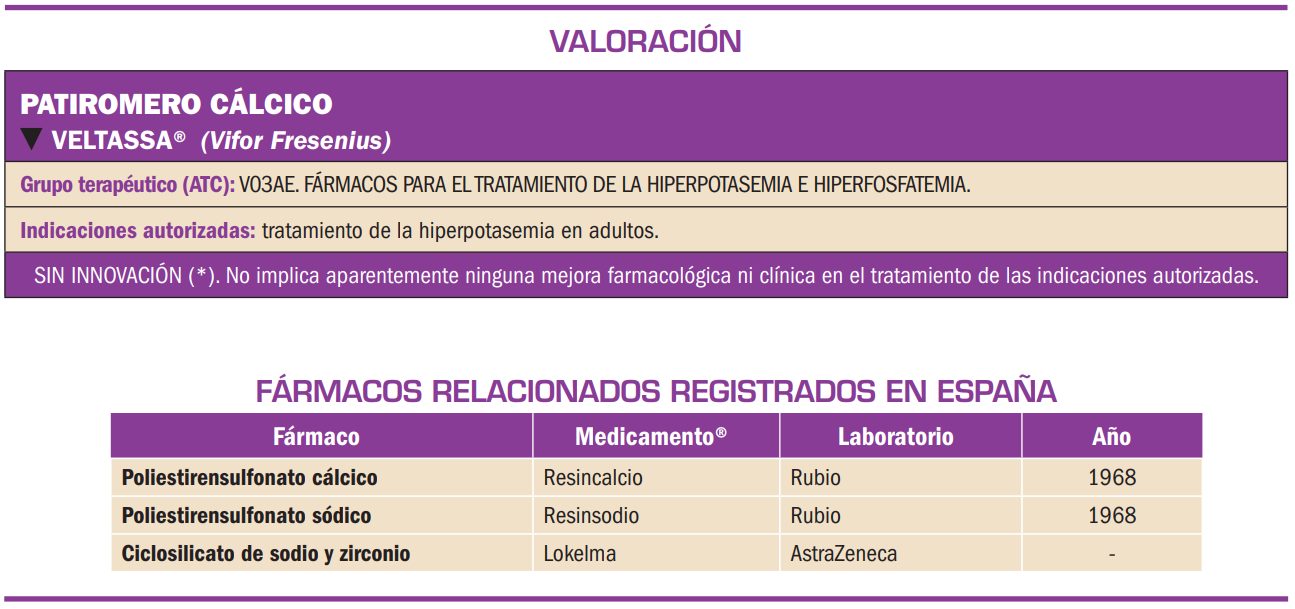
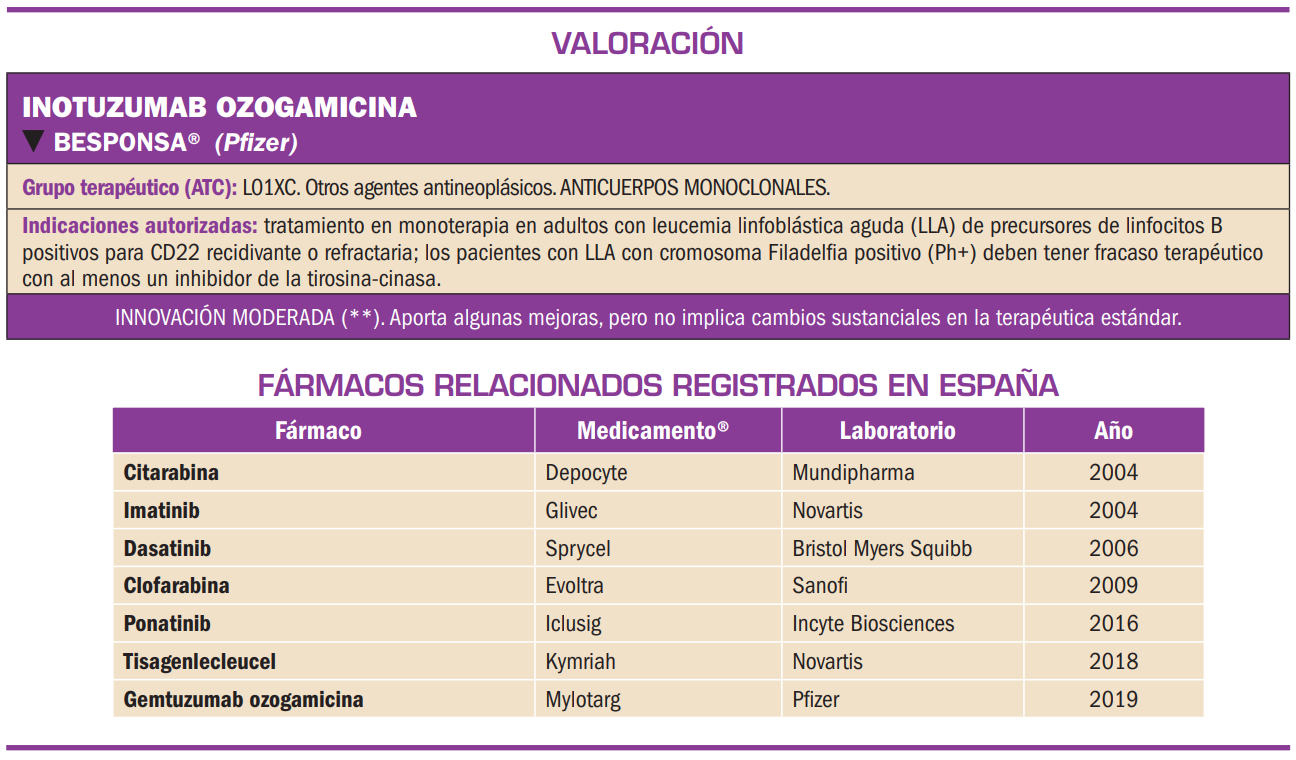
Resumen
El patiromero cálcico es un anión polimérico reticulado que actúa como polímero de intercambio de cationes; por sus características físico-químicas no se absorbe tras su administración oral y es capaz de unirse y quelar los iones potasio libres en la luz del tracto gastrointestinal (sobre todo, en el colon, donde K+ es el catión más abundante), con lo que aumenta la excreción en heces de dicho ión y reduce los niveles de potasio en suero. En base a dicho mecanismo, el medicamento ha sido autorizado para el tratamiento de la hiperpotasemia en adultos.
