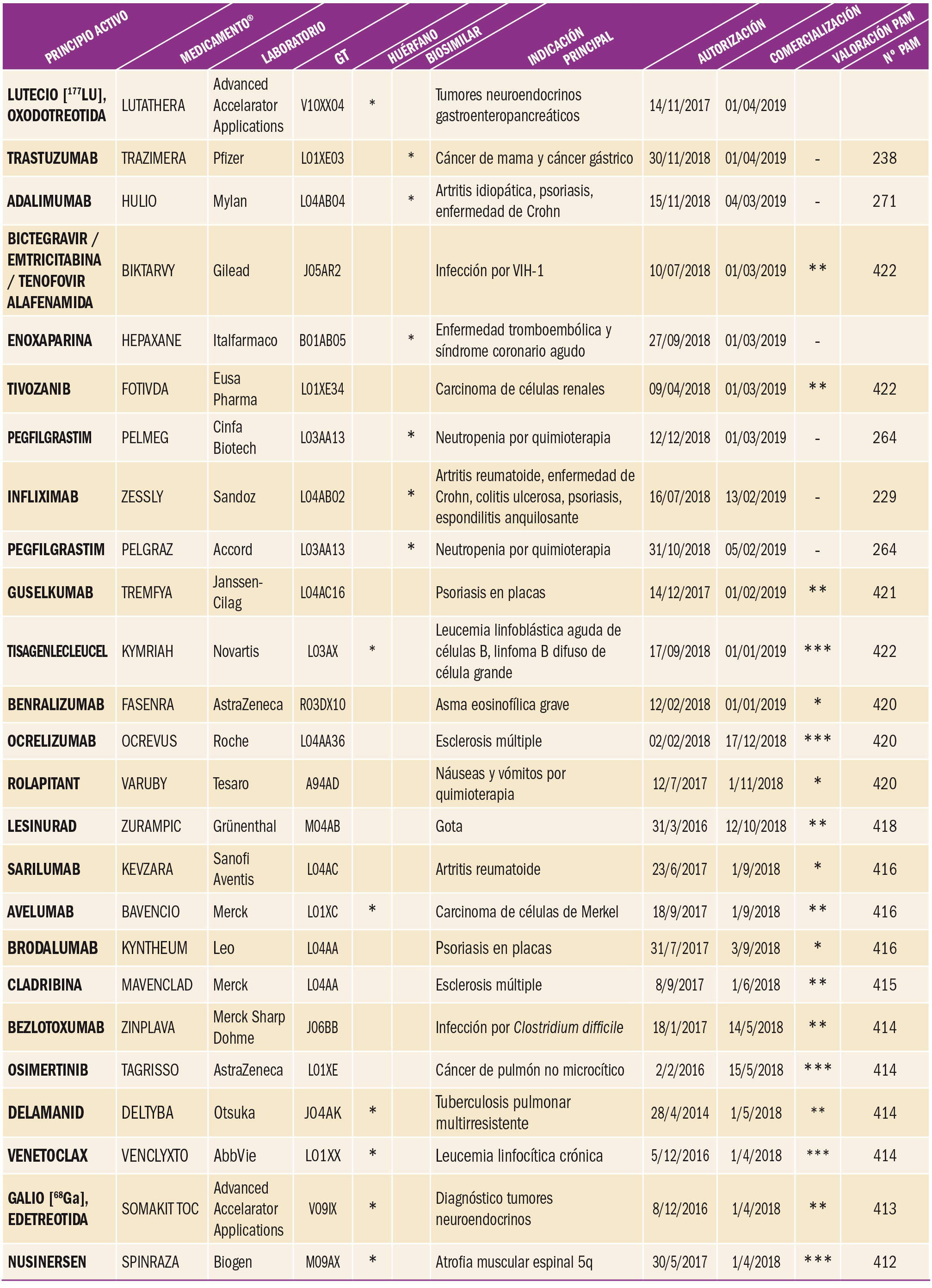
Número 422, Abril 2019
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica –sumario de características– y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta ese momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
Evidencia clínica: mediante estudios controlados, específicamente diseñados y desarrollados para demostrar la eficacia y la seguridad del nuevo medicamento, con demostración fehaciente de lo que puede ser un avance o mejora sobre la terapia estándar hasta ese momento, en el caso de que exista.
Plausibilidad científica (potencialidad): existencia de aspectos en el medicamento que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostrados mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
El tivozanib es un inhibidor potente y selectivo de tirosina cinasas, que se une con elevada afinidad a los tres receptores del factor de crecimiento endotelial vascular (VEGFR-1, 2 y 3), bloqueando diversas respuestas bioquímicas y biológicas inducidas por VEGF, entre ellas la neoangiogénesis y la vascularización de tejidos cancerígenos – procesos cruciales para el crecimiento y diseminación tumorales. También inhibe, aunque con menor potencia, la cinasa de c-kit. El medicamento ha sido oficialmente autorizado para el tratamiento de primera línea de pacientes adultos con carcinoma de células renales (CCR) avanzado y para pacientes adultos que nunca hayan recibido inhibidores del VEGFR ni de la vía mTOR, tras la progresión de la enfermedad después de un tratamiento previo con terapia con citocinas para CCR avanzado.
La eficacia y la seguridad clínicas de tivozanib han sido adecuadamente contrastadas en la indicación autorizada mediante un amplio ensayo clínico de fase 3 (TIVO-1), abierto, aleatorizado y controlado con sorafenib como comparador activo en pacientes con carcinoma renal de células claras avanzado recurrente o metastásico cuya enfermedad había progresado después de una nefrectomía completa o parcial para la escisión del tumor primario. Los resultados de la variable primaria de eficacia mostraron que tivozanib prolonga el tiempo de supervivencia sin progresión tumoral en 2,8 meses más que sorafenib (mediana de 11,9 meses vs 9,1 meses); una prolongación que, aunque modesta, es clínicamente relevante dadas las características de este tipo de pacientes. Este aumento fue incluso superior –3,6 meses– en el subgrupo de pacientes que no habían recibido tratamiento previo (naïve), lo cual evidencia la eficacia del fármaco en primera línea. Además, los pacientes tratados con tivozanib mostraron una tasa de respuesta objetiva significativamente superior a la evidenciada con el tratamiento con sorafenib (33,1% vs 23,3%) y una mayor duración de las respuesta (mediana de 15 meses con tivozanib vs 12,9 con sorafenib). Sin embargo, estos resultados no se tradujeron en un aumento de la supervivencia global, lo cual podría plantear una incertidumbre sobre el beneficio clínico real del fármaco.
En relación a la seguridad, el perfil toxicológico de tivozanib parece aceptable y manejable –aunque se asume que afecta a la calidad de vida de los pacientes–, siendo superponible con el de otros fármacos inhibidores de la tirosina cinasa utilizados en esta misma indicación. Con una frecuencia de efectos adversos emergentes durante el tratamiento similar a sorafenib, los más comunes de grado ≥3 fueron: hipertensión arterial (26% vs 18% con sorafenib), disfonía, diarrea, fatiga y astenia, dolor de espalda y pérdida de peso. También se puede destacar, por su gravedad, el riesgo de eventos isquémicos y tromboembólicos, hemorragias e insuficiencia cardíaca. La mayoría de los eventos adversos respondieron al tratamiento estándar o a una reducción de la dosis, siendo mayor el porcentaje de pacientes tratados con sorafenib que requirieron reducciones de dosis e interrupción de tratamiento en relación a tivozanib.
En definitiva, tivozanib es un nuevo inhibidor de tirosina cinasas que puede llegar a considerarse como un tratamiento de referencia en este tipo de pacientes. A falta de conocerse las consideraciones derivadas del IPT y de definir las situaciones de preferencia de su uso (en que previsiblemente pueden influir criterios económicos), se puede posicionar como una nueva alternativa a otros fármacos de primera línea (como sunitinib, pazopanib o bevacizumab/interferón) en el tratamiento de pacientes con CCR avanzado no tratados previamente o solo tratados con citocinas. Cumpliendo la condición de no-inferioridad para un nuevo fármaco, parece proporcionar un mayor beneficio en términos de eficacia clínica que algunas de esas opciones terapéuticas (sorafenib) con un perfil de seguridad similar, sin aportar adicionalmente otros elementos que supongan una innovación disruptiva.
