningún medicamento autorizado en la Unión Europea con esa indicación, en el objetivo de reparar el SNC, restaurar su función y revertir las secuelas neurológicas que originan. Sin embargo, cabe destacar que en las últimas dos décadas ha habido importantes avances en el conocimiento de los fenómenos de regeneración espontánea y en los resultados clínicos de las distintas estrategias de reparación investigadas, que todavía es demasiado limitado. Entre ellas, ha destacado como potencial terapia en los últimos años la utilización de células madre embrionarias o del estroma mesenquimal (también llamadas células madre mesenquimales o mesenquimatosas).
Ante la ausencia de opciones curativas, el enfoque del manejo clínico de pacientes afectados por lesiones medulares se orienta a facilitar su vida diaria mediante la rehabilitación física y los modernos dispositivos médicos, como sillas de ruedas de última generación, la adaptación de ordenadores, dispositivos electrónicos que puedan controlarse por voz o medios informáticos, o incluso sistemas de estimulación eléctrica funcional para controlar músculos de brazos y piernas (que incluso que les permitan caminar). También se pueden emplear diversos medicamentos para el control de algunas manifestaciones, como el dolor y la espasticidad muscular, o la disfunción sexual.
No obstante, la prevención de estas lesiones medulares es todavía probablemente el mayor reto. Para ello, se están desarrollando –y se deben seguir promoviendo– programas educativos cuyos esfuerzos se orientan a potenciar la conciencia pública sobre los riesgos y consecuencias de las lesiones medulares y, en particular, sobre la necesidad de extremar las precauciones en la conducción (especialmente cuando se toman medicamentos que puedan afectar a la capacidad para ello), de usar cascos protectores en determinados deportes (béisbol, ciclismo, esquí, etc.), de evitar deportes de contacto y de erradicar las conductas violentas de la sociedad.
Acción y mecanismo
El mecanismo de acción específico por el cual las células mesenquimales troncales (también llamadas células madre estromales) adultas autólogas que componen el medicamento NC1 (suspensión celular en plasma autólogo) pueden promover la reparación de la lesión medular no ha sido completamente elucidado. En base a resultados in vitro se ha propuesto que un posible mecanismo podría involucrar la diferenciación neural de dichas células pluripotenciales, tras ser administradas por vía intratecal y entrar en contacto con factores solubles aportados por células gliales, fenómeno que se denomina transdiferenciación biológica.
La Ficha Técnica del medicamento (AEMPS, 2019) alude a estudios preclínicos realizados en animales hembra que presentaban traumatizado el tejido del sistema nervioso, en los cuales se ha confirmado ese proceso de transdiferenciación al recibir un trasplante de células madre estromales procedentes de donantes macho. Por ejemplo, mediante técnicas de hibridación in situ e inmunohistoquímica, se pudo verificar que las células madre estromales persistían a largo plazo desde su trasplante (al menos 4 meses), mostrando signos de diferenciación fenotípica a células nerviosas, como neuronas y astrocitos. Esto sugiere la existencia de una interacción con el tejido nervioso del huésped que facilita la diferenciación nerviosa de las células trasplantadas, probablemente por acción de factores tróficos locales presentes en la zona del implante.
Diversas evidencias apuntan al hecho de que el trasplante de las células madre estromales puede desencadenar fenómenos de neurogénesis endógena, tanto a nivel de la zona ependimaria como en otras regiones de la médula lesionada. Hay que recordar que en estudios con animales lesionados medulares se ha observado que su recuperación funcional se inicia antes de que se produzca una regeneración tisular (capaz de rellenar por completo la cavidad centromedular traumática y de formar un puente que permita el paso de axones ascendentes y descendentes).
Sea como fuere, el medicamento está indicado en el tratamiento de pacientes adultos (≤65 años) con secuelas de lesión medular traumática crónica, que presenten lesiones medulares incompletas a nivel dorsal o lumbar, excluyendo lesiones medulares dorsales o lumbares completas a excepción de aquellas localizadas quísticas con cavidad centro-medular de menos de 1-3 segmentos medulares. Los estudios preclínicos sugieren que, tras la administración del medicamento en líquido cefalorraquídeo, las células pueden atravesar el parénquima medular para alcanzar las zonas de lesión traumática, sin que se detecten células fuera del SNC.