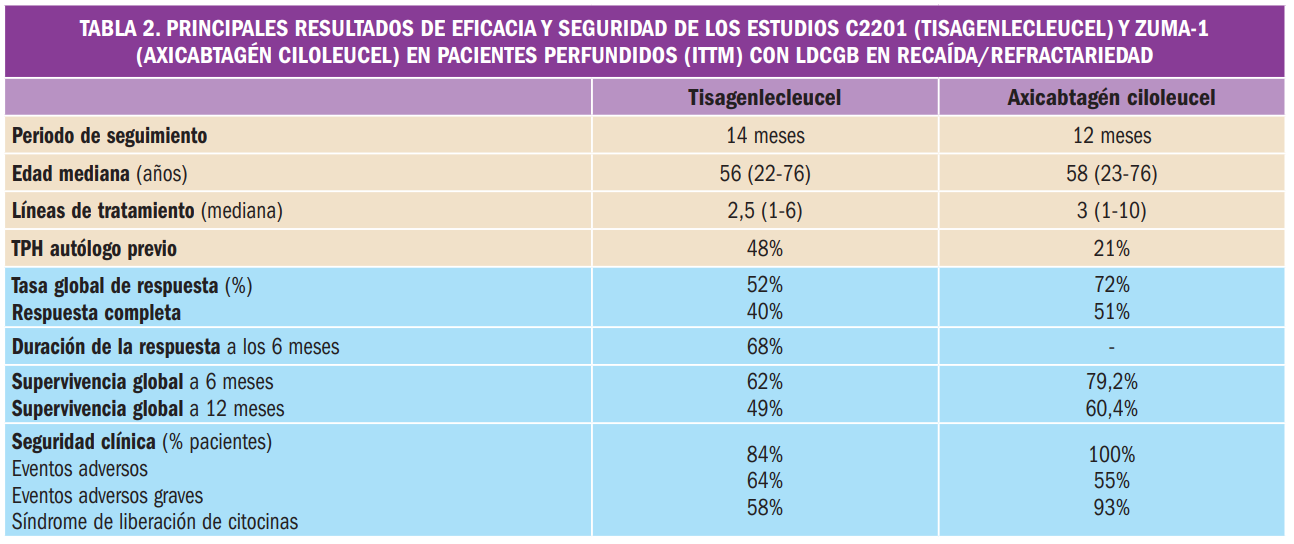
La eficacia clínica del fármaco ha sido adecuadamente contrastada en la indicación y dosis autorizada mediante un ensayo pivotal de fase 3, simple ciego, y dividido en dos partes. En la Fase 1 (N=243), no aleatorizada, abierta y de un único brazo, patiromero redujo significativamente la potasemia en -1,01 mEq/l respecto al estado basal, tras 4 semanas de tratamiento en pacientes con hiperpotasemia leve-moderada (5,1-6,4 mEq/l; promedio basal: 5,58 mEq/l) y enfermedad renal crónica, en tratamiento concomitante con inhibidores del sistema renina-angiotensina-aldosterona (IRAAs) y con una alta incidencia de comorbilidades de riesgo cardiovascular; el 76% de los pacientes alcanzó valores normales de potasemia (3,8-5,1 mEq/l). La Fase 2 del estudio, con diseño controlado por placebo y retirada aleatorizada, de 8 semanas de duración, demostró la eficacia del fármaco para prevenir recurrencias en los pacientes respondedores de la Fase 1 (N=107): la retirada de patiromero cálcico provocó un aumento significativo de la potasemia de +0,72 mEq/l (p<0,001) desde el inicio hasta la primera medida fuera de la normalidad o hasta la semana 4, con mayor proporción de pacientes hipercalémicos en el grupo placebo (60% vs. 15% en continuación de tratamiento); el patiromero cálcico permitió la continuación del tratamiento con IRAAs (proporción de retirada de 5% vs. 52% con placebo). Adicionalmente, un ensayo de soporte de fase 2 (N=304), de un único brazo, abierto y aleatorizado, demostró –como objetivo secundario– que su administración diaria reduce la potasemia en el entorno de 0,9-1 mEq/l durante al menos 1 año en el tratamiento a largo plazo o crónico en pacientes con hiperpotasemia basal leve (5,5-6,0 mEq/l).
Por otra parte, el patiromero cálcico se puede considerar como un fármaco bien tolerado, con un perfil toxicológico relativamente benigno y manejable. Solo un 20% de pacientes mostró eventos adversos leves-moderados relacionados con el fármaco (principalmente gastrointestinales –estreñimiento, diarrea dolor abdominal o flatulencia– e hipomagnesemia), similares a los descritos para otras resinas intercambiadoras de cationes. Ningún caso de evento adverso grave (8,3%) se asoció al fármaco, y hubo una baja tasa de interrupción del tratamiento (9%), incluso entre aquellos pacientes tratados durante 1 año, aunque la evidencia de seguridad a largo plazo es muy limitada. El potencial riesgo de interacciones implica la necesidad de distanciar la administración del fármaco al menos 3 horas de cualquier otro medicamento por vía oral, lo cual podría ser una limitación en tratamientos crónicos en pacientes polimedicados.