Los tumores de riñón suponen alrededor del 3% de la incidencia global enfermedades neoplásicas a escala mundial, siendo responsables de un porcentaje similar de las muertes de origen tumoral. El carcinoma de células renales (CCR; también llamado adenocarcinoma renal o simplemente cáncer de riñón) es la forma más común de cáncer renal. Mayoritariamente (75-80%), se trata de adenocarcinomas del parénquima que recubre los túbulos renales, en tanto que el resto afecta a la pelvis renal, fundamentalmente carcinomas de células transicionales. Anualmente se diagnostican más de 65.000 nuevos casos de carcinoma de células renales en la Unión Europea, con una prevalencia quinquenal de unos 230.000 casos, produciéndose cerca de 30.000 muertes relacionadas con él. En España se diagnostican cerca de 7.000 nuevos casos cada año. A nivel mundial, la incidencia anual roza los 300.000 casos. Las tasas de CCR más elevadas se han observado en el este de Asia, América del Norte y Europa y las más bajas en África.
Se suele presentar a partir de los 50 años de edad (70-75% de los casos), con una edad media en el momento del diagnóstico en torno a 64 años, siendo la incidencia doble en hombres que en mujeres; es más frecuente también en afroamericanos que en raza caucásica. El 30% de los nuevos diagnósticos se producen ya en fase metastásica; por otro lado, un 20-40% de los cuadros diagnosticados en fases tumorales localizadas acabarán desarrollando metástasis.
No se han identificado las causas concretas, si bien los factores de riesgo considerados como más importantes para el desarrollo del adenocarcinoma renal son el tabaquismo, la obesidad y la hipertensión arterial, así como la enfermedad quística renal asociada a la diálisis. Asimismo, determinadas profesiones como los trabajadores de la industria metalúrgica, los pintores y los bomberos tienen una mayor asociación con esta forma de cáncer, pues la exposición a ciertos productos químicos (cadmio, asbestos, petróleo) se considera también factor de riesgo. Además, el uso indiscriminado de analgésicos (aspirina y fenacetina) se asocian con un incremento en la incidencia, pero en este caso suponen un mayor riesgo de desarrollo de cáncer de vías urinarias.
El carcinoma renal se clasifica en función del tipo celular y el patrón de crecimiento (acinar, papilar o sarcomatoide). Frecuentemente, el tumor renal tiene más de un tipo celular, aunque es el tipo predominante el que determina la clasificación de la neoplasia. Un 2-5% de los casos tienen un origen hereditario y su forma más conocida es la asociada al síndrome de Von Hippel-Lindau1. Sin embargo, el cáncer de células renales se presenta más frecuentemente de forma esporádica (sin antecedentes familiares).
Entre los cinco tipos principales de carcinoma maligno de células renales, el carcinoma renal de células claras (CRCC) es la forma más común (en torno al 80%), siendo el carcinoma renal papilar o cromofílico el segundo tipo más común (10-15%); estos cánceres forman dedos pequeños o papilas, parecidos a proyecciones, por una parte o distribuidos por todo el tumor. Por su parte, el carcinoma renal de células cromófobas es el tercer tipo más común (5%), consistente en células pálidas, como el de células claras, aunque son mucho más grandes y con ciertas particularidades. El cuarto tipo es el carcinoma renal del túbulo colector, muy raro, cuya característica principal consiste en que las células cancerosas forman conductos irregulares. Finalmente, el restante 5% de los cánceres renales tienen una apariencia que no corresponde a ninguna de las otras categorías mencionadas.
El comportamiento y la evolución del adenocarcinoma renal son poco predecibles e incluso se han descrito remisiones espontáneas; asimismo, en algunas ocasiones el tumor no llega a expresarse clínicamente y pasa desapercibido. No obstante, en muchos casos, el tumor acaba con la vida del paciente en un periodo relativamente corto de tiempo. Entre un 20% y un 30% de los pacientes presentan metástasis en el momento del diagnóstico y otro 10-30% son portadores de micrometástasis que se manifestarán en un intervalo de tiempo variable tras la nefrectomía radical. El tumor crece de forma local, invadiendo las estructuras renales y los tejidos de alrededor. La diseminación a distancia suele producirse por vía sanguínea, generando metástasis fundamentalmente en pulmón, hígado, cápsula suprarrenal, hueso y cerebro.
El pronóstico empeora con la edad, un peor estado de salud general, y conforme aumenta el tamaño tumoral, debido a la infiltración de los órganos vecinos y la formación de metástasis distales. Los tumores confinados al riñón tienen un buen pronóstico, con una supervivencia a los cinco años en torno al 80%, que alcanza el 90% en los pacientes con tumores de un tamaño inferior a 5 cm en el momento del diagnóstico, aunque este porcentaje desciende al 50-60% en aquellos con mayor tamaño. Si hay afectación extrarrenal próxima, hacia la vena cava o los ganglios linfáticos locales, la supervivencia a los cinco años es del 40-50%, pero desciende por debajo del 15% (8-23%) en los pacientes con metástasis distales. Las localizaciones más comunes de las metástasis de origen renal son pulmones, mediastino, huesos, hígado y cerebro; de hecho, entre los tumores sólidos, el carcinoma de células renales es el segundo en frecuencia en cuanto a metástasis cerebrales.
La aparición tardía de recaídas tras la nefrectomía en muchos de los pacientes con tumores localmente localizados, así como de estabilizaciones prolongadas y de remisiones ocasionales de la enfermedad en ausencia de tratamientos sistémicos, sugiere un papel particularmente relevante de los mecanismos inmunitarios en la regulación del crecimiento celular en este tipo tumoral.
Una de las características típicas del carcinoma de células renales es la inactivación de la proteína supresora tumoral von Hippel-Landau (VHL), que se traduce en una desregulación de la vía de señalización del factor de crecimiento del endotelio vascular (VEGF, vascular endotelial growth factor) (Figura 1). Los diversos tipos de receptores del VEGF (VEGFR) contienen dominios intracelulares consistentes en tirosina cinasas y su activación –especialmente a través del VEGFR-2– promueve el crecimiento de vasos sanguíneos en los tumores sólidos (neoangiogénesis). Además de sus propiedades angiogénicas, VEGF podría también suprimir la respuesta inmunitaria antitumoral in vivo, lo cual podría deberse a una inhibición del reclutamiento y activación de las funciones de células dendríticas; todo ello sugiere que un bloqueo de su vía de señalización puede traducirse en una mejor respuesta frente al tumor. Otras vías de señalización bioquímica que aparecen frecuentemente alteradas en las células de los carcinomas de células claras renales deficientes de VHL son MET y AXL.
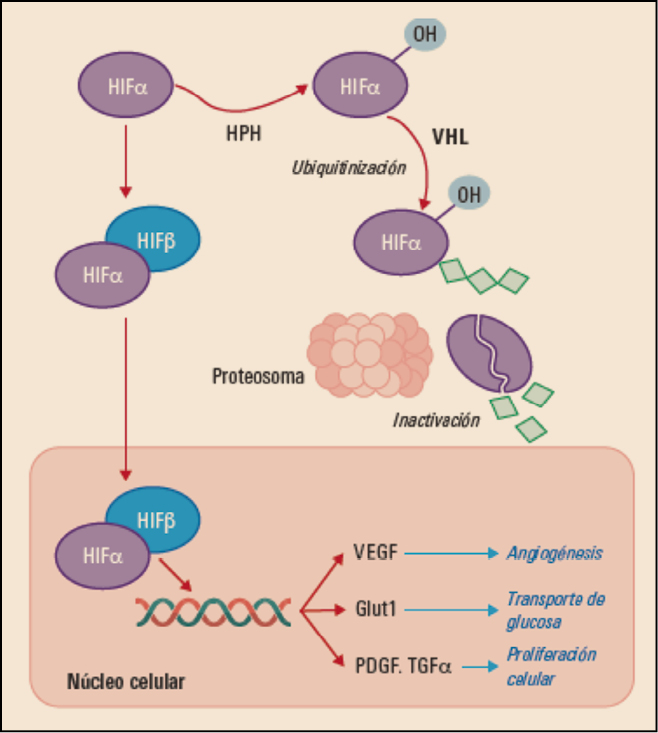
Figura 1. Mecanismo de acción normal de la proteína Von Hippel-Lindau (VHL). HIF: factor inducible por hipoxia; HPH: HIF prolil hidroxilasa; PDGF: factor de crecimiento derivado de las plaquetas; TGF: factor de crecimiento transformante; VEGF: factor de crecimiento vascular.
