El Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en la información autorizada de las fichas técnicas y de los prospectos de los medicamentos europeos por motivos de seguridad.
Una vez que se revisan y evalúan los datos de los informes periódicos de seguridad (IPS; en inglés PSUR), de forma colaboradora entre todas las 28 agencias nacionales, se presentan los cambios y se acuerdan en las reuniones mensuales del PRAC. A continuación se muestran los últimos cambios de información de seguridad acordados recientemente en el PRAC.
El Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en las fichas técnicas y los prospectos de los siguientes medicamentos, siendo los más importantes los que se describen en la Tabla 1, según informa1,2 la AEMPS en sus Boletines Mensuales de enero y de febrero de 2019.

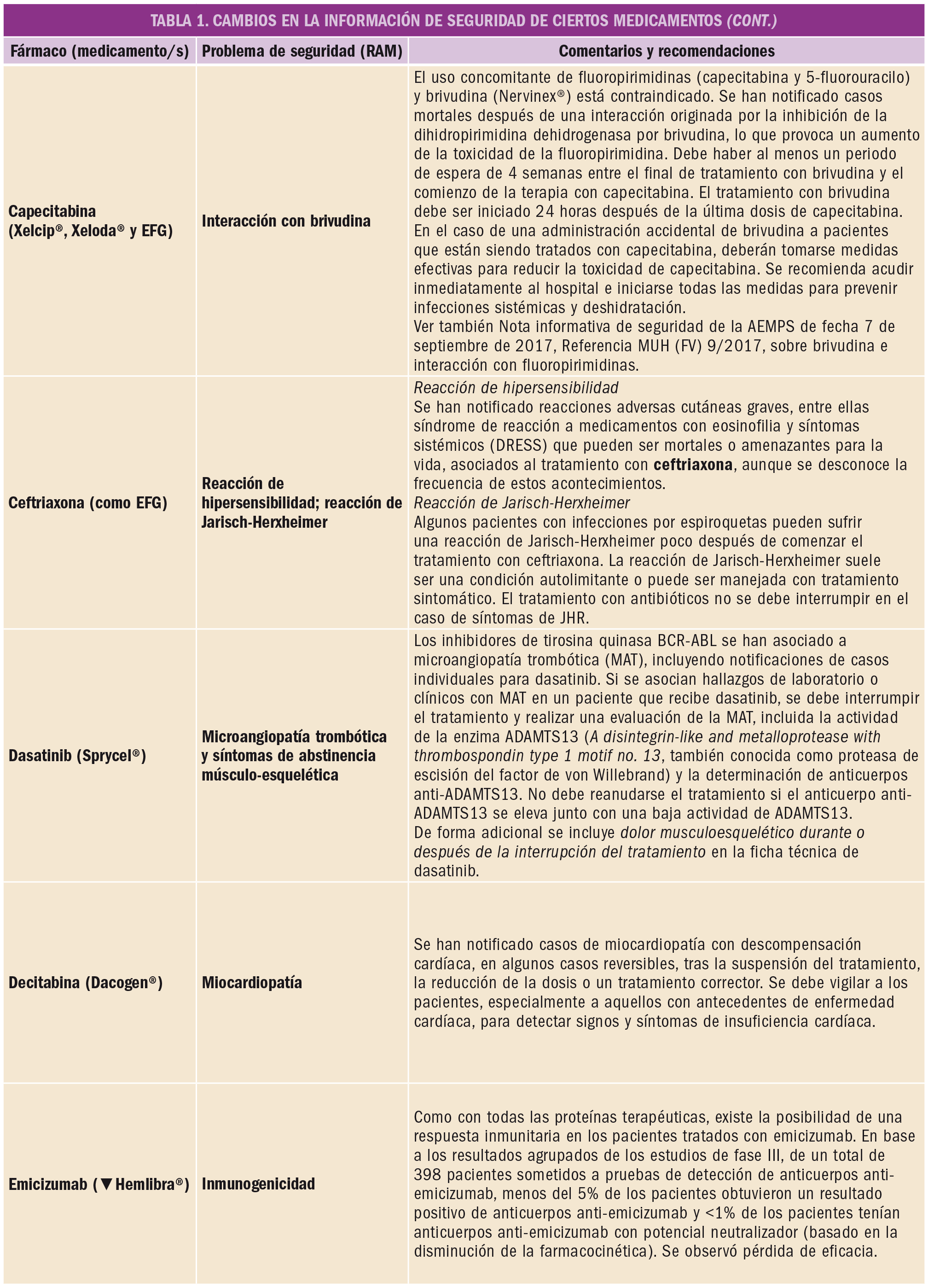
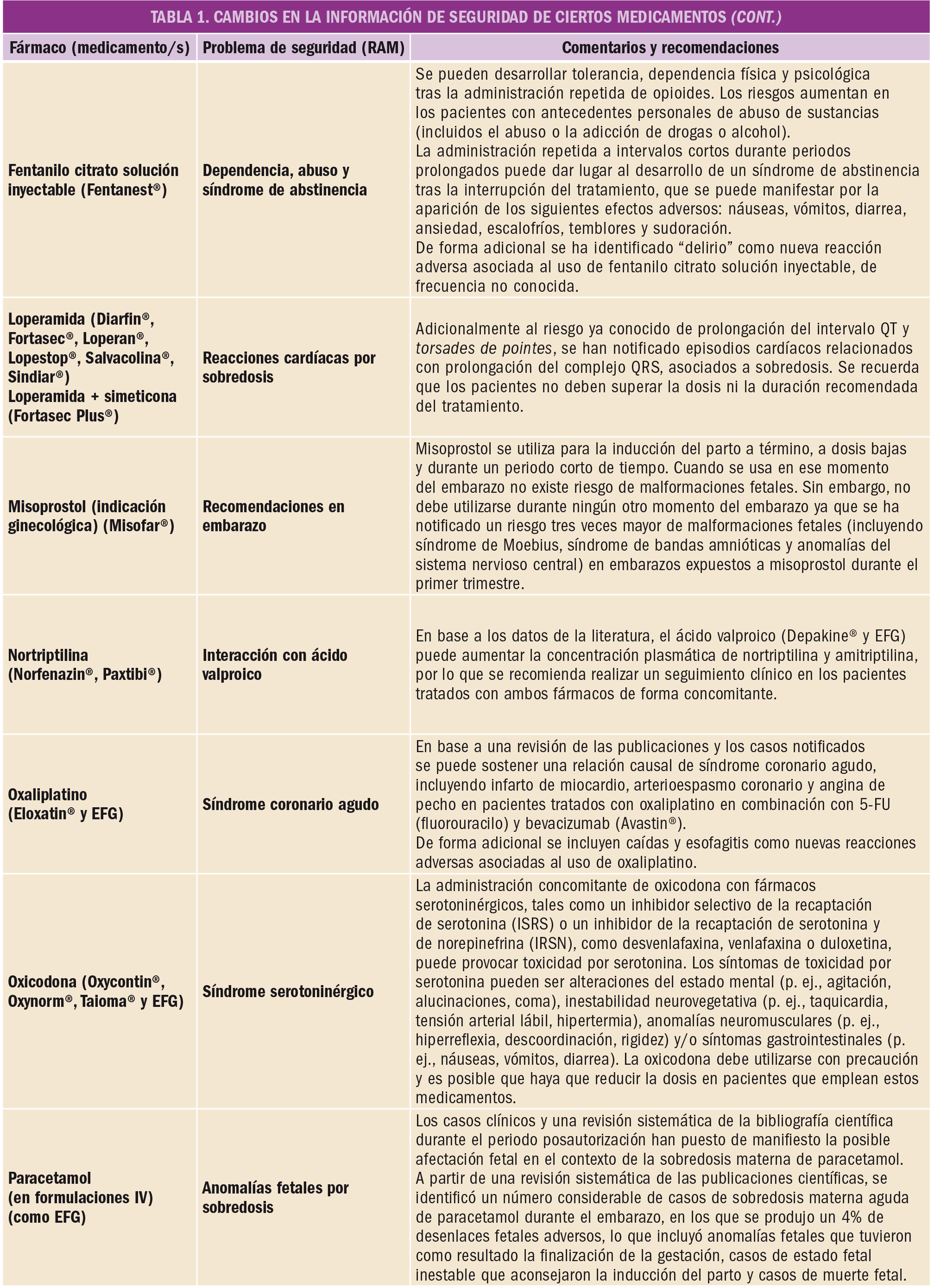
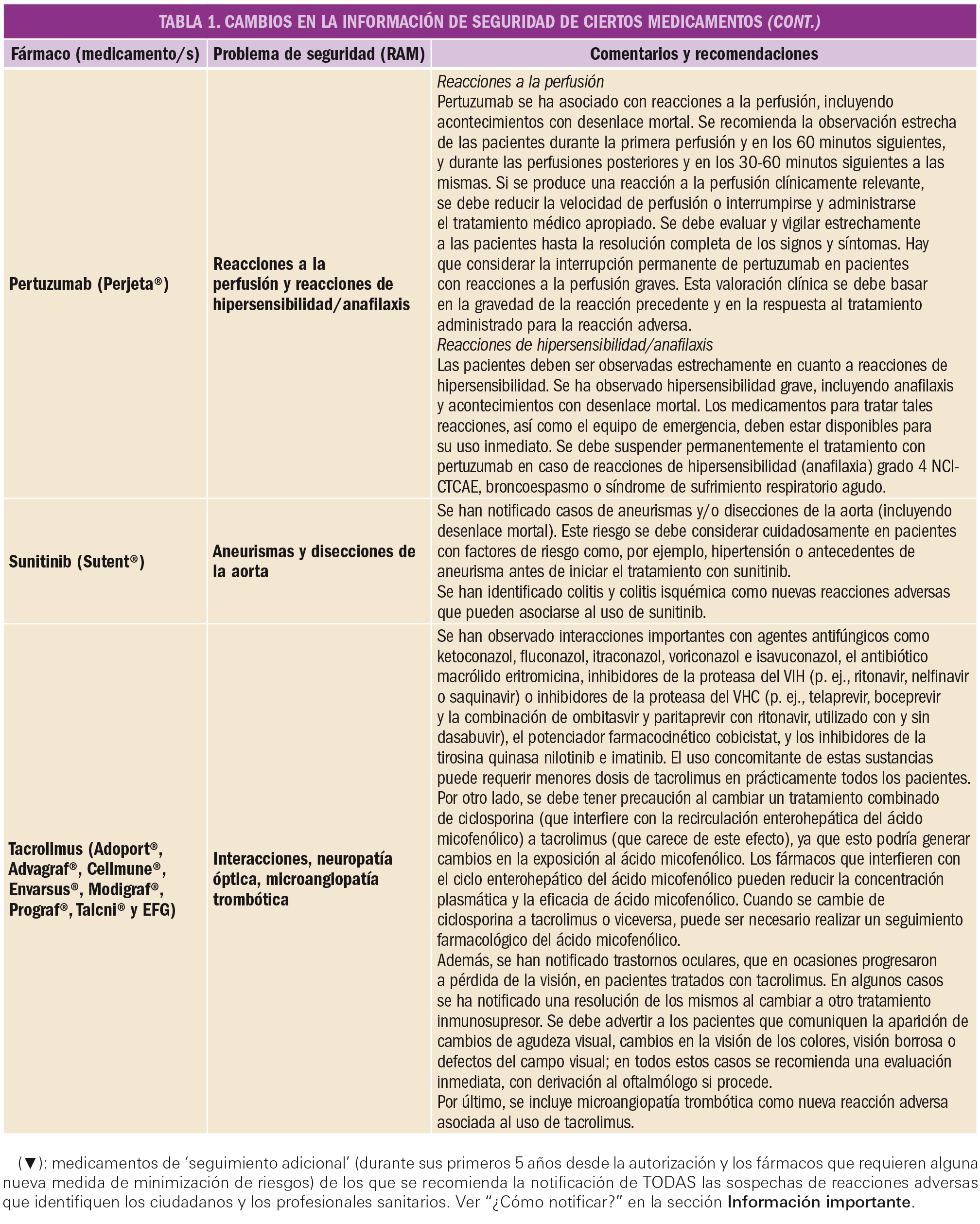
Las fichas técnicas y prospectos de los medicamentos pueden consultarse en la web de la AEMPS, dentro de la sección CIMA: Centro de Información Online de Medicamentos.