Por la ausencia de comparaciones directas del efecto a corto plazo de patiromero cálcico frente a placebo y con las resinas intercambiadoras de cationes (poliestirensulfonato de sodio o de calcio) o ciclosilicato de sodio y zirconio –las principales opciones terapéuticas, que sí han demostrado superioridad clínica frente a placebo–, no se recomienda su empleo en el tratamiento agudo de hiperpotasemia leve o moderada (5,5-6,4 mEq/l). Los datos del tratamiento a largo plazo, aunque limitados (Fase 2 del estudio pivotal), demuestran una eficacia clínicamente relevante del nuevo fármaco en el control y prevención de recaídas de la hiperpotasemia leve-moderada. En definitiva, sin aportar ninguna innovación mecanística, patiromero cálcico se posiciona como alternativa de segunda línea a las resinas de intercambio iónico en el control crónico de la hiperpotasemia, de especial interés en pacientes con enfermedad renal y riesgo cardiovascular que necesiten continuar tratamiento con IRAAs.
ASPECTOS FISIOPATOLÓGICOS
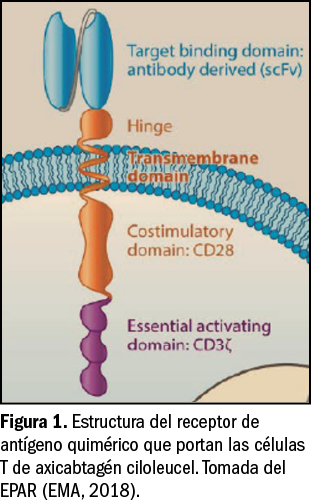
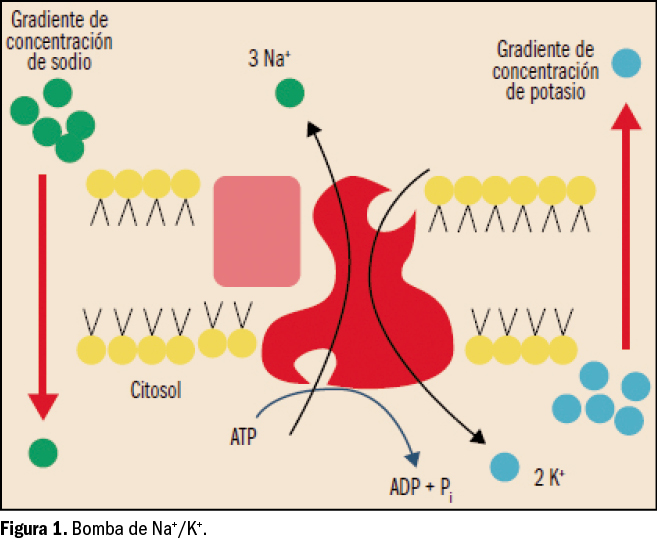
El potasio (K+) es uno de los minerales más abundantes en el organismo. Su distribución es mayoritariamente intracelular (98%), siendo su concentración normal en el interior de la célula de unos 145 mEq/l o mmol/l (5,7 g/l), en tanto que la concentración plasmática (potasemia) normal es de solo 3,5-4,5 mEq/l o mmol/l (137-175 mg/l). Esto supone una fuerte diferencia entre la concentración de potasio en el líquido intra- y extracelular, con un ratio de 35-40:1. Por tanto, para mantener el equilibrio orgánico se requiere la participación de una bomba de membrana celular dependiente de ATP o ATPasa (es decir, que consume energía) que extrae constantemente potasio fuera de la célula, a la vez que introduce sodio (Figura 1). Justamente, esta diferencia de concentración es el principal determinante del potencial de acción de membrana, y resulta fundamental para la función muscular normal (del músculo esquelético y cardiaco), así como para la neurológica.
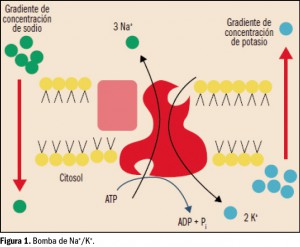
La regulación principal de la homeostasis del potasio se lleva a cabo a nivel renal y principalmente en la zona distal en las nefronas, viéndose alterada por muchos y diversos factores como las variaciones del pH, insulina, catecolaminas y aldosterona, principalmente. Las situaciones de acidosis desplazan el potasio fuera de la célula (porque este se intercambia con hidrogeniones), mientras que la alcalosis desplaza el potasio hacia el interior. Igualmente, la insulina favorece la entrada de glucosa en las células mediante una bomba de membrana que introduce potasio junto con la glucosa. La aldosterona aumenta la reabsorción de sodio a la vez que incrementa la excreción de potasio a nivel de la nefrona distal. El equilibrio de potasio también está regulado, al menos en parte, por su secreción en el colon a través de una ruta paracelular pasiva y otra de secreción activa; así, en el colon, el potasio está en alta concentración en relación con otros cationes.
El equilibrio de la potasemia puede alterarse al alza o a la baja.
La hipopotasemia (o hipocalemia) se define como la concentración plasmática –en el líquido extracelular– de K1.
Las manifestaciones clínicas de la hipopotasemia son muchas y variadas, aunque no suelen aparecer hasta que existen cifras inferiores a 3 mEq/l. Dependen, además, no solo de la cifra de potasio, sino también de la velocidad de instauración y de la existencia concomitante de otras alteraciones metabólicas. Las manifestaciones más frecuentes son la fatiga, el malestar y la debilidad muscular, sobre todo de las extremidades inferiores. La hipopotasemia intensa puede causar debilidad progresiva, con hipoventilación (por afectación de la musculatura respiratoria) e íleo paralítico (por afectación de la musculatura lisa del tubo digestivo), e incluso parálisis completa en los casos más graves.
La hipopotasemia produce también cambios electrocardiográficos bastante característicos, aunque no guardan relación directa con los niveles plasmáticos de potasio y se deben al retraso de la repolarización ventricular. Los cambios iniciales consisten en aplanamiento e inversión de la onda T, onda U prominente, depresión del segmento ST y prolongación del intervalo QT. Con hipopotasemias más intensas se produce una prolongación del intervalo PR, disminución del voltaje y ensanchamiento del complejo QRS. Esto puede dar lugar a la aparición de arritmias ventriculares, especialmente en pacientes con cardiopatía previa. Además, la hipopotasemia aumenta el riesgo de toxicidad por cardiotónicos digitálicos.