El papel potencial de las células madre mesenquimales en los procesos de reparación endógenos y en terapias basadas en células ha sido también ampliamente estudiado, entre otras, frente a patologías como la lesión renal aguda o lesión miocárdica, habiendo demostrado en estudios preclínicos que mejoran la lesión tisular y aceleran la reparación funcional. Se ha planteado que estas versátiles células se dirigen específicamente a sitios de lesión y son capaces de modular el proceso de reparación tisular mediante mecanismos protectores y regenerativos que incluyen efectos paracrinos y endocrinos, propiedades mitogénicas, antiapoptóticas, antiinflamatorias y angiogénicas (Rameshwar et al. 2018).
Aspectos celulares
A grandes rasgos, las células madre mesenquimales (o estromales) son células pluripotenciales primitivas con morfología tipo fibroblasto, que se originan a partir de la capa germinal del mesodermo y que retienen la capacidad de diferenciarse en diversos linajes celulares tanto in vitro como in vivo, tales como osteocitos, adipocitos, condrocitos, mastocitos o fibroblastos, entre otros. Son capaces de proliferar y dar lugar a células hijas con el mismo patrón de expresión génica y fenotipo y, por lo tanto, conservando es “potencialidad” de las células originales. Como células madre que son, se caracterizan por esa capacidad de autorrenovación y diferenciación.
Su uso con fines terapéuticos se ve fundamentalmente facilitado por su fácil aislamiento y expansión. La fuente más conocida de células madre mesenquimales en adultos es el estroma de la médula ósea, donde además de éstas se hallan células endoteliales y del linaje hematopoyético; también se han identificado otras fuentes de estas células, como el tejido adiposo o la sangre del cordón umbilical, y hasta se ha postulado su presencia en sangre periférica. Para el aislamiento (previo a una posible diferenciación, en su caso, en un linaje celular deseado), suelen usarse técnicas como el inmunoaislamiento o inmunoisolación (selección por marcadores de superficie celular) o la inmunodepleción o selección negativa (enriquecimiento de la población celular por lavado de otras células diferentes –sobre todo hematopoyéticas– marcadas con anticuerpos); más recientemente se han empleado técnicas que combinan ambas estrategias para aislar poblaciones más específicas y puras.
Eficacia y seguridad clínicas
La eficacia clínica del nuevo medicamento a la dosis autorizada ha sido adecuadamente contrastada en ensayos clínicos prospectivos monocéntricos, 4 de los cuales –2 de fase 1 y otros 2 de fase 2– han sido completados desde 2013; otros 2 estudios están aún en desarrollo. Todos ellos tuvieron un diseño no controlado, no aleatorizado y de seguimiento abierto, y excluyeron población pediátrica (<18 años).
En el primer ensayo de fase 1, 12 pacientes con paraplejia crónica por lesión medular completa (media de edad de 40,5 años y de tiempo desde la lesión de 13,9 años) recibieron una administración –microinyección– intratecal e intralesional de entre 100-230×106 células mesenquimales estromales de la médula ósea en plasma autólogo, con una segunda administración de 30×106 células. Los resultados se evaluaron cada 3 meses hasta los 12 de seguimiento y revelaron una eficacia dosis-dependiente e independiente del tiempo de cronificación de la paraplejia; al final de dicho periodo, todos los pacientes mostraron una mejora notable del grado de discapacidad, de moderado a leve de acuerdo a la escala IANR-SCIFRS4 .
Además de que el procedimiento de trasplante fue seguro, entre los resultados de eficacia cabe destacar que (Vaquero et al. 2016):
- Todos los pacientes experimentaban una mejora clínica con recuperación de la sensibilidad por debajo del área de la lesión (alguno de ellos, recuperación completa). Según la clasificación por la escala ASIA5, más del 30% de los pacientes (N= 4) mejoraron desde una lesión completa a una lesión incompleta, con aumentos de entre 17 y 108 puntos al final del período de seguimiento (promedio de 47,4 puntos, desde una media de 165,9 puntos en el estado basal a 213,3 puntos a los 12 meses; p= 0,002).
- Se produjo una disminución del volumen y la intensidad de las lesiones intramedulares en el 60% de los pacientes.
- Más de la mitad de los pacientes alcanzaron actividad motora intralesional, es decir, recuperaron el movimiento de los miembros inferiores, bien según pruebas clínicas o neurofisiológicas. Diez pacientes (el 83,3%) mostraron potenciales polifásicos en los músculos infralesionales, sugerentes de un proceso de reinervación activa.
- Hubo una mejora significativa de la función sexual y, sobre todo, del control de los esfínteres según evaluación por la escala de Geffner (el 90% de pacientes recuperó el control de la vejiga urinaria, uno de ellos completamente). Además, se reportó una reducción en los espasmos y la espasticidad.