El supresor tumoral VHL se inactiva por mutación o silenciamiento epigenético en aproximadamente el 80% de los casos esporádicos de carcinoma renal de células claras (CRCC). La deficiencia de VHL en CRCC impulsa la angiogénesis, la invasión local y la metástasis mediante la activación de la respuesta hipóxica regulada por el factor inducible por hipoxia (hypoxia-inducible factor, HIF). A concentraciones bajas de oxígeno, los factores de transcripción HIF1α/HIF2α inducen la expresión de genes de respuesta hipóxica incluyendo factores angiogénicos tales como VEGF y PDGF (factor de crecimiento derivado de plaquetas; Platelet-derived Growth Factor), proteínas pro-invasoras que promueven la transición epitelial-mesenquimal y enzimas que facilitan el metabolismo anaeróbico. En condiciones normales (normoxia), VHL canaliza a HIF1α/HIF2α para su degradación dependiente de ubiquitina por el proteasoma. Sin embargo, la pérdida de la función VHL conduce a la estabilización de los factores de transcripción HIF y la consiguiente expresión de sus genes diana. En células tumorales deficientes en VHL y células endoteliales, el HIF2α es considerado como el principal regulador positivo de la tumorigénesis y la angiogénesis. Precisamente, la activación de la señalización de VEGFR y PDGF como resultado de la deficiencia de VHL subyace a la actividad clínica de agentes antiangiogénicos en CRCC.
MET (c-Met) es la proteína receptora –se trata también de una tirosina cinasa– del factor de crecimiento de hepatocitos (hepatocyte growth factor, HGF), el cual induce la activación del gen GAB1, que codifica la proteína 1 asociada a GRB2, que a su vez es un importante mediador de la tubulogénesis celular y juega un papel central en el crecimiento, transformación y apoptosis celular. MET se expresa normalmente las células de origen epitelial, mientras que la expresión de HGF se limita a las células de origen mesenquimal. Normalmente, solo las células pluripotenciales (células madre) y las células progenitoras expresan MET, lo que permite que estas células crezcan naturalmente de forma invasiva con el fin de generar nuevos tejidos en un embrión o regenerar los tejidos dañados en un adulto. Sin embargo, se piensa que las células madre tumorales secuestran esta capacidad de las células madre normales para expresar el MET, favoreciendo la persistencia tumoral y la propagación a otros sitios en el cuerpo. La activación anormal del MET en el cáncer se correlaciona con un mal pronóstico, ya que la vía MET aberrantemente activa desencadena el crecimiento tumoral, la formación de nuevos vasos sanguíneos tumorales (neoangiogénesis) y la diseminación a otros órganos (metástasis); de hecho, se ha confirmado que la vía MET está desregulada en muchos tipos de neoplasias humanas, incluyendo cánceres de riñón, hígado, estómago, mama y cerebro.
Por su parte, el gen AXL codifica una proteína receptora con actividad tirosina cinasa que traslada señales bioquímicas de la matriz extracelular al interior celular mediante la unión a la proteína GAS6 (Growth arrest-specific 6), implicada en la estimulación de la proliferación, supervivencia, angiogénesis y apoptosis tumoral. La proteína AXL fue identificada en primer lugar en pacientes con leucemia mielógena crónica, aunque su sobreexpresión ha sido observada igualmente en glioblastomas, melanomas, osteosarcomas, leucemias eritroides y megacariocíticas, así como en cánceres de útero, colon, próstata, tiroides, ovario e hígado. Igualmente, la sobreexpresión de la AXL se correlaciona con las metástasis e invasiones de un cierto número de tumores, incluyendo el carcinoma de células renales, los glioblastoma, y los cánceres de mama, pulmón y próstata. Por otro lado, la expresión de la AXL aumenta como respuesta a los fármacos quimioterápicos, confiriendo resistencia a estos en los tumores de estromas gastrointestinales (GIST) y en la leucemia mieloide aguda. Además, la AXL juega también un importante papel en la transición de epitelial a mesenquimatoso en el cáncer de mama, una transición clave para la inducción de metástasis (Alonso, 2017).
El tratamiento del cáncer renal de células claras depende de la extensión de la enfermedad. En los estadios localizados (T1 y T2) o con afectación de vena renal o grasa perirrenal (T3), la cirugía, ya sea radical (extirpación completa del riñón, la grasa perirrenal, la suprarrenal homolateral y los ganglios regionales) o parcial (en tumores menores de 4 cm), continúa siendo el único tratamiento curativo. La posibilidad de realizar la cirugía con técnica laparoscópica aporta una disminución del dolor posoperatorio, un tiempo de hospitalización menor y una recuperación más rápida. La radioterapia no es efectiva en ninguno de los estadios del cáncer renal y solo se puede plantear con finalidad paliativa (control del dolor por el tumor primario o las metástasis óseas o del sangrado).
El objetivo del tratamiento en los pacientes con un carcinoma renal con enfermedad tumoral irresecable o metastásica es conseguir un mejor control de los síntomas relacionados con la diseminación tumoral, una mejor calidad de vida y una prolongación de su supervivencia. Por tanto, la utilidad de la quimioterapia se centra en los pacientes con tumores que presentan metástasis en el momento del diagnóstico y en los pacientes en los que el tumor reaparece incluso años después de haber sido tratados con cirugía. Sin embargo, la quimioterapia convencional es prácticamente ineficaz en el cáncer renal, por lo que este tumor es considerado como quimiorresistente; así, los resultados terapéuticos en casos de CCR avanzado han sido históricamente pobres, con escasas opciones de tratamiento.
Los avances recientes en la biología molecular del CCR en las últimas décadas han conducido al desarrollo terapéutico notable. Pueden seguirse dos estrategias: la inmunoterapia o la terapia dirigida. En el caso de enfermedad avanzada o metastásica, el cáncer de riñón es el único tumor en el que está indicada la realización de cirugía para reducir masa tumoral antes de realizar tratamiento con fármacos, siempre que el estado general del paciente lo permita.
Como es bien sabido, la inmunoterapia es un tipo de terapia biológica que estimula el sistema inmunitario del paciente para que actúe contra las células tumorales. Se emplean sustancias elaboradas por el propio organismo o administradas por vía parenteral, así como el empleo de vacunas dendríticas alogénicas (todavía en fase experimental). La inmunoterapia con interferón alfa y/o interleucina 2 (IL-2), iniciada hace más de 20 años y que era el único tratamiento disponible hasta 2005, despertó muchas esperanzas, alcanzando respuestas objetivas en alrededor del 20% de los casos con cáncer metastásico; sin embargo, su tasa de respuesta objetiva es baja y poco persistente; además, son tratamientos –en especial, la aldesleucina– particularmente tóxicos. El interferón alfa induce la producción de citocinas que estimulan la actividad antitumoral de las células citotóxicas mientras que la interleucina 2 (IL-2; aldesleucina) es un factor de crecimiento y diferenciación para los linfocitos T y las células NK (natural killers) que inducen y mantienen la citotoxicidad no específica contra las células tumorales (inmunidad inespecífica).
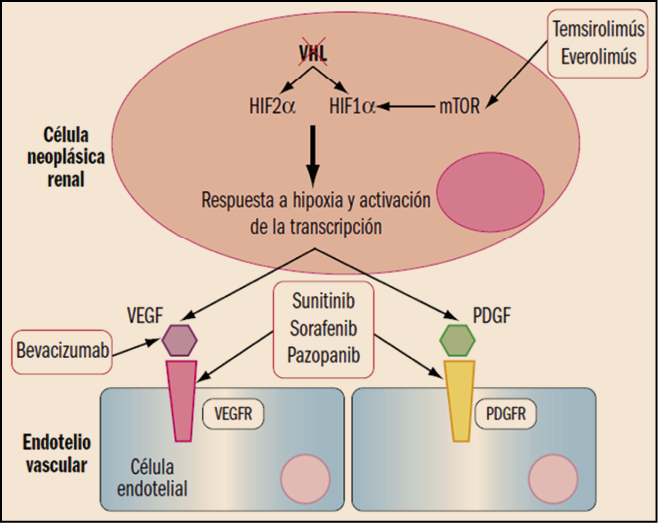
Figura 2. Situación de inactivación de VHL en la célula tumoral renal. Se refleja el mecanismo de acción de algunos antitumorales utilizados en el tratamiento del carcinoma de células renales. mTOR: proteína diana de la rapamicina en mamíferos; PDGFR: receptor del factor de crecimiento derivado de plaquetas (PDGF); VEGFR: receptor del factor de crecimiento endotelial vascular (VEGF); VHL: proteína de von Hippel-Lindau.