En el lado contrario, la hiperpotasemia (o hipercalemia) se define como una concentración plasmática de K+ superior a 5,0 mEq/l o, mayoritariamente, a 5,5 mEq/l (nivel límite variable dependiendo del autor consultado). Se puede clasificar, en función de su gravedad, como leve (5,5-5,9 mEq/l), moderada (6,0-6,4 mEq/l) o grave (>6,5 mEq/l). Se produce como consecuencia de la salida de K+ al exterior de la célula (líquido extracelular) o por disminución de las pérdidas renales como causa mayoritaria. El aumento de la ingesta de potasio es excepcional como causa de hiperpotasemia, aunque sí puede serlo en caso de insuficiencia renal o por administración excesiva de potasio por vía i.v. Por su parte, la pseudohiperpotasemia consiste en una concentración artificialmente elevada de K+ por salida de las células en el momento de la venopunción o extracción de muestra sanguínea para estudio; esto se produce por una posible mala técnica en la extracción (principalmente por un excesivo traumatismo).
La elevación de la concentración plasmática de potasio disminuye el ratio de K2 (que se emplean precisamente en el tratamiento de la ERC, la hipertensión y la insuficiencia cardiaca congestiva). En la ERC, la excreción urinaria de potasio disminuye y, además, la secreción colónica de potasio aumenta sustancialmente. La diabetes mellitus (concretamente, la cetoacidosis diabética o el estado hiperglucémico hiperosmolar, que provoca la salida no controlada de potasio desde el espacio intracelular) y el uso de betabloqueantes también pueden aumentar el riesgo de hiperpotasemia.
Así pues, la hiperpotasemia es poco frecuente en individuos sanos con función renal normal, pero su prevalencia en pacientes con insuficiencia renal (IR) o enfermedad renal crónica varía del 5% al 50%, aumentando a medida que disminuye la función renal y la excreción urinaria de potasio es menor. Se estima que cada año sufren hiperpotasemia hasta 3,8 millones de pacientes en la Unión Europea y unos 2,4 millones en los Estados Unidos. Se ha descrito que el riesgo de muerte por hipercalemia es mayor en pacientes con ERC.
El tratamiento de la hiperpotasemia tiene como objetivo primordial el de antagonizar los perjudiciales efectos cardiacos de este trastorno y el riesgo de mortalidad cardiovascular asociado, bien mediante la promoción del paso de potasio desde el espacio extracelular al intracelular y/o forzando la eliminación de potasio.
La mayor o menor urgencia del tratamiento vendrá determinada por la presencia de síntomas, la gravedad de la potasemia y su causa. Cuando la hipercalemia se presenta como consecuencia de afecciones clínicas agudas que ocurren en un entorno hospitalario, puede ser necesario un tratamiento inmediato, particularmente cuando el grado de hipercalemia es grave (K+ sérico ≥6,5 mEq/l) y/o se asocia a trastornos de la repolarización cardiaca. El manejo terapéutico difiere en caso de pacientes cuya hipercalemia tienen un carácter persistente y prolongado, derivada de condiciones subyacentes crónicas.
A grandes rasgos, los pacientes se pueden clasificar en tres niveles de tratamiento:
- Los pacientes con hiperpotasemia aguda severa (K+ sérico ≥7 mEq/l, o al menos >6,5 mEq/l) o alteraciones en el electrocardiograma y con presencia de síntomas relacionados con hiperpotasemia (debilidad o parálisis muscular y arritmias o alteraciones de la conducción cardiaca, entre otros), que generalmente están hospitalizados o son enviados al hospital, se consideran una emergencia de tratamiento. En estos casos, el manejo clínico debe buscar la estabilización temporal del miocardio y un cambio relativamente rápido y a corto plazo en el potasio intracelular, para reducir el riesgo de eventos arrítmicos fatales.
Suelen administrarse: a) tratamientos que antagonicen los efectos del potasio sobre el miocardio con un inicio de acción rápida (<10 min), como es el caso del calcio intravenoso (cloruro o gluconato cálcico) y del suero salino hipertónico, este último solo eficaz en pacientes hiponatrémicos; b) insulina intravenosa, para favorecer la entrada de potasio al espacio intracelular (administrada concomitantemente con glucosa para evitar la hipoglucemia); c) bicarbonato sódico intravenoso si además hay presencia de acidosis metabólica; y d) agonistas β-adrenérgicos como salbutamol (uso off label), capaces de estimular la bomba Na+/K+-ATPasa para promover la entrada de potasio al interior celular y de promover la secreción de insulina por las células β pancreáticas.
También se administrarán fármacos que favorecen la eliminación de potasio, tales como diuréticos del asa, si la función renal no está muy alterada, o resinas de intercambio catiónico (poliestirensulfonato de sodio o de poliestirensulfonato de calcio, con un inicio de acción a las 1-2 h y duración de hasta 6 h) y el empleo de técnicas de sustitución renal (hemodiálisis), si la afectación renal es relevante.
- Los pacientes en que la hiperpotasemia es leve o moderada (K3.
- El caso intermedio lo constituyen pacientes con hiperpotasemia leve-moderada que están asintomáticos pero que tienen una necesidad de inicio temprano (6-12 h) del tratamiento –no urgente– por insuficiencia renal severa o cirugía próxima. Éstos suelen manejarse con medidas como la hemodiálisis, la administración de bicarbonato intravenoso, la infusión de glucosa nocturna (para estimular la liberación de insulina), o el empleo de resinas de intercambio catiónico, ciclosilicato de sodio y zirconio o diuréticos del asa.
Sin embargo, estos fármacos no son bien tolerados y su uso se ha asociado en ocasiones con eventos adversos potencialmente mortales, incluida la necrosis intestinal. El uso de poliestirensulfonato de sodio se ha asociado, además, a una sobrecarga de sodio que obliga a extremar la precaución de uso en pacientes sensibles a hiponatremia, lo cual dificulta el uso de este fármaco en periodos prolongados. Por los riesgos de seguridad, las dos resinas de intercambio catiónico están contraindicadas en pacientes con niveles de potasio sérico <5,0 mEq/l y requieren frecuentes interrupciones de dosificación, complicando aún más los tratamientos crónicos. Así pues, existe la necesidad de nuevas terapias para la hiperpotasemia cuya eficacia y seguridad estén bien caracterizadas y puedan administrarse a largo plazo (EMA, 2017).
