Un estudio prospectivo de cohortes ha demostrado que el inicio del tratamiento con estatinas (pravastatina) en la infancia en pacientes con hipercolesterolemia familiar disminuye a largo plazo –seguimiento de 20 años– los niveles medios de colesterol LDL en un 32%, alcanzándose los objetivos terapéuticos (< 100 mg/dl sangre) en un 20% de pacientes; también permite atenuar la progresión media del grosor íntima-media de la carótida. Todo ello se relaciona con un riesgo significativamente reducido de eventos cardiovasculares y de muerte por causas cardiovasculares en la edad adulta.
La hipercolesterolemia familiar es un trastorno del metabolismo de lipoproteínas de herencia autosómica dominante, caracterizado por la presencia de mutaciones en genes que codifican proteínas implicadas en los procesos de endocitosis y reciclaje del receptor de lipoproteínas de baja densidad (LDL), que resulta en una menor captación celular y niveles plasmáticos elevados de estas LDL, causantes de una enfermedad cardiovascular prematura. La eficacia del tratamiento a corto plazo con estatinas (que reducen la síntesis de colesterol mediante la inhibición de la enzima hidroximetil-glutaril-coenzima A reductasa) ha sido adecuadamente caracterizada en ensayos clínicos y en la práctica real en pacientes pediátricos con hipercolesterolemia familiar, pero son escasos e inciertos los datos clínicos de su eficacia a largo plazo sobre el riesgo de patología cardiovascular.
Un reciente estudio prospectivo de cohortes desarrollado por investigadores holandeses ha arrojado luz sobre esta cuestión. Se seleccionaron un total de 214 pacientes con hipercolesterolemia familiar (confirmación genética en el 98% de los pacientes) que habían participado previamente en un ensayo controlado con placebo que investigó la eficacia y seguridad del tratamiento de 2 años con pravastatina. Estos iniciaron, junto con sus 95 hermanos no afectados (como grupo control), un periodo de tratamiento y seguimiento de 20 años en que los pacientes se sometieron periódicamente a pruebas de muestras sanguíneas y mediciones del grosor de las capas íntima-media de la arteria carótida. La media de edad de los pacientes fue de 13 (± 3) años al inicio del estudio y de 32 (± 3) años al final del mismo. El 86% de los pacientes de la cohorte experimental fueron evaluados durante el seguimiento, si bien los datos sobre eventos cardiovasculares y de muertes por causas cardiovasculares estaban disponibles, respectivamente, para el 95% y el 100% de los mismos.
Los resultados muestran que el tratamiento prolongado con estatinas redujo el nivel medio de colesterol LDL de 237,3 a 160,7 mg/dl, esto es, una disminución del 32% desde el nivel basal, alcanzándose los objetivos del tratamiento (colesterol LDL <100 mg/dl) en 37 pacientes (20%). La progresión media del grosor íntima-media de la carótida durante todo el período de seguimiento fue de 0,0056 mm por año en pacientes con hipercolesterolemia familiar y de 0,0057 mm/año en el grupo control de hermanos no afectados (diferencia de medias ajustada por sexo de -0,0001 mm/año; IC95% de −0,0010 a 0,0008). Se observó, además, que la incidencia acumulada de eventos cardiovasculares y de muerte por causas cardiovasculares a los 39 años de edad era menor entre los pacientes con hipercolesterolemia familiar tratados desde la infancia que entre sus padres afectados (tasas de incidencia de 1% vs. 26% y 0% vs. 7%, respectivamente).
En resumen, en base a los resultados de este estudio, parece evidente que el inicio de la terapia con estatinas durante la infancia en pacientes con hipercolesterolemia familiar va a permitir reducir el riesgo cardiovascular de estos pacientes cuando alcancen la edad adulta.