En general, el uso de interferón alfa y de aldesleucina ha ido trasladándose sustancialmente hacia la farmacoterapia dirigida, debido a una mejor y más persistente respuesta antitumoral, así como a disponer de perfiles toxicológicos significativamente mejor tolerados por los pacientes. La terapia dirigida se realiza con fármacos antiangiogénicos diseñados para actuar específicamente sobre puntos concretos del ciclo biológico de las células renales tumorales, limitando sus efectos adversos sobre las células normales. Se pueden dividir en 3 grupos:
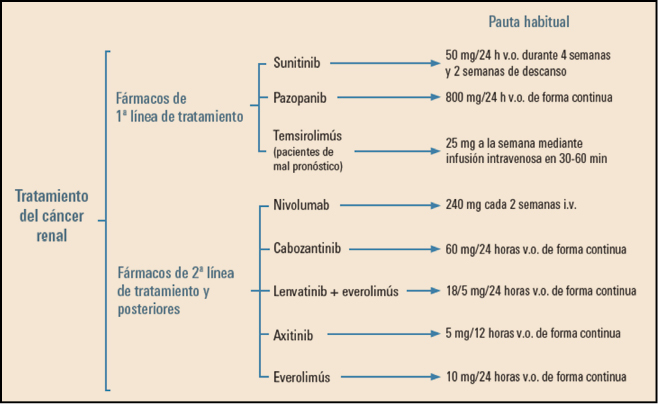
Figura 3. Fármacos de primera y segunda línea en el cáncer renal. i.v.: intravenoso; v.o.: vía oral.
Algunos autores proponen un esquema de tratamiento con distintas líneas (Figura 3). En terceras y cuartas líneas de tratamiento, se recomienda administrar aquel fármaco que no se dio previamente, pero no se conocen biomarcadores certeros que puedan guiar en la mejor secuencia terapéutica.
El tivozanib es un inhibidor potente y selectivo de tirosina cinasas, que se une con elevada afinidad a los tres receptores del factor de crecimiento endotelial vascular (VEGFR-1, 2 y 3), bloqueando diversas respuestas bioquímicas y biológicas inducidas por VEGF, entre ellas la neoangiogénesis y la vascularización de tejidos tumorales; también inhibe, aunque con menor potencia, la cinasa de c-kit. El medicamento ha sido oficialmente autorizado para el tratamiento de primera línea de pacientes adultos con carcinoma de células renales (CCR) avanzado y para pacientes adultos que nunca hayan recibido inhibidores del VEGFR ni de la vía mTOR, tras la progresión de la enfermedad después de un tratamiento previo con terapia con citocinas para CCR avanzado.
Como se ha sugerido anteriormente, una de las características típicas del carcinoma de células renales es la inactivación de la proteína supresora tumoral von Hippel-Landau (VHL), que se traduce en una desregulación de la vía de señalización del VEGF (factor de crecimiento del endotelio vascular). VEGF es un potente factor mitógeno que desempeña un papel fundamental en la angiogénesis y la permeabilidad vascular de los tejidos tumorales. Por su mecanismo de acción, tivozanib –a concentraciones picomolares– impide la fosforilación inducida por el ligando VEGF en los tres VEGFR y la proliferación de células endoteliales humanas.
La siguiente cinasa inhibida con más potencia es c-kit, que representa el receptor celular del factor de células madre (codificado por el proto-oncogen c-kit) y suele encontrarse mutado y sobre-expresado en diversos tipos celulares tumorales, especialmente en tumores del estroma gastrointestinal y el melanoma. Si bien esta tirosina cinasa es 8 veces menos sensible a la inhibición por tivozanib en comparación con VEGFR 1, 2 y 3, el bloqueo de c-kit impedirá el crecimiento de ciertos tipos de células sanguíneas.
Se ha demostrado que el tratamiento con tivozanib genera un aumento rápido de los niveles séricos de VEGF y, concomitantemente, los niveles de VEGFR-2 soluble disminuyen de forma aparentemente dosis-dependiente. El receptor VEGFR-2 soluble es un inhibidor de la linfangiogénesis inducida por VEGF, proceso crucial en la progresión tumoral. Teniendo en cuenta que el VEGFR-2 soluble puede funcionar como un “señuelo”, su reducción y el aumento de VEGF parece ser un mecanismo compensatorio a la interferencia de la inhibición de la tirosina cinasa con la señalización angiogénica.
Por otro lado, en el glioblastoma, tivozanib ha demostrado ejercer una detención del ciclo celular en la fase G2/M mediante la inhibición de la vía de la cinasa-1 tipo polo y la regulación a la baja de las cinasas Aurora A y B, ciclina B1 y CDC25C. Además, la administración de tivozanib fue capaz de disminuir las propiedades adhesivas celulares debido a la reducción de la ICAM-1 (molécula de adhesión intercelular-1) y la VCAM-1 (molécula de adhesión de células vasculares 1). Por tanto, la invasión celular se reduce por el deterioro del sistema catepsina B/activador del plasminógeno tipo uroquinasa/metaloproteinasa matricial-2 (Santoni, 2018).
El tivozanib está estrechamente relacionado estructural y farmacológicamente con otros miembros de la serie de inhibidores de proteína cinasas y, en particular con el cabozantinib y el vandetanib (Figura 4). Como otros miembros de la serie de inhibidores de proteína cinasas, es el resultado de la optimización funcional mediante modelización molecular a partir de una serie de 2-fenilaminopirimidinas, de donde surgió el imatinib, cabeza de serie del grupo. En cualquier caso, todos ellos guardan –en mayor o menor grado– una familiaridad química con la molécula de ATP (o, en su caso, con la de GTP, como sucede en las cinasas MAPK), con la que compiten para provocar el bloqueo de la cinasa correspondiente. Se han desarrollado modelos moleculares de relación estructura-actividad para este grupo de sustancias. En todos los casos, las interacciones estéricas y electrostáticas han demostrado ser las más determinantes para el efecto inhibitorio sobre la tirosina cinasa.
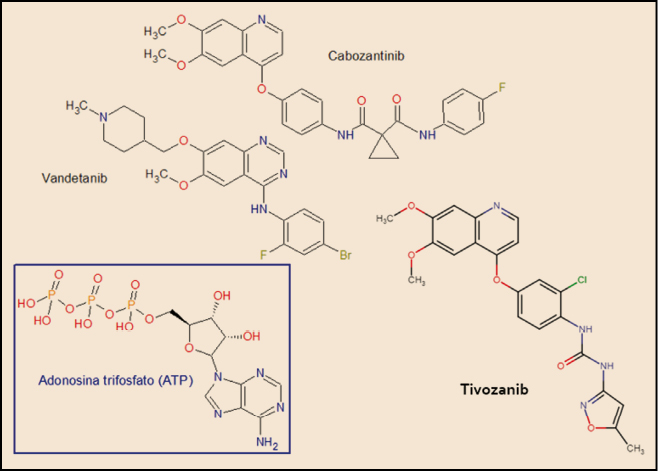
Figura 4. Estructura química de tivozanib y fármacos relacionados.
El nombre químico del clorhidrato de tivozanib monohidrato (forma química en que se presenta en el medicamento) es el 1-{2-cloro-4-[(6,7-dimetoxiquinolin-4-il)oxi]fenil}-3-(5-metilisoxazol-3-il) urea hidrocloruro hidrato. La fórmula química de la molécula es C22H19ClN4O5 y su masa molecular es 454,87 g/mol.
Estudios previos han identificado que la unión de los inhibidores de VEGFR se produce a nivel de dos conformaciones del dominio yuxtamembrana de VEGFR. Se ha sugerido también que el rendimiento clínico de un inhibidor de VEGFR está relacionado con la lipofilia del ligando, lo cual puede explicar la capacidad de tivozanib para inhibir de forma potente y selectiva la fosforilación intracelular inducida por VEGF en los VEGFR-1, -2 y -3, con valores de IC50 de 0,21, 0,16, y 0,24 nM, respectivamente.
La eficacia y la seguridad clínicas del tivozanib han sido adecuadamente contrastadas en la indicación autorizada mediante un único ensayo pivotal de fase 3 (AV-951-09-301; TIVO-1) –confirmatorio de seguridad y eficacia– abierto, de 2 años de duración, aleatorizado, multicéntrico y multinacional, y controlado con sorafenib en pacientes con carcinoma renal de células claras avanzado recurrente o metastásico cuya enfermedad había progresado después de una nefrectomía completa o parcial para la escisión del tumor primario (Motzer, 2013).