Por su parte, el ciclosilicato de sodio y zirconio (Lokelma®) es un fármaco de más reciente autorización, que no está aún disponible comercialmente en España. Es un polvo inorgánico no polimérico y no absorbible, con una estructura uniforme de microporos que captura de forma selectiva el potasio en el tracto gastrointestinal y lo intercambia por cationes de hidrógeno y sodio, aumentando la excreción fecal de potasio, con lo que disminuye sus niveles séricos. La eficacia de este fármaco ha sido contrastada en dos ensayos clínicos en pacientes con ERC, en tratamiento con fármacos IRAAs y/o diabetes; la mayoría de los pacientes presentaba potasemia basal <5,5 mEq/L. El ciclosilicato de sodio y zirconio demostró ser tras estadísticamente superior a placebo en la reducción del potasio plasmático tanto en la fase aguda (48 horas) como en la de mantenimiento (tras 12-28 días de tratamiento).
ACCIÓN Y MECANISMO
El patiromero cálcico es un polímero de intercambio de cationes que no se absorbe y es capaz de unirse y quelar los iones potasio libres en la luz del tracto gastrointestinal (sobre todo, en el colon, donde K+ es el catión más abundante), de manera que aumenta la excreción en heces de dicho ión y reduce los niveles de potasio en suero. En base a dicho mecanismo, el medicamento ha sido autorizado para el tratamiento de la hiperpotasemia en adultos.
En ensayos in vitro, bajo condiciones que mimetizaban el pH y el contenido en potasio del colon, la capacidad de patiromero de unión a potasio fue de 1,5 a 2,5 veces mayor que la de los polímeros de intercambio catiónico tradicionales, incluido el poliestirensulfonato de sodio. En un modelo de hiperpotasemia en ratas, el patiromero aumentó significativamente la excreción fecal de potasio, llevando a la normalización de sus niveles séricos.
En personas adultas sanas, patiromero (a dosis de 4,2, 8,4 y 16,8 g tres veces al día) demostró un efecto dosis-dependiente sobre el incremento de la excreción de potasio en heces, con una correspondiente reducción de su excreción en orina. Se ha postulado que, por su acción en el intestino, patiromero también puede afectar potencialmente –en menor medida– a la homeostasis de otros iones; de hecho, en individuos sanos indujo descensos dosis-dependientes de los niveles de sodio, magnesio y fosfato en orina, pero un incremento de calcio urinario también dependiente de la dosis (Blair, 2018). Adicionalmente, los estudios de interacción in vitro han demostrado que patiromero puede afectar a la biodisponibilidad de diversos fármacos (de los 28 principios activos evaluados, aproximadamente la mitad mostraron interacción positiva); así, por el potencial riesgo existente, la ficha técnica del medicamento recomienda que la administración de cualquier otro medicamento por vía oral se haga al menos 3 horas antes o después del empleo de patiromero.
Los ensayos clínicos han demostrado que, a una dosis de 25,2 g/día durante 6 días, el fármaco aumentó la excreción de potasio en heces en 1.283 mg/día, disminuyendo su excreción urinaria una media de 1.438 mg/día; la excreción diaria de calcio en orina aumentó respecto al valor inicial en 53 mg/día. Otro estudio demostró que la administración de 15 g/día de patiromero cálcico durante 7 días resultó en una disminución media de 0,40 (± 0,44) mEq/l en la concentración de K+ en suero respecto al estado basal en pacientes con insuficiencia renal, siendo ligeramente superior en días sin diálisis respecto a los días con diálisis. El tratamiento durante 7 días resultó en un aumento estadísticamente significativo en la excreción de potasio fecal (media de 359 ± 277 mg/día).
En cuanto al inicio de su efecto, la reducción significativa del potasio en suero en pacientes con hiperpotasemia parece detectarse a las 7 horas desde la primera dosis, mientras que los niveles de potasemia permanecen estables durante 24 h tras última dosis en caso de suspensión del tratamiento, volviendo a aumentar posteriormente durante al menos 4 días.
Aspectos moleculares
El patiromero cálcico es un anión polimérico reticulado formado por la unión de patiromero a un complejo de calcio-sorbitol como contraión. El dominio activo de la molécula, patiromero, es un polímero de intercambio catiónico que no es absorbible una vez ingerido por vía oral, pues se fabrica con un diseño de partículas esféricas de flujo libre de aproximadamente 100 micrómetros de diámetro, tamaño que impide su paso a través de las células de la pared intestinal.
El nombre químico del patiromero cálcico es poli[(D-glucitol-calcio)2-fluoroacrilato-co-dietenilbenceno-co-octa-1,7-dieno] y su fórmula, 2C6H14O6)m(C3H2FO2)4m(C10H10)4n(C8H14)4p. La compleja estructura molecular del fármaco no implica un orden regular de los monómeros, que se unen aleatoriamente para formar las cadenas reticuladas del polímero. Está compuesto de tres dominios o unidades de repetición: una unidad de 2-fluoro-2-propenoato cargada negativamente y dos moléculas difuncionales (divinilbenceno y 1,7-octadieno), que se entrelazan mediante enlaces covalentes para formar el polímero, el cual se acompleja posteriormente con calcio-sorbitol.
El fármaco está asociado a moléculas de agua (15%) y se presenta como un polvo no higroscópico, amarillo-marrón, amorfo, de flujo libre, compuesto de partículas esféricas individuales que tienen un peso molecular de 5,6×1017 g/mol. La sustancia activa es insoluble en agua, en metanol y en heptano.