El otro gran ensayo clínico de fase 1 incluyó 10 pacientes con lesión medular incompleta a nivel dorsal o cervical, con una edad media de 42 años (rango 34-59) y tiempo medio desde la lesión de 14,2 años (rango 2,4-34,6), que recibieron cuatro administraciones subaracnoideas de 30×106 células mesenquimales autólogas cada 3 meses (alcanzando una dosis total de 120×106 células). Entre los principales resultados de eficacia, medidos al mes 12, sobresalen los siguientes (Vaquero et al. 2017):
- Se confirmó una mejora importante de la sensibilidad en todos los pacientes (evidente desde el mes 2), con incremento de las medias de puntuaciones en la escala ASIA, desde 188,2 puntos en el estado basal a los 235,5 puntos postratamiento (p= 0,0053).
- Mejoría temprana de la movilidad de todos los pacientes: el valor basal promedio fue de 53,0 puntos, mientras que al final del tratamiento el valor era de 59,2 puntos (p= 0,008). En el 60% de los casos esa significación estadística se detectó desde el mes 2 (media de 55,1 puntos; p= 0,03). Además, al final del periodo de seguimiento, 9 pacientes mostraban registros electromiográficos por debajo del sitio de la lesión, sugerentes de reinervación muscular.
- La escala IANR-SCIFRS reveló una mejoría notable de la función medular: al inicio, 5, 3 y 2 pacientes mostraban un grado de discapacidad funcional leve, moderado y grave, respectivamente, pero tras el tratamiento, 6 tenían una discapacidad leve y 4 moderada.
- La función sexual mejoró significativamente en 2 de los 8 pacientes varones, y según la escala de Geffner, hubo una mejoría relevante (p= 0,024) en el control de los esfínteres en 8 de los 9 pacientes que al inicio mostraban disfunción de la vejiga urinaria.
- El dolor neuropático desapareció en 2 de los 4 pacientes que lo mostraban al inicio del estudio, y se redujo de forma destacable en otro. En cuanto a los espasmos musculares, 2 de los 7 que los padecían, se beneficiaron clínicamente, de forma similar a 3 de los 9 que sufrían grados variables de espasticidad.
- Adicionalmente, el tratamiento reveló una mejora de las capacidades para desarrollar las actividades de la vida diaria, según las dos escalas empleadas de FIM (p= 0,027) y Barthel (p= 0,039).
El tercer y último estudio sobre el que se han divulgado resultados (Vaquero et al. 2018) es un ensayo de fase 2 que evaluó la administración intratecal –en el espacio subaracnoideo por punción lumbar– de un régimen de 3 dosis (1 cada 3 meses) de 100×106 células mesenquimales autólogas en 11 pacientes adultos con lesión crónica de la médula espinal (media de edad de 44,9 años y de tiempo desde la lesión de 13,7 años); de ellos, 4 pacientes tenían la lesión a nivel cervical, otros 4 a nivel dorsal y 3 a nivel dorsolumbar.
A los 10 meses de seguimiento tras la primera dosis, todos los pacientes mostraron un grado variable de mejoría clínica, independientemente de la edad, del nivel o la gravedad de la lesión medular o el tiempo desde la misma. El análisis de eficacia, con los datos de 9 pacientes, reveló los siguientes resultados (Vaquero et al. 2018):
- De acuerdo a la escala ASIA, hubo una mejoría notable en la sensibilidad por debajo de la lesión (3 pacientes mejoraron un grado en la clasificación de esta escala), con significación estadística desde la segunda dosis al mes 4 y hasta el final del seguimiento. Esto se tradujo en un aumento promedio de 35 puntos en la puntuación ASIA desde el nivel basal, desde una media de 181,6 puntos hasta 216,6 al mes 10 (p= 0,021).
- Recuperación significativa de las capacidades motoras en todos los pacientes excepto uno con lesión a nivel cervical. El valor medio basal de todos los pacientes fue de 53,0 puntos, aumentando hasta los 56,0 puntos al mes 10 (p= 0,028). La mejora de la movilidad de las extremidades superiores en los pacientes con lesión medular a nivel cervical fue más moderada (<2 puntos).
- La puntuación global de la escala IANR-SCIFRS reveló una funcionalidad medular potenciada en todos los pacientes al final del estudio (36,2 vs. 27,3 puntos de media; p= 0,009): 7 pacientes mostraban discapacidad leve y otros 2, moderada, mientras que al inicio eran 2, 5 y 2 pacientes los que referían una discapacidad leve, moderada y grave, respectivamente.