Los criterios de elegibilidad incluyeron: edad ≥ 18 años, enfermedad recurrente o metastásica con componente histológico/citológico confirmado de células claras, nefrectomía previa, enfermedad medible según RECIST2, un índice de ECOG3 de 0 o 1, y una esperanza de vida de al menos 3 meses. Los pacientes podían no haber recibido ningún tratamiento sistémico previo (naïve), o solo uno (que podía ser inmunoterapia, quimioterapia, terapia hormonal o un agente experimental). Se excluyeron aquellos pacientes en los que ese tratamiento previo se había realizado con inhibidores de la vía del VEGF (fármacos inhibidores de tirosina cinasa) o con inhibidores de mTOR (everolimús, tacrolimús), así como pacientes que sufrían tumores o metástasis a nivel del sistema nervioso central, anormalidades hematológicas relevantes y otras comorbilidades (patología cardiovascular, alteraciones gastrointestinales, inmunosupresión, etc.) graves.
El estudio fue diseñado para proporcionar el poder estadístico adecuado para la evaluación tanto de la variable clínica primaria de eficacia, supervivencia libre de progresión tumoral (SLP), como las secundarias de supervivencia global (SG) y tasa de respuesta objetiva (TRO), todas ellas según revisión radiológica independiente enmascarada. Un total de 517 pacientes fueron aleatorizados (1:1) a recibir, en ciclos de 4 semanas, bien tivozanib hidrocloruro (una dosis oral de 1,5 mg/día durante 3 semanas seguido de una semana de descanso) o bien sorafenib (400 mg/12 h por vía oral durante 4 semanas sin periodo de descanso) hasta progresión de la enfermedad, muerte o toxicidad inaceptable. En el periodo de seguimiento se permitían interrupciones del tratamiento experimental para la administración de dosis limitadas de radioterapia.
Las características basales de la población incluida en el estudio TIVO-1 estuvieron bastante equilibradas entre los dos grupos de tratamiento de tivozanib y sorafenib en relación a la edad (58,2 frente a 58,4 años de media, respectivamente), sexo (71,2% frente a 73,5% varones, respectivamente), peso y altura (80,7 kg y 171,4 cm frente a 80,1 kg y 171,3 cm de media), raza (95,8% frente a 96,9% blancos), región geográfica (88,1% frente a 88,7% de Europa Central/Oriental) y tratamiento previo para CCR metastásico (69,6% frente a 70,8% sin tratamiento previo, respectivamente). El tratamiento previo más frecuente era la monoterapia con interferón-alfa, que fue empleado para 75 pacientes (28,5%) en el grupo de tivozanib y para 62 pacientes (24,1%) en el grupo de sorafenib. Los pulmones y los ganglios linfáticos fueron los sitios más frecuentes de metástasis. Las características de riesgo alto, intermedio y bajo se presentaron en el 27%, 67% y 7% de los pacientes tratados con tivozanib y en el 34%, 62% y 4% con sorafenib, respectivamente.
El análisis primario de los datos del ensayo (análisis por intención de tratar4) arrojó los resultados que se resumen en la Tabla 1. Tivozanib mostró una mejoría estadísticamente significativa en la supervivencia libre de progresión (SLP) y en la tasa de respuesta objetiva (TRO). No hubo diferencias relevantes en términos de supervivencia global (SG) a los dos años de seguimiento del estudio.
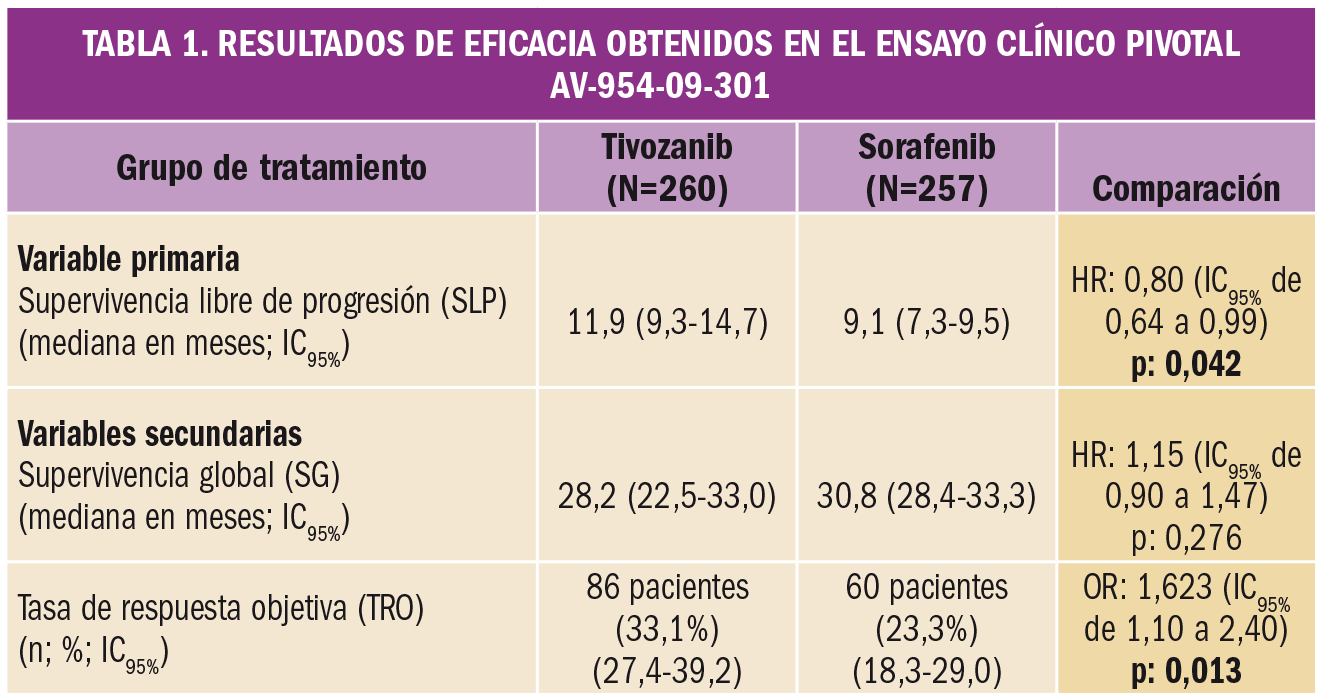
El subanálisis por grupos de pacientes reveló que tivozanib aportaba un mayor beneficio clínico en pacientes que no habían recibido tratamiento previo para el cáncer metastásico: en el grupo tratado con tivozanib la mediana de la SLP fue de 12,7 meses frente a los 9,1 meses en el grupo tratado con sorafenib (HR: 0,76; p: 0,037). En pacientes con un tratamiento previo, la diferencia en SLP entre tivozanib (11,9 meses) y sorafenib (9,1 meses) no fue estadísticamente significativa (HR: 0,88; p: 0,52). Por otro lado, en base a los datos del ensayo AV-954-09-301 (en que el 25% de los pacientes que recibieron tivozanib tenían ≥65 años de edad), no se observaron diferencias globales en la eficacia de tivozanib entre pacientes ancianos y más jóvenes.
Cabe destacar, de forma interesante, que a aquellos pacientes que habían sido aleatorizados a recibir sorafenib y sufrieron evolución de su enfermedad medible por RECIST (N=161) se les ofreció la posibilidad de cambiar su tratamiento a tivozanib –mismo régimen posológico que en el ensayo pivotal– al entrar en un ensayo de extensión de fase 3 abierto (AV-951-09-902). Los datos de este estudio de soporte confirmaron el beneficio clínico aportado por tivozanib según los resultados previamente comentados. Incluyendo los datos del ensayo pivotal, la mediana de SLP fue de 14,7 meses para el grupo tratado con tivozanib (N=249) frente a 9,7 meses para los tratados únicamente con sorafenib (N=28) (HR: 0,76; p: 0,006).
Para los 161 pacientes que cambiaron de sorafenib a tivozanib, la TRO fue del 18,0%, siendo parciales todas las respuestas; la mediana de duración de la respuesta fue de 15,2 meses. De entre ellos, un total de 108 pacientes (67,1%) sufrieron progresión de la enfermedad o murieron en el estudio, resultando una mediana de la SLP de 11,0 meses. La mediana de la SG desde el inicio de la primera dosis en este estudio fue de 21,6 meses. Para los pacientes que permanecieron con tivozanib (N=88) o sorafenib (N=28) según la aleatorización inicial del ensayo pivotal, la TRO fue de 55,7% y 57,1%, respectivamente (EMA, 2017).
