El análisis primario de los datos derivados de dos ensayos clínicos de fase 3 demuestra que el tratamiento de la anemia en pacientes con enfermedad renal crónica con roxadustat por vía oral (un inhibidor de la prolil hidroxilasa del factor inducible por hipoxia) genera un incremento significativo de los niveles de hemoglobina tanto en pacientes dializados, en los que ha demostrado no-inferioridad frente a epoteina alfa, como en aquellos no sometidos a diálisis (superior a placebo). Esto, unido al efecto adicional sobre otros parámetros bioquímicos y su perfil toxicológico tolerable, sugiere que puede ser una alternativa terapéutica eficaz en el tratamiento de la anemia en este tipo de pacientes.
Roxadustat es un inhibidor oral de la prolil hidroxilasa del factor inducible por hipoxia (HIF) que estimula la eritropoyesis y regula el metabolismo del hierro; fue autorizado por primera vez en China a finales de 2018 para el tratamiento de la anemia en pacientes con insuficiencia renal crónica dependientes de diálisis. En los ensayos de fase 2 en estos pacientes, demostró su capacidad de aumentar los niveles de eritropoyetina endógena hasta el rango fisiológico, junto con el aumento de los niveles de hemoglobina y la mejora de la homeostasis del hierro.
Recientemente se han divulgado los resultados de dos ensayos clínicos de fase 3 desarrollados a fin de obtener datos clínicos confirmatorios de eficacia y seguridad del fármaco en el tratamiento de la anemia en pacientes con enfermedad renal crónica, tanto aquellos que se someten a diálisis (N= 305) como los que no (N= 154). Se trata de dos ensayos aleatorizados (2:1), doblemente ciegos, de grupos paralelos y multicéntricos, que compararon la eficacia de roxadustat –administrado 3 veces/semana– con epoetina alfa (tratamiento estándar de la anemia en pacientes sometidos a diálisis) y con placebo (en el caso de pacientes no dializados), y que determinaron como variable primaria el cambio medio en los niveles de hemoglobina respecto al nivel basal, a las semanas 23-27 y a las semanas 7-9 de tratamiento, respectivamente.
Los análisis primarios de los datos demostraron que roxadustat inducía un incremento medio de hemoglobina de 0,7 ± 1,1 g/dl en pacientes sometidos a diálisis (vs. 0,5 ± 1,0 g/dl con epoetina alfa) y de 1,9 ± 1,2 g/dl en pacientes no sometidos a diálisis (vs. descenso de 0,4 ± 0,8 g/dl en el grupo placebo), lo que se traduce en una probada no-inferioridad de eficacia frente a epoetina alfa en los primeros y una superioridad clínica frente a placebo en los segundos. En ambos grupos de pacientes, roxadustat indujo una reducción media de los niveles de hepcidina –asociado a una mayor disponibilidad de hierro– significativamente superior a los comparadores (30,2 ng/ml vs. 2,3 ng/ml con epoetina y 56,1 ng/ml vs. 15,1 ng/ml con placebo). El tratamiento experimental se asoció, además, con reducciones de los niveles de colesterol en ambos tipos de pacientes (significativamente mayores que las inducidas por los comparadores) y con aumentos de los niveles séricos de transferrina y mantenimiento de los de hierro en aquellos dializados. Si bien es un fármaco bien tolerado, entre los principales eventos adversos del tratamiento se describe una mayor incidencia de hipercalemia, acidosis metabólica e infecciones del tracto respiratorio superior, pero atenúa el riesgo de hipertensión de epoetina alfa.
De manera interesante, una fase abierta de 18 semanas del estudio con pacientes no dializados permitió confirmar que la eficacia del nuevo fármaco en la corrección de niveles de hemoglobina se mantenía en el tiempo. Por tanto, se puede considerar que este fármaco será una alternativa eficaz para el tratamiento de la anemia en pacientes con enfermedad renal crónica tanto en aquellos que están sometidos a diálisis (como alternativa a la epoetina alfa) como en los que no lo están.