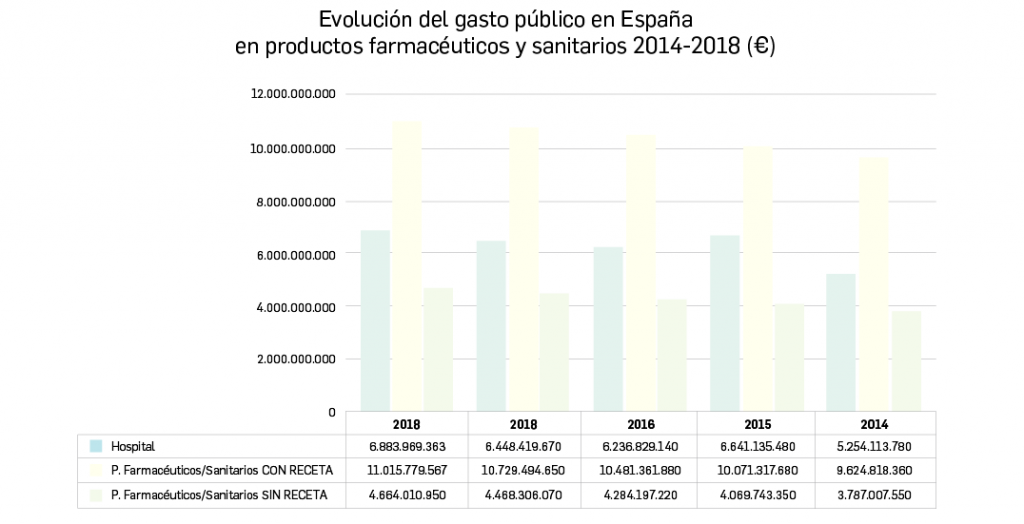
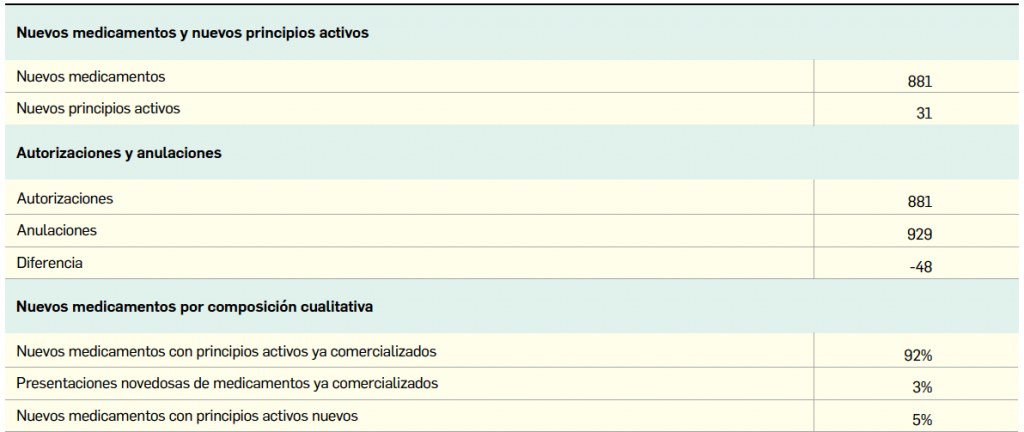
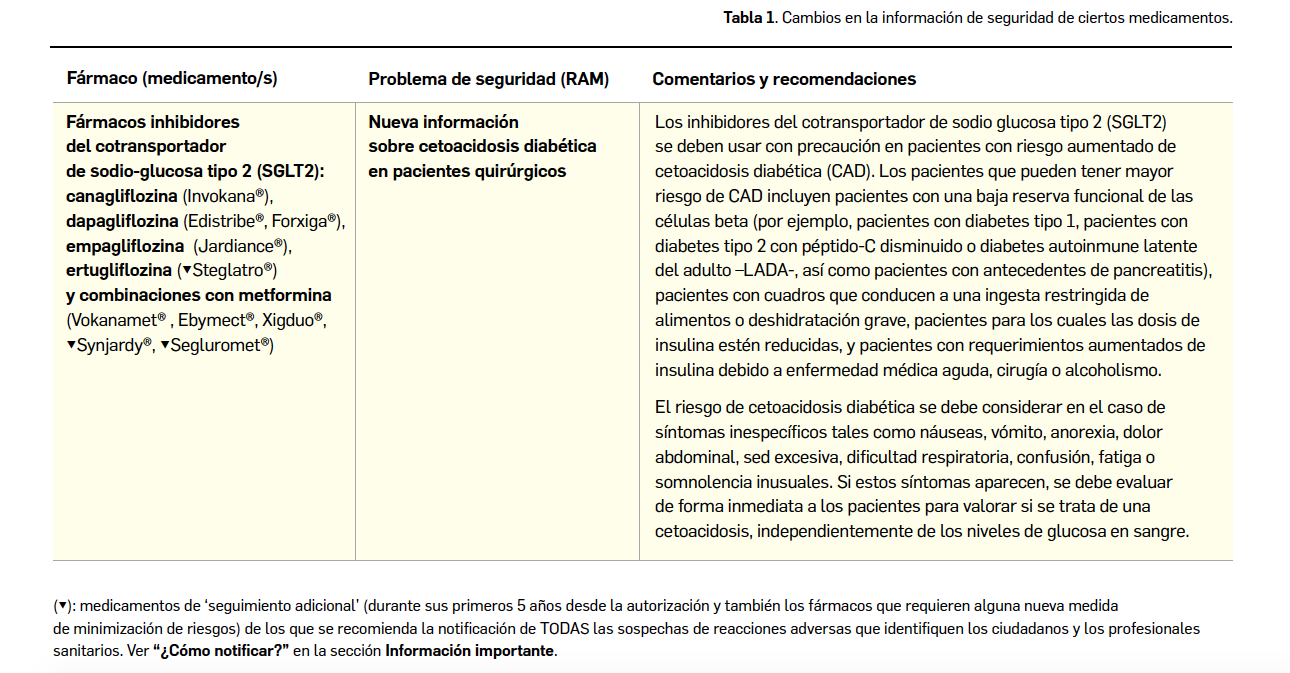