- Pese a que había una tendencia numéricamente positiva, no se verificaron cambios estadísticamente significativos en la función sexual según la escala IANR-SCIFRS. En cambio, esta escala puso de manifiesto una mejora importante en el control de los esfínteres respecto al nivel basal (3,3 vs. 2,0 puntos de media; p= 0,018); así, dos tercios de los pacientes mostraban una disminución en el residuo posmiccional y mejora en el control vesical.
- Según la escala VAS, el dolor neuropático, que estaba presente al inicio en 8 pacientes, desapareció o sufrió una reducción significativa en 7 de ellos (p= 0,012).
- Los estudios neurofisiológicos mostraron una mejoría de los potenciales evocados somatosensoriales o motores en 7 pacientes, 4 de los cuales presentaban también una mejora en la conducción nerviosa periférica y en el control de la contracción muscular voluntaria; en esos 4 pacientes se detectaron potenciales motores polifásicos por debajo del sitio de lesión, sugerentes de reinervación muscular.
Con respecto a la seguridad de la suspensión de células mesenquimales, parece que su administración repetida en el espacio subaracnoideo es bien tolerada por los pacientes. De todos los eventos adversos notificados en los tres ensayos clínicos referidos (69, 20 y 9 eventos, respectivamente), se relacionaron –posiblemente– con el tratamiento entre un 32-40% de los mismos, todos de intensidad leve (en torno a un 80%) y moderada. Por su frecuencia, sobresalen los eventos adversos asociados con el procedimiento de punción lumbar: cefalea, dolor postoperatorio (especialmente en el lugar de administración), dolor ciático (hasta en el 38% de pacientes en un estudio), hipertermia transitoria o infiltración subcutánea en el área de la herida quirúrgica. Cabe destacar que, en uno de los estudios, ningún evento adverso se relacionó con el tratamiento (Vaquero et al. 2018); de igual modo, los eventos adversos descritos como graves (por ejemplo, un caso de bronquitis aguda) tampoco se relacionaron con el tratamiento.
En cualquier caso, conviene subrayar que se desconocen los riesgos potenciales a largo plazo tras la administración del medicamento. Aunque en los estudios con seguimientos de 1 año no se describió ningún caso, hay referencias en la literatura científica que apuntan a un posible riesgo de carcinogénesis asociado al tratamiento con células mesenquimales. Es por ello que se ha establecido un plan de seguimiento de los pacientes en los 3 años posteriores a la administración del fármaco, y no se recomienda para pacientes con antecedentes recientes (<3 años) de neoplasia activa.
Aspectos innovadores
Se trata de un novedoso medicamento de terapia avanzada cuyo principio activo son células mesenquimales troncales adultas obtenidas de la médula ósea del propio paciente (autólogas) y expandidas in vitro (fuera del organismo), que son resuspendidas previamente a su administración en plasma del mismo paciente a una concentración de 100.000 células/μl. Sin haberse esclarecido por completo el mecanismo de acción específico, se ha descrito que estas células pluripotenciales, inyectadas –por punción lumbar en el espacio subaracnoideo– en el lugar de la lesión medular traumática, atraviesan el parénquima medular para alcanzar las zonas de lesión, donde podrían posiblemente sufrir el proceso de transdiferenciación biológica, o sea, diferenciación a células nerviosas al entrar en contacto con factores tróficos solubles liberados localmente por células gliales del tejido hospedador; podrían, además, desencadenar fenómenos de neurogénesis endógena y regeneración tisular en la región medular lesionada.
En base a ello, la AEMPS ha otorgado al Hospital Universitario Puerta de Hierro de Majadahonda la primera autorización de uso para la fabricación y administración del medicamento, con indicación en el tratamiento de pacientes adultos (≤65 años) con secuelas de lesión medular traumática crónica, que presenten lesiones incompletas a nivel dorsal o lumbar; se excluyen pacientes con lesiones medulares completas, a excepción de las lesiones localizadas quísticas con cavidad centro-medular que no se extienda más de 1-3 segmentos medulares.
Tras más de 20 años de investigaciones, parece que el beneficio clínico del fármaco ha sido adecuadamente contrastado. A día de hoy, se dispone fundamentalmente de los resultados de 3 ensayos clínicos de fase 1/2 en paciente con lesión medular completa o incompleta, con otros tantos estudios ya finalizados o aún en marcha. No se han realizado ensayos aleatorizados y controlados de fase 3 (que sería la situación óptima) y los grupos de pacientes tratados no son amplios (N= 9-11), pero se puede considerar aceptable el diseño de los estudios por tratarse de una patología en la que no se dispone de comparadores activos (y en la que el uso de placebo podría cuestionarse éticamente) o por la limitación de las poblaciones de pacientes disponibles.
