Además de los listados mensuales que podemos consultar en PAM, en Bot PLUS se incorpora la información que publica la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) relativa a notificaciones sobre seguridad y/o calidad de los medicamentos. Mediante un pictograma específico se pueden visualizar de forma rápida medicamentos afectados por alguna alerta de seguridad o de defectos de calidad, con tan solo entrar en su ficha
Cómo localizar alertas de calidad con retiradas de lotes en Bot PLUS
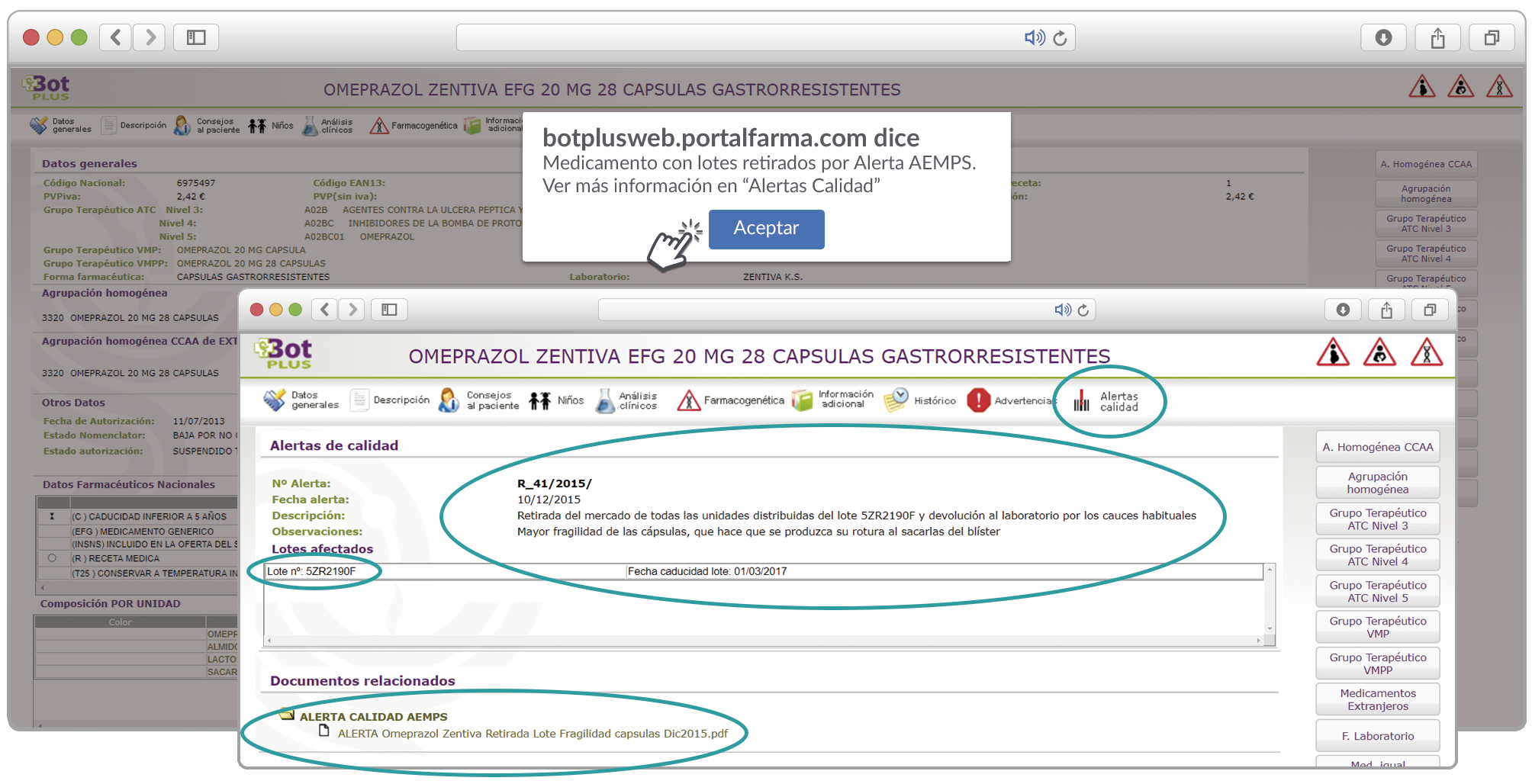
Al acceder a la ficha de un medicamento afectado por una retirada, se visualiza un mensaje con la advertencia “Medicamento con lotes retirados por Alerta AEMPS. Ver más información en “Alertas Calidad”.

Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.

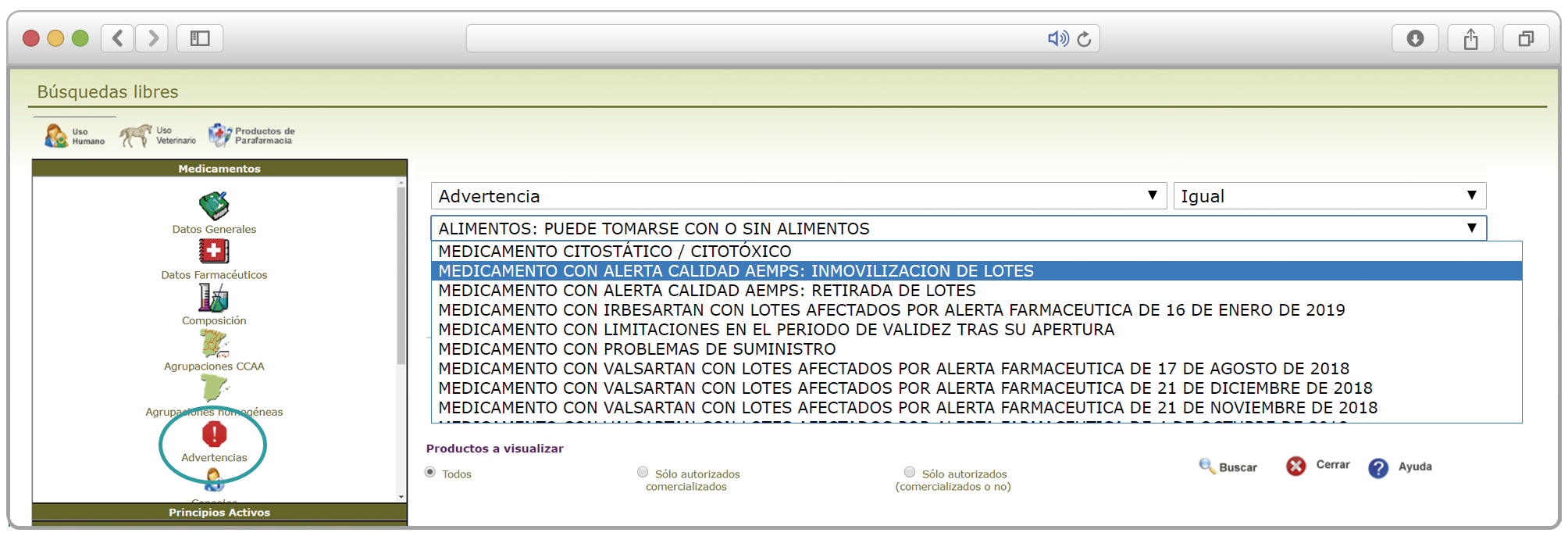
De forma interesante, dicha información se puede explotar a través de la Búsqueda Libre de Bot PLUS para obtener listados de todos los medicamentos afectados por alertas de calidad que implica la retirada (o también la inmovilización) de sus lotes en un momento dado.
Esta codificación de los lotes retirados es una información puesta a disposición de todos los usuarios, con el objetivo de ofrecer una nueva información capaz de integrarse con otros sistemas de información y mejorar la gestión e identificación de estos medicamentos, en los que la labor asistencial y de control del farmacéutico es fundamental.





{kind=link}
{kind=link}
{kind=link}