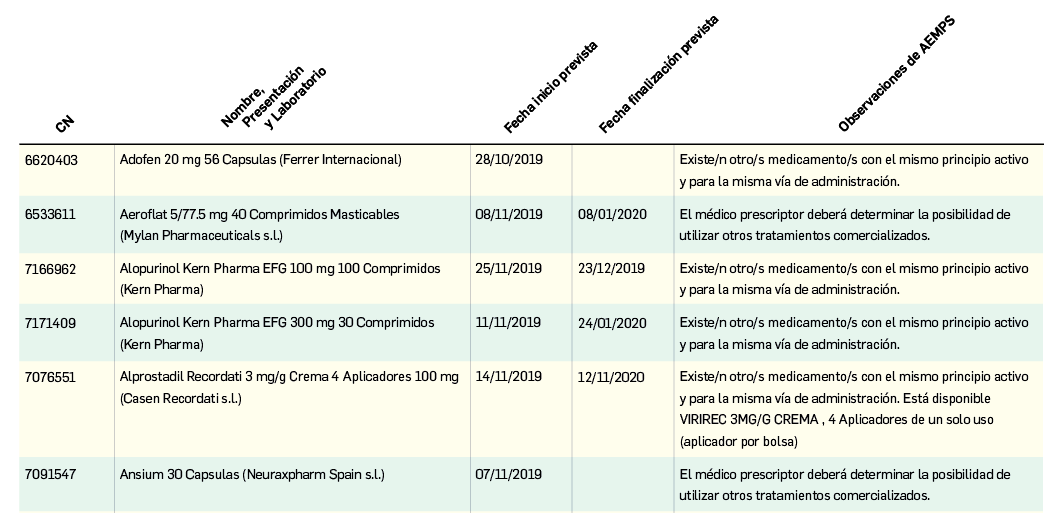








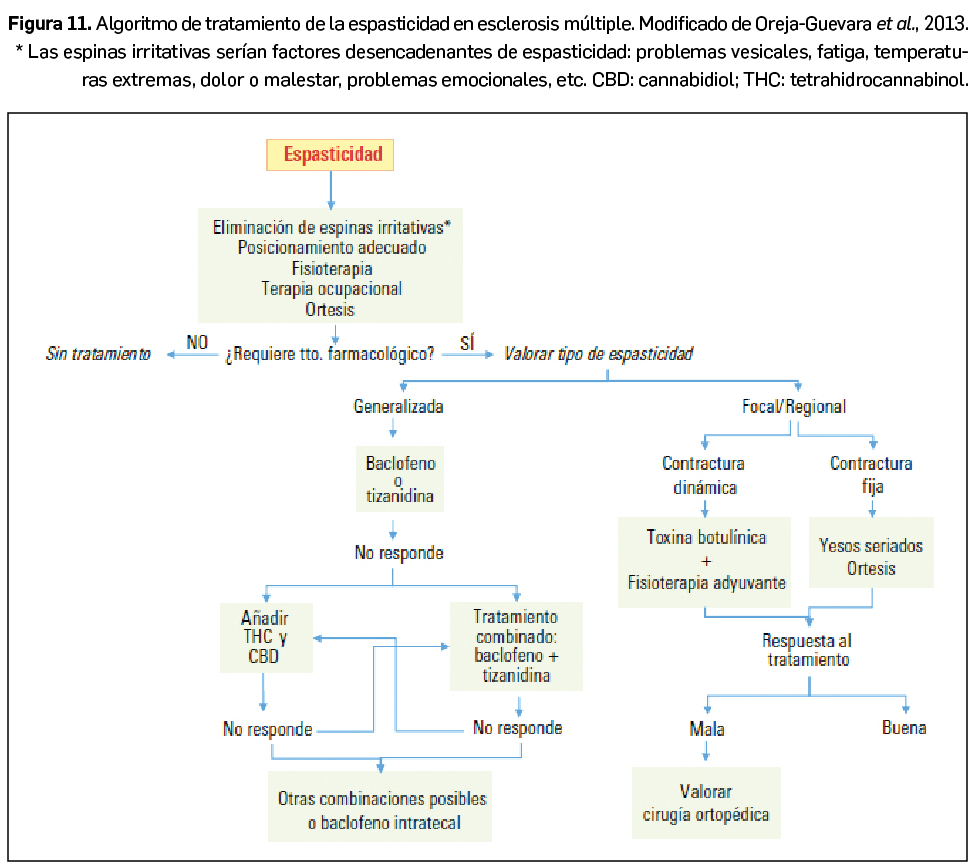
{kind=link}
{kind=link}