EFICACIA Y SEGURIDAD CLÍNICAS
La farmacodinamia clínica del patiromero cálcico ha sido evaluada en su dosis e indicación autorizadas mediante un amplio ensayo pivotal de fase 3 (RLY506-301), confirmatorio de seguridad y eficacia, que presentó un diseño simple ciego (los investigadores no eran ciegos al tratamiento) y que se estructuró en dos partes (Weir et al., 2015).
En una primera parte de 4 semanas de duración (Fase 1), el estudio fue abierto, de un solo brazo, y no aleatorizado. Bajo la hipótesis de la superioridad sobre la ausencia de cambios intra-individuo, evaluó el efecto del patiromero cálcico en un total de 243 pacientes adultos con hiperpotasemia leve-moderada (5,1-6,4 mEq/l) y enfermedad renal crónica (tasa de filtración glomerular de 15 a 60 ml/min/1,73 m2), que estaban siendo tratados con dosis estables de al menos un fármaco IRAA (inhibidores de la enzima convertidora de angiotensina, antagonistas de la aldosterona o antagonistas de los receptores de la angiotensina II). Se excluyeron pacientes con patologías (diabetes, hipertensión o insuficiencia cardiaca) no controladas.
La edad media de la población de estudio fue de 64 años (29-80 años), con un 54% y un 17% de pacientes con ≥65 y ≥75 años, respectivamente. El 58% de los pacientes fueron varones, el 98% de raza blanca, y casi todos presentaban al menos una comorbilidad cardiovascular: el 97% de los pacientes eran hipertensos, el 57% padecía diabetes de tipo 2, el 42% insuficiencia cardiaca de clase II (de la NYHA) y el 25% tenía antecedente de infarto de miocardio. En cuanto a la disfunción renal, la ERC se encontraba en estadio 2, 3a, 3b y 4 en el 9%, 20%, 26% y 45% de pacientes, respectivamente.
La dosis inicial del fármaco se ajustó al nivel de K+ sérico, de forma que quienes tenían niveles entre 5,1 y 5,5 mEq/l recibieron una dosis de 8,4 g/día (grupo A; N=92) y aquellos pacientes con valores de 5,5 a 6,5 mEq/l recibieron 16,8 g/día (grupo B; N=147), en todos los casos como dosis dividida. A partir del día 3 de tratamiento y hasta el final de la Fase 1, las dosis se fueron ajustando, hasta un máximo de 50,4 g/día a fin de mantener el nivel de potasio en suero en el intervalo prefijado de 3,8-5,1 mEq/l, alcanzándose unas dosis medias de 13 y 21 g/día de patiromero cálcico en los grupos A y B, respectivamente. Se suspendía el tratamiento con IRAAs si la potasemia se mantenía descontrolada (por encima de 6,5 mEq/l, o de 5,1 mEq/l si se usaban dosis máximas del fármaco). Un total de 219 pacientes (92% en el grupo A y 89% en el grupo B) completaron la primera fase.
La media del cambio en el nivel de la potasemia tras 4 semanas de tratamiento (variable principal de eficacia) fue de -0,65 ± 0,05 mEq/l (IC95% -0,74, -0,55) en el grupo A y de -1,23 ± 0,04 mEq/l (IC95% -1,31, -1,16) en el grupo B, respecto a los niveles basales promedio de potasemia (5,31 mEq/l y 5,74 mEq/l, respectivamente). En el global de pacientes, el cambio medio fue de -1,01 ± 0,03 mEq/l (IC95% -1,07, -0,95) respecto a la media de 5,58 mEq/l de potasemia basal, lo cual representó un efecto con robustez estadística (p<0,001). Adicionalmente, el porcentaje de pacientes –respondedores– con potasemia en el rango objetivo entre 3,8 y 5,1 mEq/l a la semana 4 (variable secundaria) fue del 76% (IC95% 70-81) en la población global; esta variable tomó valores similares en ambos grupos de dosis de patiromero cálcico: 74% para el grupo A y 77% para el grupo B.
La segunda fase del estudio pivotal tuvo una duración de 8 semanas (Fase 2), presentó un diseño controlado con placebo, y evaluó el efecto de la retirada aleatorizada de patiromero cálcico en el control de la potasemia con el objetivo de valorar la eficacia del tratamiento crónico con el fármaco en la prevención de recurrencias de hiperpotasemia. A esta fase accedieron los 107 pacientes que presentaban un nivel basal de potasio de 5,5 a 6,5 mEq/l al inicio de la primera fase y habían cumplido las 4 semanas de tratamiento de ésta, situándose su potasemia dentro del intervalo objetivo (3,8-5,1 mEq/l) al final de dicho periodo; además, debían continuar el tratamiento con IRAAs durante toda la Fase 2. De ellos, el 54% de pacientes fueron hombres, el 100% de raza blanca, la media de edad se mantenía en 65 años (32-80), y los porcentajes de pacientes con comorbilidades cardiovasculares y estadios de patología renal eran muy similares a los de la Fase 1.
Los pacientes fueron asignados al azar (1:1) a seguir recibiendo patiromero cálcico, a una dosis media diaria de 21 g/día (N=55), o a cambiar a placebo (N=52), estratificándose según los niveles basales de potasio en la Fase 1 (5,5-5,8 y >5,8 mEq/l) y el diagnóstico de diabetes mellitus tipo 2. La dosis de patiromero cálcico podía aumentarse, así como reducirse la de IRAAs en ambos grupos, en caso de potasemias superiores a 5,5 mEq/l o 5,1 mEq/l en las primeras o segundas 4 semanas, respectivamente. Se suspendía el tratamiento con IRAAs si la potasemia se mantenía descontrolada a pesar de tales medidas. Una proporción relevante de pacientes –18% y 42% en los brazos experimental y placebo, respectivamente– no completó el periodo de tratamiento.
En dicha Fase 2, la mediana del cambio del potasio sérico desde el inicio hasta la primera medida fuera del intervalo de 3,8-5,5 mEq/l o hasta la semana 4 en caso de que la potasemia permaneciera en ese intervalo (variable primaria de eficacia) fue de +0,72 mEq/l en el grupo placebo y de 0,00 mEq/l en el grupo de tratamiento con patiromero cálcico; es decir, había una diferencia significativa de 0,72 mEq/l (IC95% 0,46-0,99) entre ambos grupos (p<0,001).