En términos generales, la eficacia del fármaco parece ser dosis-dependiente e independiente de factores como la edad del paciente, del nivel o la gravedad de la lesión medular o tiempo desde la misma. La administración intratecal de las células mesenquimales ha demostrado, tras 10 meses-1 año de seguimiento, una mejora estadísticamente significativa (p<0,05) en la sensibilidad por debajo de la lesión en todos los pacientes evaluados, con un aumento de las puntuaciones promedio de la escala ASIA sobre 35-47 puntos en comparación con el estado basal; de forma interesante, esa mayor sensibilidad se hace evidente poco tiempo después de la cirugía (desde la primera medida a los 3-4 meses). Además, se verificó una mejora notable en las capacidades motoras de casi todos los pacientes (salvo uno con lesión medular incompleta a nivel cervical y otros con lesión completa), describiéndose aumentos de entre 2,6 y 6 puntos de media en el dominio específico de la escala ASIA, que se traducían en recuperaciones parciales de la movilidad por debajo de la lesión (sobre todo, de los miembros inferiores), con indicios de reinervación muscular en algunos pacientes según registros electromiográficos.
Los resultados de la escala IANR-SCIFRS también parecen concluyentes: una gran mayoría de los pacientes cambiaron de grado de discapacidad funcional, mayoritariamente de moderado a leve, pero también algunos de grave a moderado, con incrementos de la puntuación promedio desde el estado basal en el entorno de los 8 puntos (rango de 7,8-8,9 puntos en los 3 estudios comentados). Si bien solo algunos pacientes experimentaron mejorías en la función sexual tras el tratamiento, uno de los principales beneficios en la práctica totalidad de pacientes se refiere al control de los esfínteres y la función vesical. Además, en algunos pacientes se han descrito reducciones significativas en los espasmos y las espasticidad muscular, así como en la intensidad del dolor neuropático.
Por otra parte, la administración del medicamento es bien tolerada. La mayoría de los eventos adversos notificados fueron de gravedad leve-moderada, y solo un porcentaje relativamente bajo (32-40%) de los mismos se relacionó con el tratamiento (ninguno grave) y, fundamentalmente, con el procedimiento de punción lumbar. Por su frecuencia, destacan: cefalea, dolor postoperatorio, dolor ciático, hipertermia transitoria o infiltración subcutánea en el área de la herida quirúrgica. No obstante, los seguimientos máximos de 1 año postratamiento no permiten descartar los riesgos a largo plazo, principalmente de carcinogénesis (sugerido en la bibliografía), que deberán ser esclarecidos por el plan de seguimiento de 3 años establecido para los pacientes tratados.
Desde el punto de vista mecanístico, este medicamento inaugura una prometedora vía de tratamiento en su indicación. Si bien el tipo de células empleadas en NC1 podría recordar al recientemente comercializado darvadstrocel (células madre de origen mesenquimal), cabe destacar el distinto origen de las mismas: mientras que en darvadstrocel son extraídas de tejido adiposo de un donante, en NC1 son de origen medular (estroma) y del propio paciente a tratar.
En resumen, estamos ante el primer medicamento de terapia avanzada (terapia celular) que demuestra un beneficio clínico en la reversión –al menos parcial– de las secuelas de una lesión medular traumática. Aporta una importante mejoría en aspectos tan trascendentes para estos pacientes como la sensibilidad y la capacidad motora en las extremidades y el control de los esfínteres y la funcionalidad vesical, que indudablemente va a suponer una mejora en la calidad de vida y abre una puerta a la esperanza para pacientes que hasta ahora no tenían ningún tratamiento eficaz. Supone una innovación terapéutica disruptiva tanto a nivel nacional como internacional.
A pesar de la circunstancia de condicionalidad de la autorización y dada la mejora del conocimiento científico que se producirá con la experiencia de uso en la práctica real (que debe en primer lugar verificar la replicación de los resultados obtenidos en ensayos clínicos), podría no ser disparatado plantearse si ¿puede ser el principio de la curación de la discapacidad que generan las lesiones medulares? Por ahora, se han excluido de la indicación autorizada los pacientes con lesiones medulares traumáticas completas (salvo algún tipo concreto), habida cuenta de que parece razonable que los pacientes con lesión incompleta puedan ser quienes se beneficien en mayor medida del tratamiento; en todo caso, la evidencia sugiere que futuros estudios más amplios y la experiencia clínica podrían permitir ampliar la indicación a lesiones completas.