Con relación a la seguridad clínica de tivozanib, se han evaluado los datos de un total de 835 pacientes con CCR que han recibido tivozanib en monoterapia, de entre los cuales 674 pacientes aleatorizados en cinco ensayos clínicos diferentes recibieron tivozanib como primera línea de tratamiento. El análisis de todos los eventos adversos emergentes (EAE) durante el tratamiento demostró se producían reacciones adversas de cualquier grado en el 92% de los pacientes, siendo las reacciones adversas más frecuentes la hipertensión (47,6%), disfonía (26,9%), cansancio (25,8%) y diarrea (25,5%). Además, el 64% de los pacientes que recibieron tivozanib experimentaron un evento adverso grave de grado 3/4 (vs 70% para los que recibieron sorafenib). En cuanto a estos EAE graves, la hipertensión –quizá el más relevante– se produjo en un 26% de los pacientes (vs 18% con sorafenib), la fatiga y astenia en un 5% (vs 4% con sorafenib), el dolor de espalda en un 3% (vs 2% con sorafenib), pérdida de peso en un 3% (vs 4% con sorafenib) y diarrea en un 2% (vs 7% con sorafenib).
En el estudio pivotal en monoterapia en CCR, se suspendió tivozanib en un total de 20 pacientes (3%) debido a reacciones adversas, generalmente por hipertensión (0,4%), hipertensión grave persistente (0,3%) o infarto agudo de miocardio (0,3%). Las reacciones adversas más frecuentes que condujeron a reducción de la dosis/interrupción de tivozanib fueron hipertensión (4,7%), diarrea (3,1%), cansancio (1,8%). Un mayor número de pacientes necesitaron una reducción de la dosis por EAE en el brazo de sorafenib (37,4%) que en el brazo de tivozanib (11,6%), así como una interrupción del tratamiento (37,0% vs 22,4%). Las muertes relacionadas con el tratamiento fueron raras: entre los pacientes que recibieron tivozanib como tratamiento inicial, hubo 3 EAE con resultado de muerte (una fue hipertensión no controlada con sospecha de sobredosis y dos se notificaron simplemente como muertes).
Desde el punto de vista cualitativo (sin tener en cuenta la frecuencia de aparición), además de la hipertensión, el EPAR de la EMA advierte sobre el riesgo de desarrollo de: encefalopatía o delirio (de etiología no infecciosa), eventos trombo-embólicos arteriales o venosos, isquemia cerebrovascular, hemorragia, insuficiencia cardíaca, hipotiroidismo y prolongación del intervalo QT del electrocardiograma.
El tivozanib es un inhibidor potente y selectivo de tirosina cinasas, que se une con elevada afinidad a los tres receptores del factor de crecimiento endotelial vascular (VEGFR-1, 2 y 3), bloqueando diversas respuestas bioquímicas y biológicas inducidas por VEGF, entre ellas la neoangiogénesis y la vascularización de tejidos cancerígenos, procesos cruciales para el crecimiento y diseminación tumorales. También inhibe, aunque con menor potencia, la cinasa de c-kit. El medicamento ha sido oficialmente autorizado para el tratamiento de primera línea de pacientes adultos con carcinoma de células renales (CCR) avanzado y para pacientes adultos que nunca hayan recibido inhibidores del VEGFR ni de la vía mTOR, tras la progresión de la enfermedad después de un tratamiento previo con terapia con citocinas para CCR avanzado.
La eficacia y la seguridad clínicas de tivozanib han sido adecuadamente contrastadas en la indicación autorizada mediante un amplio ensayo clínico de fase 3 (TIVO-1), abierto, aleatorizado y controlado con sorafenib, comparándolos en pacientes con carcinoma renal de células claras avanzado recurrente o metastásico cuya enfermedad había progresado después de una nefrectomía completa o parcial para la escisión del tumor primario. Los resultados de la variable primaria de eficacia mostraron que tivozanib prolonga el tiempo de supervivencia sin progresión tumoral en 2,8 meses más que sorafenib (11,9 vs 9,1); una prolongación que, aunque modesta, es clínicamente relevante dadas las características de este tipo de pacientes. Este aumento fue incluso superior –3,6 meses– en el subgrupo de pacientes que no habían recibido tratamiento previo (naïve), lo cual evidencia la eficacia del fármaco en primera línea.
En relación a las variables secundarias de eficacia, los pacientes tratados con tivozanib mostraron una tasa de respuesta objetiva significativamente superior a la evidenciada con el tratamiento con sorafenib (33,1% vs 23,3%). Esto, junto a datos de duración de la respuesta (mediana de 15 meses con tivozanib vs 12,9 con sorafenib) y otros resultados de ensayos de fase 2 (que, por ejemplo, verificaron su eficacia en pacientes con CCR sin componente de células claras), permiten justificar la utilidad clínica de tivozanib. Además, los resultados del estudio de extensión (AV-951-09-902) sugieren cierta eficacia como fármaco de segunda línea tras refractariedad a un tratamiento con fármacos dirigidos a tirosina cinasas, aunque esta indicación precisaría de un estudio más amplio.
Sin embargo, el hecho de que tivozanib no ejerciera un incremento significativo, con respecto a sorafenib, en términos de supervivencia global en el periodo de seguimiento del ensayo pivotal plantea incertidumbres sobre el beneficio clínico real aportado por el fármaco, al no confirmar ni correlacionarse con los resultados de supervivencia libre de progresión. Así lo refleja la posición divergente de los evaluadores noruegos sobre el balance beneficio-riesgo recogida en el EPAR del medicamento (EMA, 2017).
Con muchos de los pacientes incluidos en los ensayos clínicos expuestos a tivozanib durante un periodo de 2 años (incluso más largo en el ensayo de extensión), su seguridad clínica se cree bien caracterizada. En general, el perfil toxicológico de tivozanib parece aceptable –aunque se asume que afectará a la calidad de vida de los pacientes–, siendo superponible con el de otros fármacos inhibidores de la tirosina cinasa utilizados en esta misma indicación. Así, los eventos adversos emergentes durante el tratamiento tuvieron aproximadamente la misma frecuencia con tivozanib que con sorafenib. En este sentido, los eventos adversos graves (de grado ≥3) acontecieron en el 64% de los pacientes tratados con tivozanib frente al 70% en el brazo de sorafenib. Un mayor porcentaje de pacientes tratados con sorafenib precisaron reducciones de dosis e interrupción del tratamiento en relación a tivozanib, si bien la hipertensión5 grave (el efecto adverso más común con ambos fármacos) fue más frecuente en los pacientes tratados con tivozanib (26% vs 18% con sorafenib). Además, puesto que se excluyeron del ensayo pivotal pacientes con patología cardiovascular grave, el riesgo de eventos adversos graves de este tipo con el uso del fármaco en la población real puede verse incrementado y debe ser vigilado.
Otras reacciones adversas de gravedad a destacar con el empleo tivozanib incluyen, por su elevada frecuencia, la disfonía, la diarrea, la fatiga y la astenia, el dolor de espalda o la pérdida de peso; desde el punto de vista cualitativo por el riesgo para el paciente, se deben mencionar los eventos isquémicos y tromboembólicos, hemorragias e insuficiencia cardíaca; riesgos todos ellos en línea con otros inhibidores de VEGF. La mayoría de los eventos adversos respondieron a tratamiento estándar o a reducción de la dosis.
En la actualidad, la farmacoterapia dirigida es de elección en el tratamiento de la patología que aquí nos ocupa. En la Unión Europea se encuentran autorizados varios fármacos para el tratamiento en primera línea del CCR avanzado, tales como otros inhibidores de VEGF (axitinib, bevacizumab), inhibidores múltiples de tirosina cinasas (sorafenib, sunitinib, y pazopanib) e inhibidores de m-TOR (temsirolimus). A pesar de que no se dispone aún de resultados directamente comparativos con otros agentes antitumorales diferentes a sorafenib –particularmente con los inhibidores de tirosina cinasa sunitinib y pazopanib o con la combinación bevacizumab/interferón-α–, las comparaciones indirectas con resultados de ensayos previos podrían sugerir una eficacia superior o como mínimo parecida para tivozanib (Hepgur, 2013). No parece competir con termsirolimus, debido a que éste fármaco se limita a pacientes con especial riesgo pronóstico. No obstante, se debe recordar que cualquier comparación de estudios en paralelo –y, por tanto, indirecta– no permite sacar conclusiones sólidas al no contar con las mismas condiciones y circunstancias (perfil fisiopatológico de los pacientes, duración del tratamiento, incidencia de eventos adversos, etc.).
