Las recomendaciones de tratamiento de la diabetes mellitus tipo 2 (DM2) señalan a metformina (MET) como terapia inicial/de elección en caso de enfermedad cardiovascular establecida; si es preciso intensificar el tratamiento se recomienda la adición de un inhibidor del cotransportador de sodio-glucosa tipo 2 (SGLT2, como canagliflozina, dapagliflozina y empagliflozina) o un agonista del receptor del péptido similar a glucagón tipo 1 (liraglutida, exanatida). Si no hay patología cardiovascular, la intensificación de MET se puede realizar con inhibidores de la enzima dipeptidil peptidasa 4 (DPP-4, como sitagliptina, saxagliptina o linagliptina), inhibidores de SGLT2, tiazolidindionas (rosiglitazona o pioglitazona), sulfonilureas (SU, como gliclazida, glimepirida, glipizida), agonistas del receptor del péptido similar a glucagón tipo 1 o insulina basal. Si, a pesar del tratamiento con dos fármacos en combinación, es preciso una intensificación posterior, se recurriría a la administración de terapias triples compuestas por tres de las opciones mencionadas.
Una terapia de intensificación habitual es la combinación de MET con SU; sin embargo, se observa un mayor riesgo de hipoglucemia y ganancia de peso con dicha combinación. Los fármacos más nuevos, como los inhibidores de DPP-4 o de SGLT2, han mostrado mejores perfiles. Por ello, los autores3 analizaron el impacto en resultados de salud y costes de diversas rutas de intensificación del tratamiento de DM2 en el ámbito de EE.UU.
El estudio utiliza el modelo CDM (CORE Diabetes Model), el cual simula la evolución de una cohorte de 1.000 pacientes con DM2 durante toda la vida (limitando a 40 años) y estima el coste así como la tasa de las complicaciones micro y macrovasculares y de muerte. Se analizaron dos rutas alternativas de intensificación: a) tras fracaso a MET se adicionaban inhibidores de DPP-4 seguido de la adición de inhibidores de SGLT2, todo ello antes de administrar insulina; y b) a MET se adicionaba en principio una SU y, posteriormente, se incluía insulina. Los costes incluidos comprendieron fármacos, tiras reactivas de automonitorización de glucemia, jeringas, etc. En el caso de complicaciones o eventos adversos, se restó el efecto negativo correspondiente a éstos a la utilidad de cada estado de salud. Finalmente, a partir de los estudios clínicos publicados, se consideraron también los valores de eficacia (variación de la HbA1c y modificación del peso o de la tasa de hipoglucemias).
Los resultados demostraron que la alternativa (a) mejoró ligeramente la supervivencia, aumentando los años de vida ganados (AVG); asimismo, mejoró la calidad de vida respecto de la otra alternativa, contribuyendo a aumentar el número de años de vida ajustados a calidad (AVAC). Por otra parte, el coste total fue mayor, si bien se compensó parcialmente con una reducción del coste de las complicaciones, asociado a un mejor control. Finalmente, se estimó un valor del ratio coste-efectividad incremental (RCEI) inferior al habitualmente admitido en EE.UU., de 100.000 $/AVAC (Tabla 3). Los análisis de sensibilidad univariantes mostraron la robustez del resultado: únicamente si la edad de los pacientes era superior a 65 años o la HbA1c inicial era ≥7% el RCEI aumentaba hasta valores próximos a 100.000 $/AVAC. El análisis de sensibilidad probabilístico estimó una probabilidad del 60% de que la alternativa (a) sería coste-efectiva para una disposición a pagar de 100.000 $/AVAC.
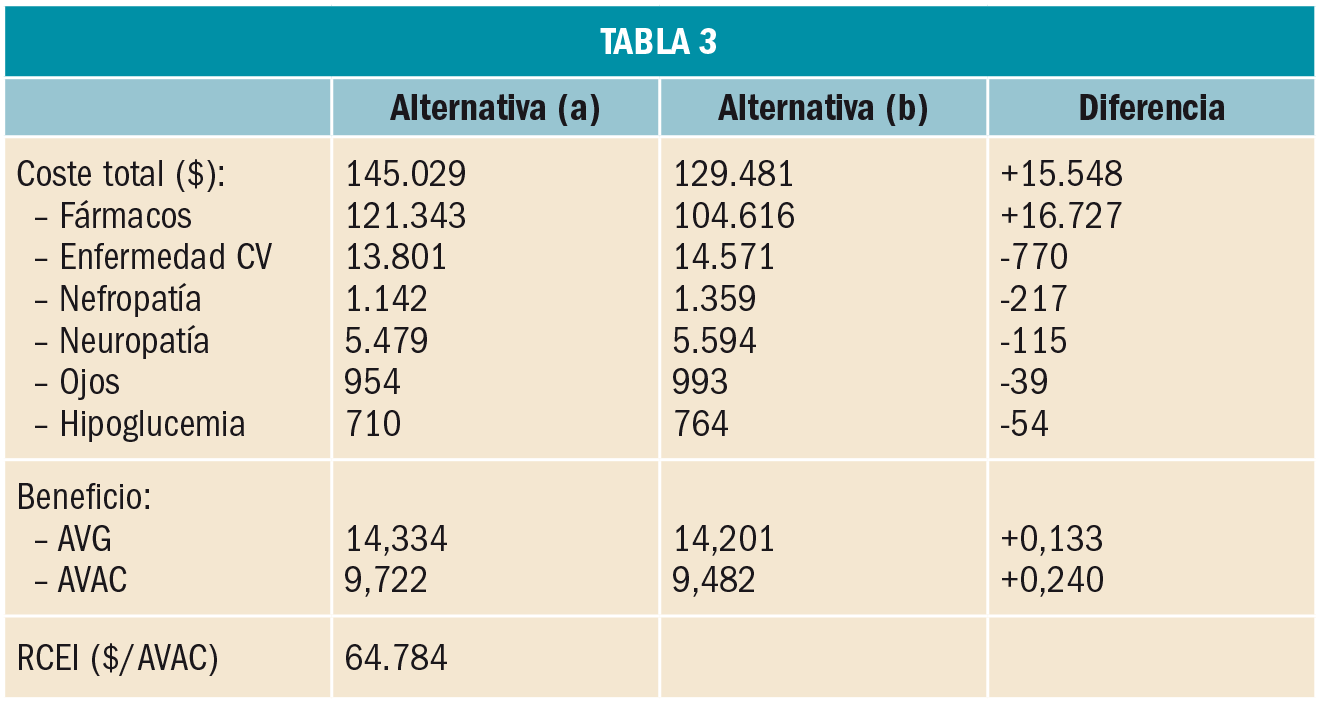
Los autores concluyen que la adición secuencial de inhibidores de SGLT2 a los inhibidores de DPP-4, antes de administrar insulina, puede ser considerada como coste-efectiva en comparación con el tratamiento habitual con en pacientes que han fracasado en la consecución del objetivo glucémico con metformina en EE.UU.
{kind=link}