Con respecto a las variables secundarias, el porcentaje de pacientes que alcanzaron valores de potasemia superiores a 5,5 mEq/l en cualquier momento fue del 60% (IC95% 47-74) y del 15% (IC95% 6-24) en los grupos placebo y experimental, respectivamente; para valores superiores a 5,1 mEq/l, esos porcentajes se situaron en el 91% (IC95% 83-99) y el 43% (IC95% 30-56), respectivamente. Así pues, las diferencias entre el grupo placebo y el grupo de tratamiento con patiromero cálcico hasta la semana 8 de tratamiento fueron estadísticamente significativas en ambos supuestos, con diferencias estimadas del 45% y el 48%. Un menor número de pacientes tratados con patiromero cálcico (5%) tuvo que interrumpir el tratamiento con IRAAs en comparación con aquellos tratados con placebo (52%).
Por otra parte, se dispone de los datos de eficacia de un estudio de soporte (RLY5016-2015), un ensayo de fase 2, con diseño abierto, aleatorizado y de 52 semanas de duración (las 8 primeras de búsqueda de dosis). Incluyó un total de 304 pacientes de raza blanca con hiperpotasemia, hipertensión y nefropatía diabética, en tratamiento con IRAAs y con o sin espironolactona, cuya media de edad fue de 66 años (37-80; un 60% de pacientes >65 años) y de los cuales la mayoría eran hombres (63%). Éstos fueron divididos en base a su potasemia basal en dos estratos: 1) niveles entre 5,0 y 5,5 mEq/l (N=219), que recibieron aleatoriamente dosis de 8,4-25,2 g/día de patiromero cálcico (dosis media de 14 g/día), y 2) niveles de potasio sérico entre 5,5 y 6,0 mEq/l (N=84), quienes recibieron 16,8-33,6 g/día del fármaco (dosis media de 20 g/día). Las dosis se titularon a fin de alcanzar valores de potasemia entre 4,0 y 5,0 mEq/l las 8 primeras semanas y de 3,8-5,0 mEq/l en adelante.
A grandes rasgos, se observaron reducciones significativas de la potasemia (p<0,001) en comparación con los niveles basales tras 4 semanas de tratamiento (variable primaria) para todas las dosis de patiromero cálcico evaluadas en ambos grupos; dicho efecto era constatable desde el tercer día de tratamiento, con la administración de al menos 4 dosis del fármaco. La disminución media de la potasemia fue mayor en el estrato 2 (entre -0,88 y -0,90 mEq/l) que en estrato 1 (entre -0,35 y -0,53 mEq/l). La proporción de pacientes respondedores cuya potasemia se situó en el rango objetivo de 4,0-5,0 mEq/l fue del 85% y 73% en los estratos 1 y 2, respectivamente. Las modificaciones de la potasemia respecto al estado basal en las semanas 8 y 52 (variables secundarias) se mantuvieron muy similares a las de la semana 4. En definitiva, con una baja incidencia de hipopotasemia (2,3%), la mayoría de los participantes (98%) alcanzó y mantuvo los niveles deseados de potasio sérico durante aproximadamente el 80% del tiempo del periodo de seguimiento de un año (EMA, 2017).
La seguridad clínica de patiromero cálcico ha sido definida de acuerdo a los datos derivados de al menos 666 pacientes incluidos en hasta 4 ensayos y que han recibido al menos una dosis en el rango de 8,4-50,4 g/día; el periodo de tratamiento con el fármaco fue >4 semanas para 584 pacientes y de >6 meses para 219 pacientes. Se notificaron eventos adversos en el 61% de los pacientes, pero solo en el 20% se relacionaron con el fármaco; el número de pacientes que interrumpió el tratamiento por dichos eventos adversos fue bajo (9,0%). Los trastornos gastrointestinales fueron las reacciones adversas más frecuentes: estreñimiento (6,2%), diarrea (3%), dolor abdominal (2,9%), flatulencia (1,8%). También se reportaron casos de hipomagnesemia (5,3%), insuficiencia renal crónica, anemia, hiperglucemia o extrasístoles; los casos de hipopotasemia (4,7%) fueron leves. Todas ellas fueron leves-moderadas, no se identificó una relación de dosis-dependencia y tuvieron carácter autolimitado o se resolvieron con tratamiento.
Los eventos adversos graves se notificaron en un 8,3% de pacientes tratados con patiromero cálcico, aunque ningún caso se consideró relacionado con el fármaco. De éstos, destacan las alteraciones cardiacas (2,4%), que no obstante acontecieron en pacientes con antecedentes cardiovasculares y factores de riesgo, o los casos de fallo renal crónico (1,1%) y agudo (0,5%). Ninguno de los casos de muerte notificados (19) se relacionó con el fármaco o los niveles de potasio.
ASPECTOS INNOVADORES
El patiromero cálcico es un anión polimérico reticulado que actúa como polímero de intercambio de cationes. Por sus características físico-químicas, no se absorbe, y es capaz de unirse y quelar los iones potasio libres en la luz del tracto gastrointestinal (sobre todo, en el colon, donde K+ es el catión más abundante), de manera que aumenta la excreción en heces de dicho ión y reduce los niveles de potasio en suero. En base a dicho mecanismo, el medicamento ha sido autorizado para el tratamiento de la hiperpotasemia en adultos.