Valoración



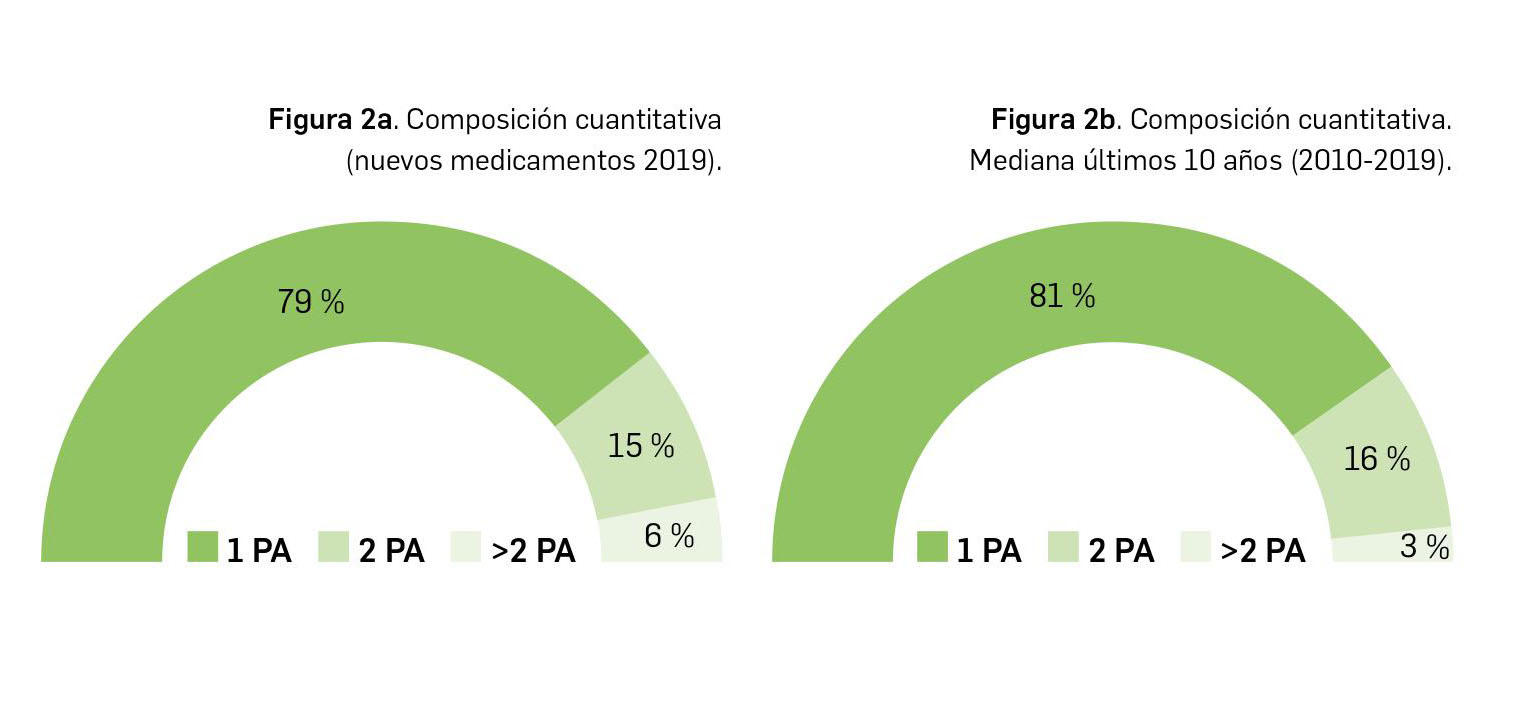
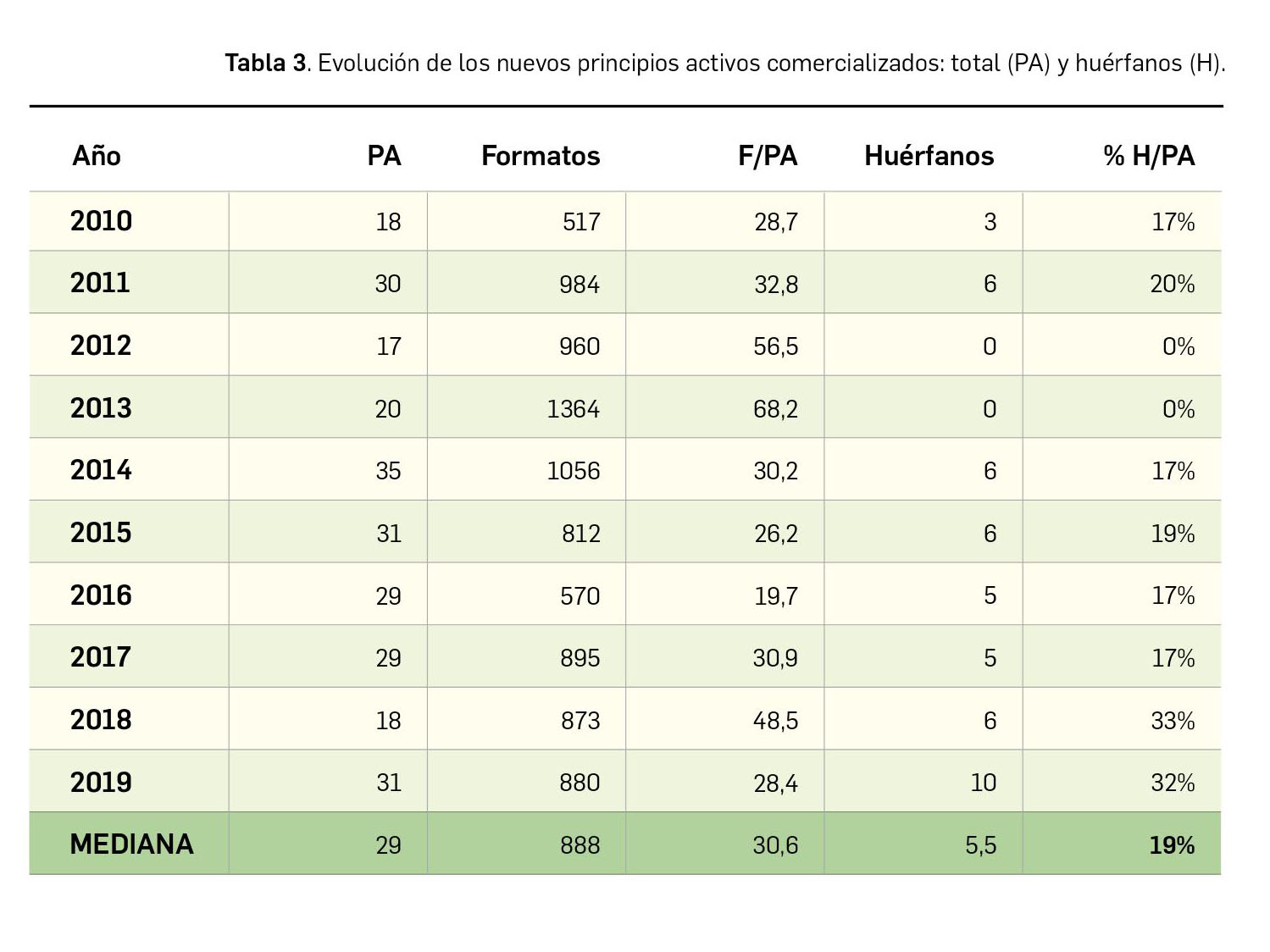