En resumen, el empleo de un comparador activo durante el desarrollo clínico de tivozanib permite situar este fármaco como una nueva alternativa en el tratamiento en primera línea de pacientes con CCR avanzado, más eficaz que sorafenib (cumpliendo, con seguridad, la condición de no inferioridad) y con un perfil de seguridad similar (quizás con una menor incidencia de diarrea y síndrome de eritrodisestesia palmar-plantar6). En este sentido, podría considerarse como un tratamiento de referencia en este tipo de pacientes, aunque cabe indicar que no se ha evaluado en pacientes con metástasis cerebrales, considerando que la segunda causa de éstas son los tumores renales. No aporta beneficios adicionales en términos de adherencia terapéutica –de forma similar a otros inhibidores de tirosina cinasa, es de administración por vía oral un vez al día– ni de perfil de interacciones farmacológicas (metabolizado mayoritariamente por CYP3A4, puede también interaccionar con los fármacos que induzcan, inhiban o se metabolicen por esta vía).
Por tanto, a falta de conocer las consideraciones derivadas del Informe de Posicionamiento Terapéutico de la AEMPS, tivozanib es un nuevo inhibidor de tirosina cinasas que se puede posicionar como una alternativa en primera línea para el tratamiento de CCR avanzado. A falta de definir las situaciones en las que el uso de tivozanib sería preferente frente al resto de fármacos de primera línea (lo cual previsiblemente puede verse influido por criterios económicos), parece aportar ciertos beneficios clínicos frente a algunos de ellos (sorafenib), sin aportar adicionalmente otros elementos que supongan una innovación disruptiva.
Tisagenlecleucel es un novedoso tratamiento de inmunoterapia consistente en células T autólogas modificadas genéticamente ex vivo mediante el empleo de un vector lentiviral para expresar un receptor de antígeno quimérico (CAR) anti–CD19. Tras la unión de los linfocitos T reprogramados que expresan CAR a las células que expresan CD19 –células del linaje B desde etapas tempranas de su desarrollo a células plasmáticas, tanto malignas como normales–, la proteína quimérica transmite las señales intracelulares necesarias para activar la citotoxicidad frente a esas células CD19+, así como una señal que favorece la expansión de las células T y la persistencia de tisagenlecleucel. Se trata de la primera terapia celular CAR-T disponible comercialmente en España.
El medicamento ha sido oficialmente autorizado como medicamento huérfano para el tratamiento, en perfusión única, de pacientes pediátricos y adultos jóvenes de hasta 25 años de edad con leucemia linfoblástica aguda (LLA) de células B refractaria, en recaída postrasplante o en segunda o posterior recaída, y de pacientes adultos con linfoma difuso de células grandes B (LBDCG) en recaída o refractario tras dos o más líneas de tratamiento sistémico. Ambas indicaciones refieren a situaciones clínicas con limitadas alternativas terapéuticas (de resultados clínicos pobres) y de pronóstico desfavorable, representando necesidades médicas no cubiertas.
La eficacia y seguridad clínicas de tisagenlecleucel en cada indicación autorizada han sido evaluadas mediante un ensayo pivotal de fase 2, de un solo brazo y abierto (sin comparador directo), con un número reducido de pacientes (N<160). Para la indicación de LLA se dispone, además, de datos de dos ensayos clínicos fase 2 de soporte. A nivel general, tisagenlecleucel ha mostrado respuestas clínicamente relevantes en términos de tasa de respuesta global (del 40 al 60%) como variable primaria de eficacia. Si bien los periodos de seguimiento de los pacientes han sido muy limitados y es difícil establecer la duración de la respuesta, se han obtenido datos de supervivencia global a 12 meses en el rango de 49-76%. Esto, unido a la escasa experiencia poscomercialización, impide aún considerarlo como un tratamiento curativo; no obstante, los futuros resultados con un mayor seguimiento clarificarán si existe beneficio en términos de supervivencia libre de enfermedad a largo plazo.
En cuanto a la seguridad clínica, el perfil toxicológico de tisagenlecleucel (influido por la quimioterapia previa a la perfusión) en ambas indicaciones es importante, con un alto porcentaje de pacientes que sufren reacciones adversas específicas graves (grado ≥3) o potencialmente mortales en el periodo posperfusión. El síndrome de liberación de citoquinas, las infecciones graves y los efectos adversos neurológicos son quizá los aspectos cruciales del manejo de su toxicidad, que debe ser abordado por profesionales sanitarios especialmente capacitados para el uso de estas terapias.
Por su fundamento, mecanismo de acción y resultados clínicos prometedores, supone una innovación disruptiva en el área clínica de la onco-hematología, que viene a posicionarse como una herramienta terapéutica con gran utilidad potencial en dos indicaciones con escasas posibilidades terapéuticas. La dificultad para realizar comparaciones indirectas con esas alternativas, los restrictivos criterios de inclusión de los ensayos clínicos y su coste económico pueden limitar el acceso al fármaco a determinados pacientes con relativo buen estado funcional. Por tanto, se requieren futuros resultados de eficacia y seguridad a largo plazo para comprender su impacto real y el beneficio de su uso clínico.
Las neoplasias hematológicas engloban a todos aquellos procesos de origen tumoral que afectan al tejido hematopoyético y al sistema linfoide. En general, se considera tejido hematopoyético a la médula ósea y todo su complejo sistema celular. Respecto al sistema linfoide, integra a los ganglios, tejido linfoide de diferentes órganos y bazo fundamentalmente, incluyendo sobre todo los procesos que afectan a elementos celulares, como son los linfocitos B y T, y a las células plasmáticas (Figura 1).
Grosso modo, se pueden clasificar las neoplasias hematológicas siguiendo el esquema de la Tabla 1.
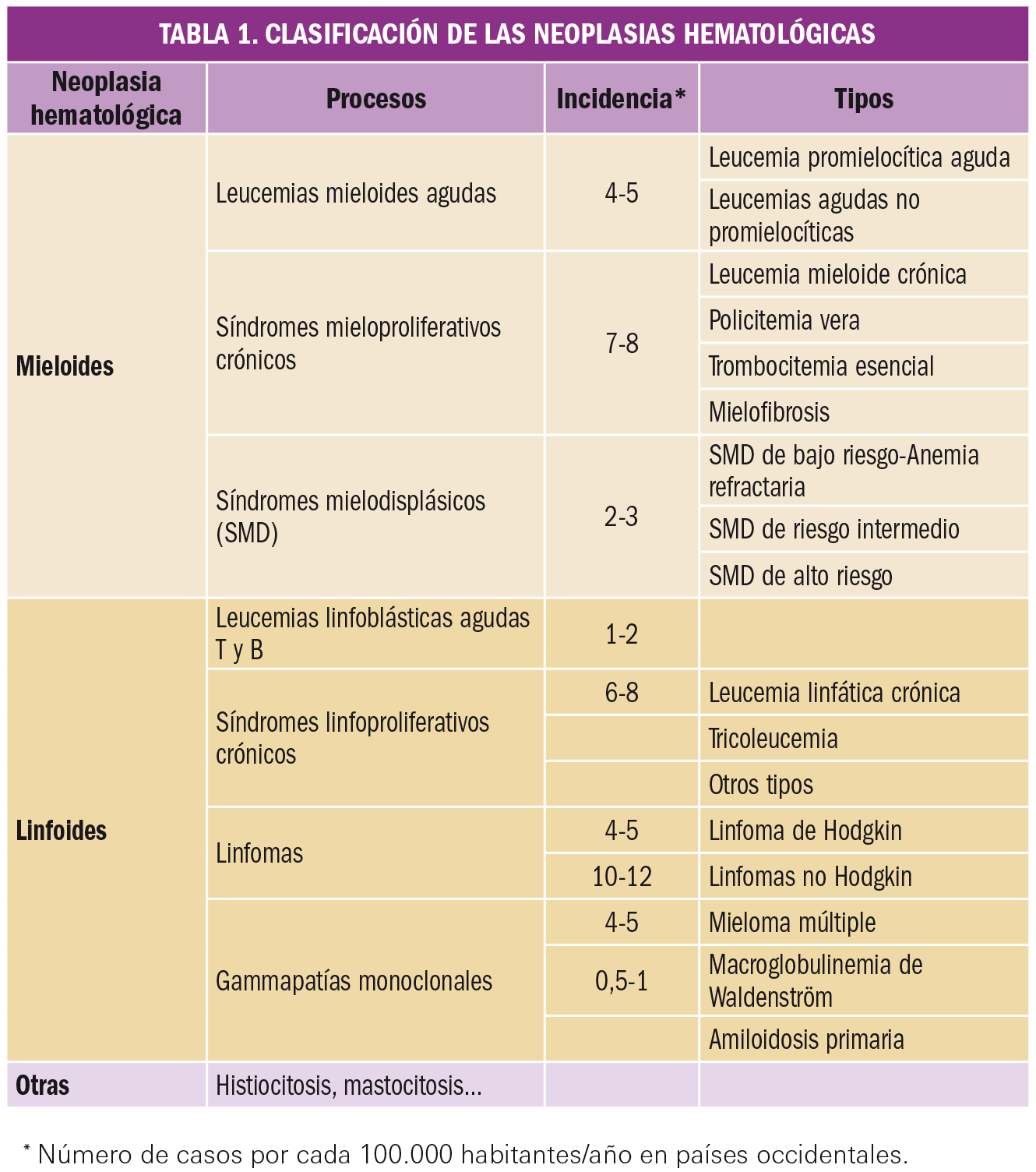
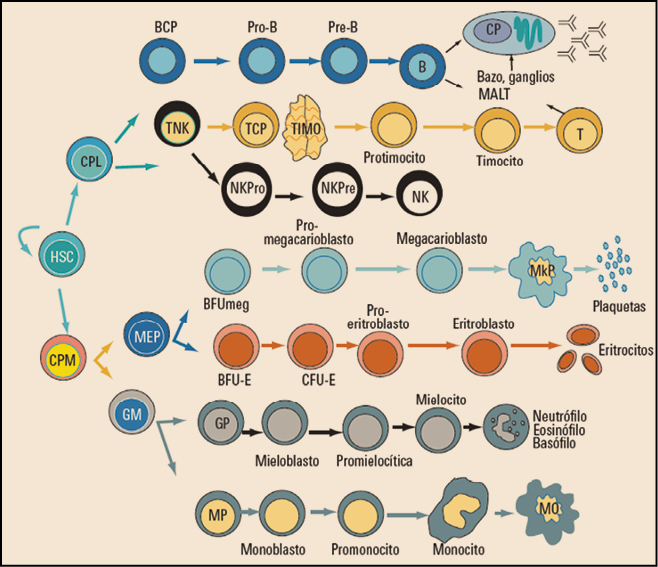