La eficacia clínica del fármaco ha sido adecuadamente contrastada en la indicación y dosis autorizada mediante un ensayo pivotal de fase 3, simple ciego, y que constó de dos partes diferenciadas. En la Fase 1 (N=243), no aleatorizada, abierta y de un único brazo, patiromero fue capaz de reducir significativamente el nivel de potasio sérico en -1,01 (± 0,03) mEq/l –respecto al estado basal promedio (5,58 mEq/l)– tras 4 semanas de tratamiento en pacientes con enfermedad renal crónica (mayoritariamente en estadios 3 y 4), tratamiento concomitante con inhibidores del sistema renina-angiotensina-aldosterona y con una alta incidencia de hipertensión arterial, insuficiencia cardiaca y diabetes mellitus tipo 2. El 76% de los pacientes –respondedores– alcanzó valores de potasemia dentro de la normalidad (3,8-5,1 mEq/l), siendo efecto del fármaco en el descenso de la potasemia más pronunciado –de casi el doble– en pacientes con mayores niveles basales (-1,23 mEq/l en pacientes con 5,5-6,5 mEq/l vs.-0,65 mEq/l en aquellos con <5,5 mEq/l).
La Fase 2 del estudio, con diseño controlado por placebo y de retirada aleatorizada, evaluó la eficacia del fármaco durante un periodo de 8 semanas para prevenir recurrencias de hiperpotasemia en los pacientes respondedores de la primera fase (N=107). Los resultados muestran que la retirada de patiromero cálcico provocó un aumento significativo de la potasemia de +0,72 mEq/l (p<0,001) desde el inicio, bien hasta la primera medida fuera de la normalidad o hasta la semana 4. Hubo una diferencia del 45% en la proporción de pacientes que alcanzaron niveles de hiperpotasemia (>5,5 mEq/l) en el grupo placebo (60%) respecto a quienes mantuvieron el tratamiento experimental (15%). Los resultados de esta fase también sugieren que patiromero cálcico favorece la continuación del tratamiento con IRAAs (proporción de retirada de 5% vs. 52% con placebo).
Adicionalmente, los resultados de un ensayo clínico de soporte de fase 2 –de un único brazo, abierto y aleatorizado– aportan evidencia de que la administración diaria de patiromero cálcico es capaz de mantener reducciones significativas de los niveles de potasio sérico en el entorno de 0,9-1 mEq/l durante al menos 1 año en el tratamiento a largo plazo o crónico (si bien esto se evaluó como variable secundaria del estudio) en pacientes con hiperpotasemia basal leve (5,5-6,0 mEq/l).
Entre las principales incertidumbres de los resultados de eficacia se debe apuntar al reducido número de pacientes (107) que fueron incluidos en la Fase 2 del estudio pivotal, así como la elevada proporción de los mismos que no completó dicha fase (18% en el brazo experimental y 42% en el brazo placebo). Si bien todos los pacientes enrolados en ese estudio presentaban enfermedad renal (principal causa de hiperpotasemia), parece razonable extrapolar los resultados de reducción de la potasemia a otros casos de hipercalemia debidos a causas no-renales, habida cuenta del mecanismo de acción de patiromero cálcico.
Por otra parte, el perfil toxicológico benigno del nuevo fármaco parece adecuadamente definido, y se puede considerar como un fármaco bien tolerado. Solo un 20% de pacientes incluidos en 4 ensayos de su desarrollo clínico mostraron eventos adversos leves-moderados relacionados con el fármaco (principalmente trastornos gastrointestinales –estreñimiento, diarrea dolor abdominal o flatulencia– e hipomagnesemia), que fueron similares a los descritos para el resto de resinas intercambiadoras de cationes, las cuales presentan otros problemas de seguridad adicionales, como hipernatremia o hipercalcemia.
Cabe destacar que ninguno de los casos de eventos adversos graves (8,3%) se asoció al fármaco, describiéndose una baja tasa de interrupción del tratamiento por efectos adversos (9%), incluso entre aquellos pacientes tratados durante 1 año. No obstante, la evidencia del uso a largo plazo del fármaco se limita a un único estudio de soporte, por lo que se requieren más datos que confirmen que la seguridad clínica se mantiene en tratamientos crónicos. Además, para certificar un uso seguro del fármaco, se requiere distanciar la administración del fármaco al menos 3 horas de cualquier otro medicamento por vía oral, lo cual podría representar una limitación en administraciones crónicas en pacientes polimedicados.
Según todo lo anteriormente expuesto, parece que patiromero cálcico será una alternativa a las resinas intercambiadoras de cationes (poliestirensulfonato de sodio o de calcio) y a ciclosilicato de sodio y zirconio (cuando éste se comercialice en España) para el tratamiento de la hiperpotasemia leve (5,5-5,9 mEq/l) o moderada (6,0-6,4 mEq/l), a priori tanto a corto como a largo plazo. Sin embargo, no se ha realizado una comparación directa del nuevo fármaco frente a estas opciones terapéuticas, las cuales han sí demostrado superioridad clínica a corto plazo frente a placebo en la hiperpotasemia leve a moderada. Puesto que tampoco se dispone de datos comparativos de patiromero cálcico frente a placebo en la valoración de la reducción del potasio (la Fase 1 del estudio pivotal fue no comparativa), el IPT de la AEMPS considera que no debe recomendarse el nuevo fármaco en ese escenario clínico (AEMPS, 2019).
Además, el inicio de acción “lento” (7 h) de patiromero cálcico determina que no deba emplearse en situaciones de emergencia hiperpotasémica, en las cuales no reemplazará al uso de otros tratamientos útiles. No obstante, los datos del tratamiento a largo plazo de hiperpotasemia (derivados de la Fase 2 del estudio pivotal) sí demuestran una eficacia clínicamente relevante del nuevo fármaco en la prevención de recaídas de la hiperpotasemia leve-moderada, por lo cual representará una alternativa a las resinas de intercambio catiónico en el control crónico de dicho trastorno, con especial interés en pacientes con enfermedad renal y riesgo cardiovascular que necesiten continuar tratamiento con inhibidores del sistema renina-angiotensina-aldosterona. La limitación de los datos disponibles requiere una evaluación poscomercialización cuidadosa del perfil beneficio-riesgo.
En definitiva, patiromero cálcico es un nuevo fármaco que no supone una innovación mecanística y su empleo en el manejo de la hiperpotasemia leve-moderada se considerará como alternativa de segunda línea a las resinas de intercambio iónico en el tratamiento a largo plazo.
BIBLIOGRAFÍA






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}