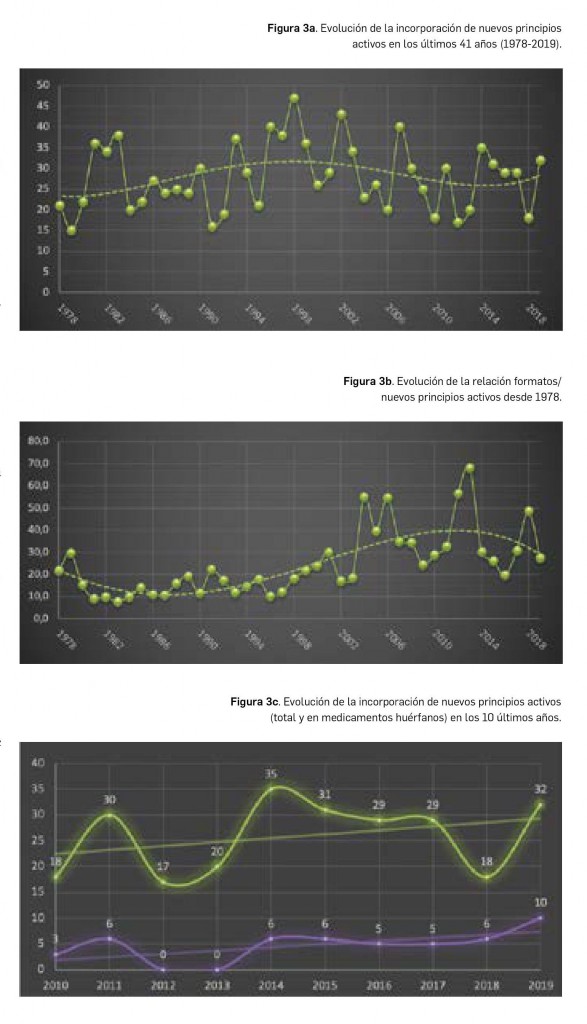