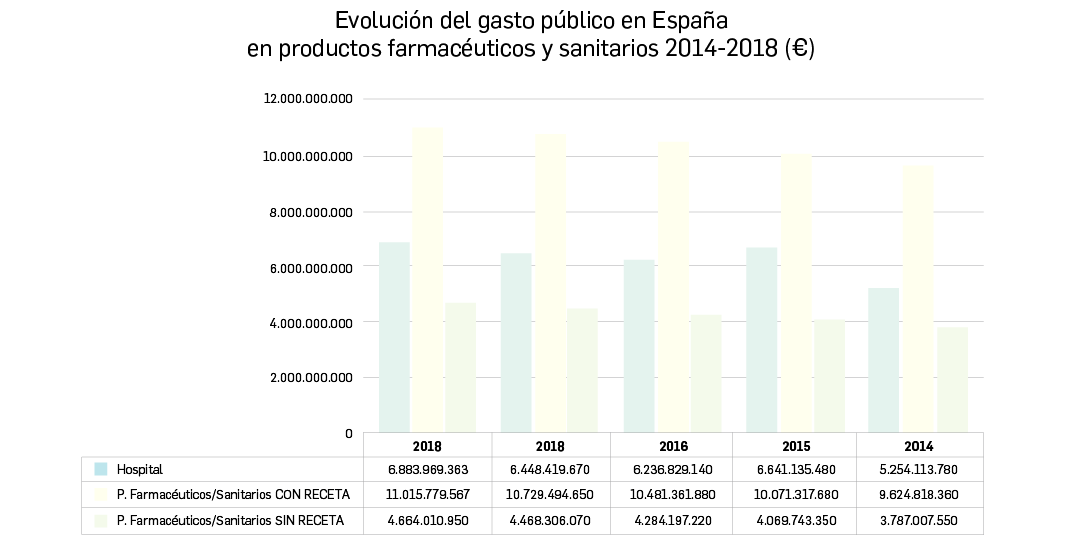{kind=link}
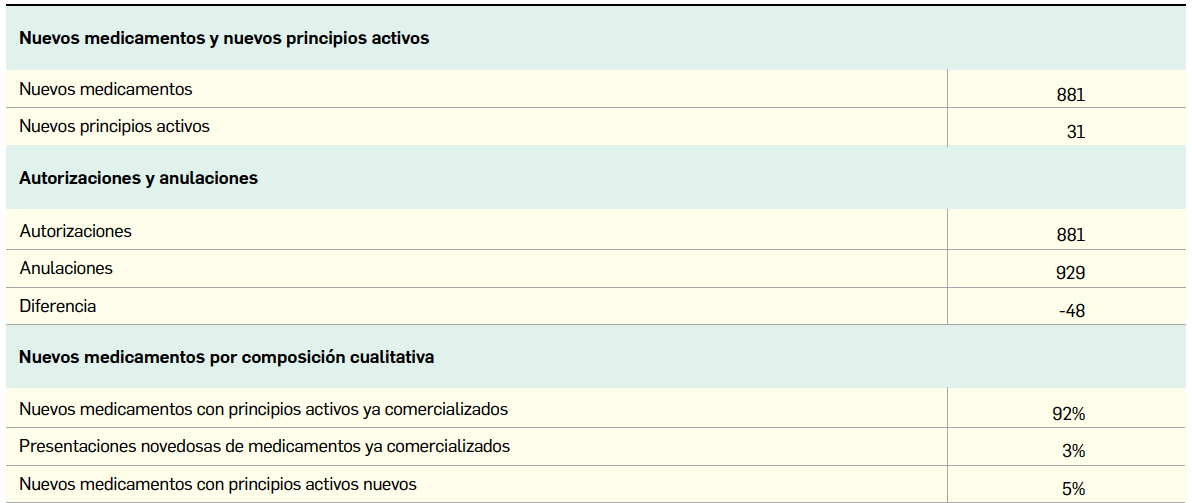{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}