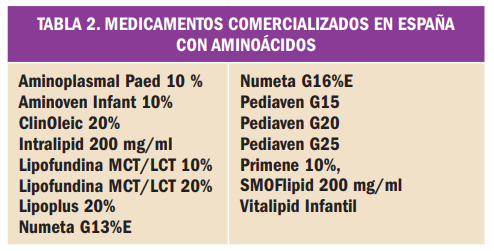
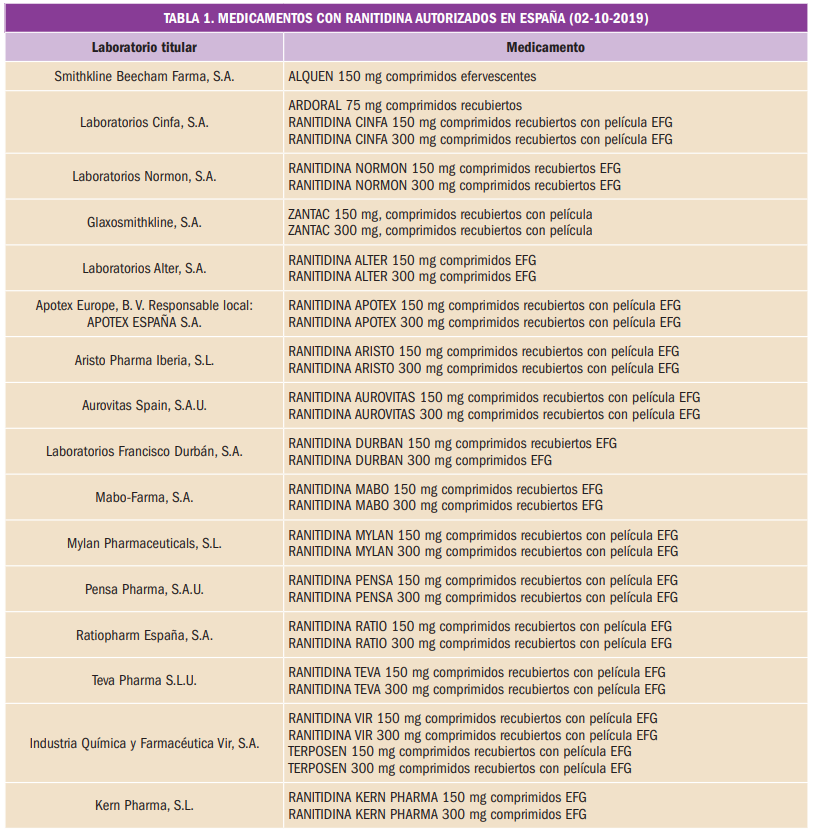
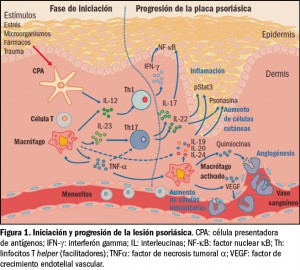
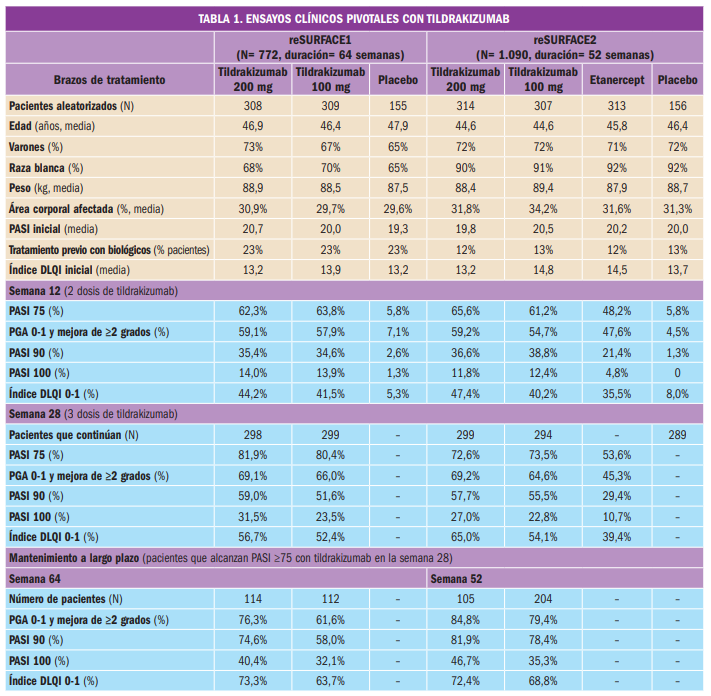



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}