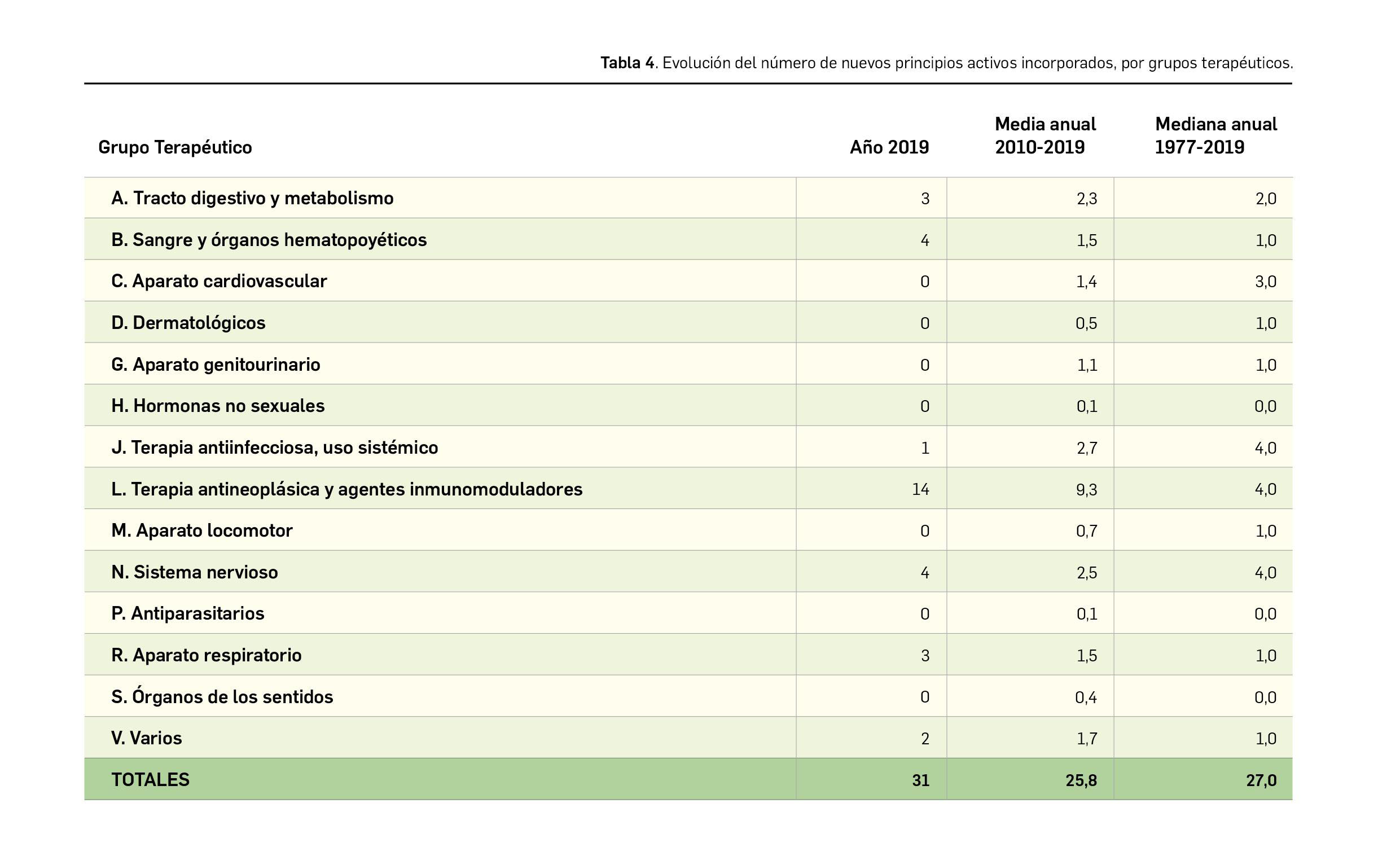
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}