Referencias
- Agencia Española de Medicamentos y Productos Sanitarios. Nueva información de seguridad procedente de la evaluación periódica de los datos de farmacovigilancia. Boletín Mensual de la AEMPS sobre Medicamentos de uso Humano, enero 2019, páginas 11 a 14. Disponible en la web de la AEMPS: https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2019/enero/docs/boletin-mensual-MUH_enero-2019.pdf (consultado 27 de mayo de 2019).
- Agencia Española de Medicamentos y Productos Sanitarios. Nueva información de seguridad procedente de la evaluación periódica de los datos de farmacovigilancia. Boletín Mensual de la AEMPS sobre Medicamentos de uso Humano, febrero 2019, páginas 8 a 10. Disponible en la web de la AEMPS: https://www.aemps.gob.es/informa/boletines-AEMPS/boletinMensual/2019/febrero/docs/boletin-mensual-MUH_febrero-2019.pdf (consultado 27 de mayo de 2019)
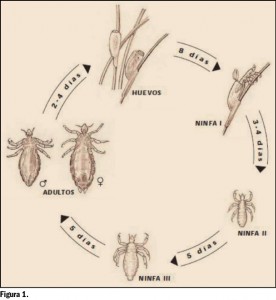

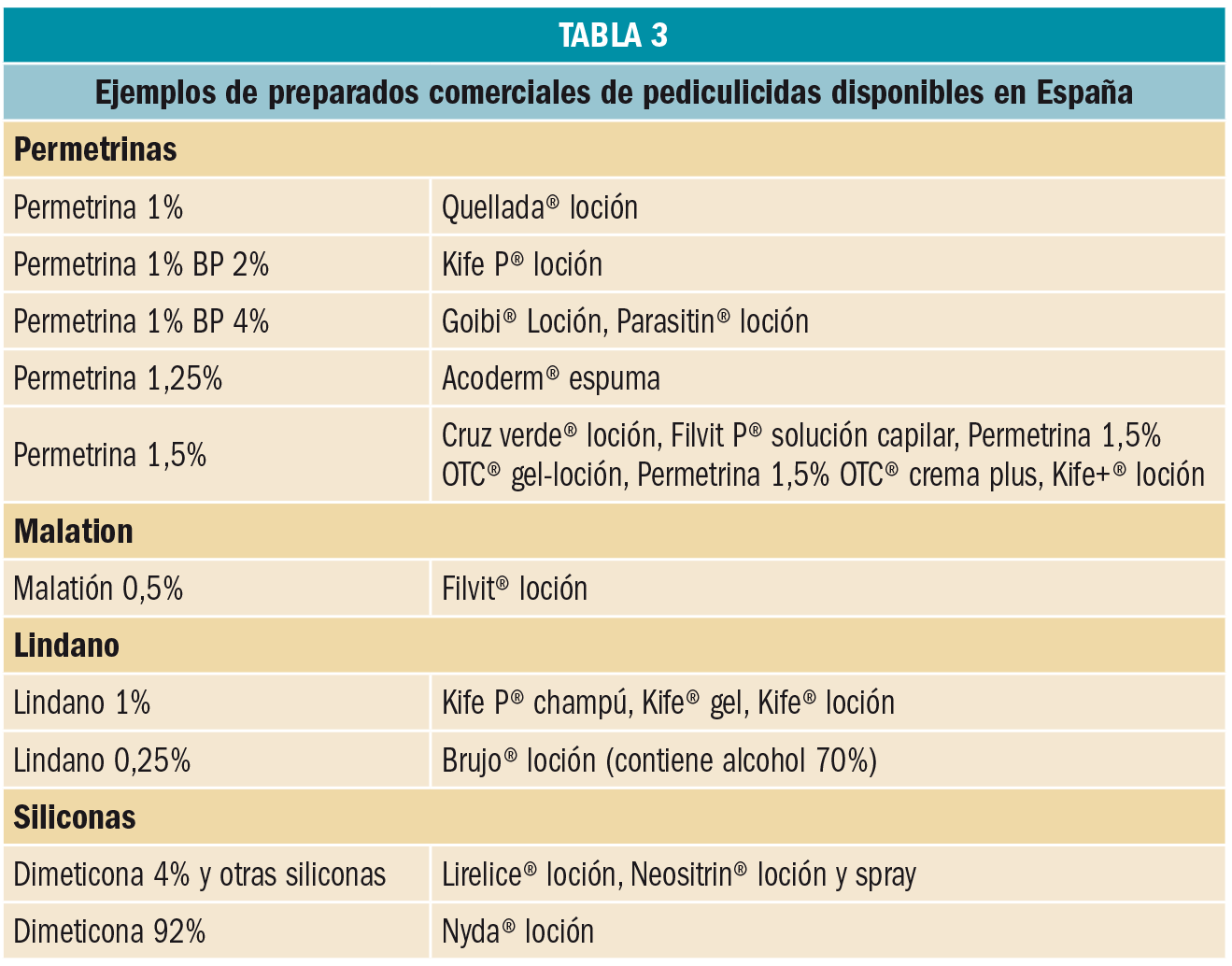
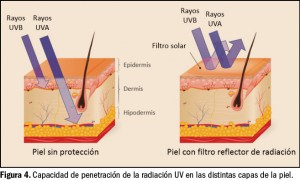
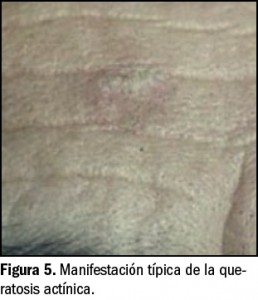
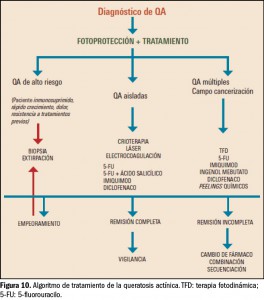
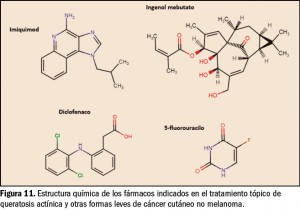
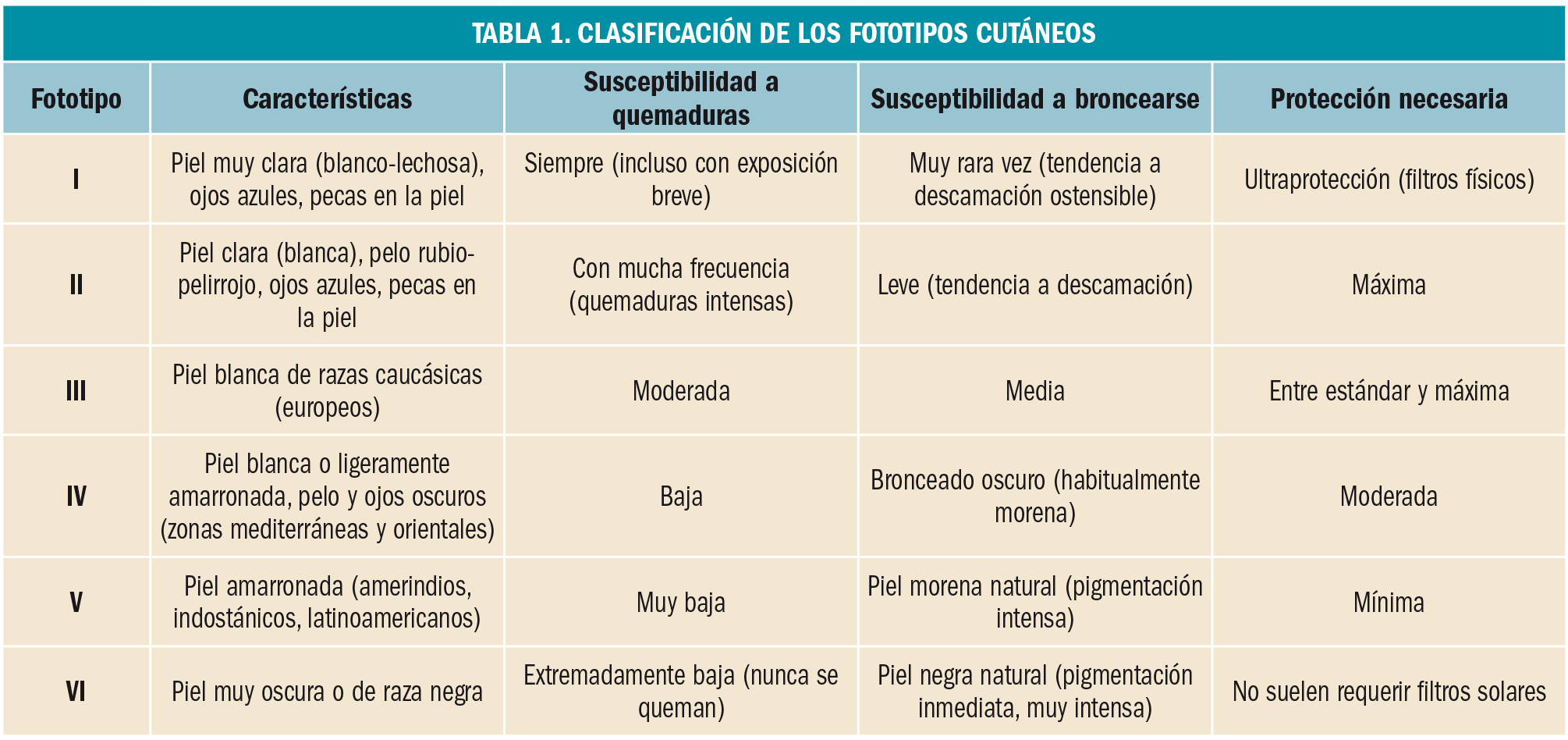

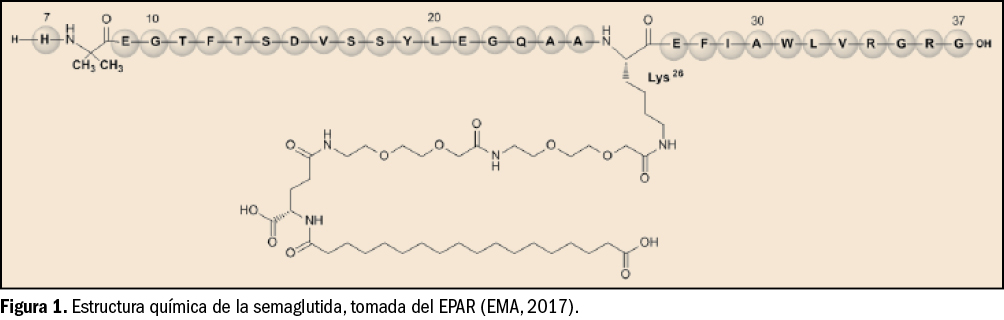{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}