Figura 1. Modelo general de hematopoyesis. BCP: células progenitoras de linfocitos B; BFU: unidad formadora de brotes; CFU: unidad formadora de colonias; CPL: células progenitoras linfoides; CPM: células progenitoras mieloides; GM: células precursoras de granulocitos y macrófagos; GP: células precursoras de granulocitos; HSC: células madre pluripotenciales; MEP: células progenitoras de megacariocitos y eritrocitos; MkP: megacariocito; MO: macrófago; MP: células precursoras de monocitos; NKPre: células precursoras de células NK; NKPro: células progenitoras de células NK; TCP: células progenitoras de linfocitos T; TNK: células progenitoras de linfocitos T y NK.
Las neoplasias hematológicas suponen algo más del 10% de los tumores en humanos. Son más frecuentes en general en la edad avanzada, con la excepción de las leucemias linfoides agudas, que son la principal causa de cáncer infantil, y el linfoma de Hodgkin (LH), que ocurre en edades medias de la vida. Esta incidencia se ve incrementada en casi todos los casos en los pacientes de edad avanzada, multiplicándose unas 10 veces a partir de los 80 años, sobre todo en leucemias agudas y gammapatías. Una excepción es la leucemia linfoblástica aguda (LLA), que es más frecuente en la infancia, siendo una de las principales neoplasias infantiles.
Al contrario que en los tumores sólidos, no existen registros exhaustivos de algunos de estos procesos. En algunas ocasiones, porque acontecen en edad muy avanzada y, en otras, porque son procesos neoplásicos poco agresivos que conviven con otras enfermedades de base del paciente y no son comunicados, como es el caso de la leucemia linfática crónica (LLC) en los estadios iniciales. Los cuadros mixtos denominados síndromes mielodisplásicos (SMD) no tienen siempre un carácter maligno y en sus primeras etapas pueden cursar solo con anemia u otras citopenias, por lo que suelen estar infradiagnosticados. Otros procesos pueden considerarse preneoplásicos, como es el caso de la gammapatía monoclonal de significado incierto (GMSI), que precisa de seguimiento periódico dado que un 1% anual puede derivar en mieloma múltiple (MM).
En general, la causa de los procesos hematológicos es multifactorial y no se conocen con exactitud causas directas. Predominan eventos oncogenéticos primarios o secundarios que originan una proliferación descontrolada de un clon celular neoplásico. Como en todas las neoplasias, se han implicado algunos factoresambientales, como las radiaciones ionizantes y ciertas sustancias químicas, como benceno o pesticidas. Lo que sí está claro es que el tratamiento previo con quimio/radioterapia por otra neoplasia o las situaciones de inmunodeficiencia conllevan un mayor riesgo de padecerlas.
No existe un mecanismo genético molecular común para estas enfermedades. Solo en algunos procesos hematológicos se conoce con exactitud el evento genético mutacional que da lugar a la enfermedad, lo cual es relevante para el abordaje terapéutico. Así, por ejemplo, en el linfoma de Burkitt se puede apreciar una translocación entre alguno de los cromosomas de los pares 8 y 14 –t(8;14)– que se traduce en la activación de un protooncogén (c-MYC); en el linfoma folicular puede detectarse una t(14;18) que sobreexpresa Bcl2, responsable del bloqueo de apoptosis de las células tumorales; finalmente, en el linfoma de células del manto, puede aparecer una t(11;14), con sobreexpresión de Bcl-1 (ciclina D1).
La leucemia linfoide, linfocítica o linfoblástica aguda (LLA) se debe a una proliferación incontrolada de un clon celular inmaduro dentro de la linfopoyesis (linfoblastos), que infiltra la médula ósea e invade la sangre periférica y diversos órganos, con el resultado de pérdida de la hematopoyesis normal y fracaso orgánico que conduce a la muerte si no se trata. De manera específica, la LLA de células B se caracteriza por el crecimiento descontrolado de linfoblastos de células B.
La actualización de la clasificación de la OMS en 2016 divide las LLA en leucemia/linfoma linfoblástica B y T, y cada uno de éstas en una amplia variedad de tipos celulares. Globalmente, la incidencia anual de LLA es 1,7 casos por cada 100.000 habitantes. Se trata de la leucemia más frecuente en niños, siendo el segundo cáncer más común en la infancia, con una incidencia de 4/100.000/año1; son más comunes entre los 2 y los 5 años. Respecto a los adultos, tiene una incidencia de 3/100.000/año y predomina en adultos jóvenes (25-30 años) y de sexo masculino. No obstante, el 60% de los pacientes son menores de 20 años, seguido de un segundo pico a partir de los 60 años.
Al igual que ocurre con el resto de leucemias agudas, los síntomas se establecen de manera rápida (no más de 3 meses antes del diagnóstico) y la clínica deriva de la infiltración de la sangre periférica y otros órganos por las células leucémicas. En esta entidad es más frecuente, respecto a las leucemias agudas mieloides, la presencia de adenopatías (inflamación de los ganglios linfáticos), hepatomegalia y esplenomegalia (aumento de tamaño del hígado y del bazo, respectivamente). Existe un sustrato molecular y citogenético que produce la evolución descontrolada del clon maligno celular.
Los principales factores pronósticos son: a) la edad, siendo más favorable en niños y adultos jóvenes; b) el recuento de leucocitos, con peor pronóstico en caso de hiperleucocitosis; c) el fenotipo, siendo desfavorables aquella de fenotipo T y la pro-B; y d) la citogenética, ya que las hiperploidías tienen mejor pronóstico y las hipoploidías y algunas alteraciones genéticas peor. La rápida respuesta al tratamiento confiere mejor pronóstico, así como lograr una disminución rápida y mantenida de la enfermedad mínima residual.
En la mayoría de los casos no se conoce la causa concreta de la enfermedad, si bien en todos ellos la LLA progresa rápidamente, llevando a la muerte a los pacientes no tratados en pocas semanas o meses. No obstante, un adecuado tratamiento dirigido a conseguir la curación proporciona porcentajes de respuesta completa cercanos al 90%, alcanzándose la curación en el 50% de los lactantes, el 80% de los niños, y el 35% de los adultos. Especialmente en niños, se estima que hasta el 70% de los pacientes tratados están libres de enfermedad (y probablemente curados) a los 5 años.
Sin embargo, es una enfermedad heterogénea con diferentes subgrupos que muestran una respuesta variable a la quimioterapia, por lo que la estrategia terapéutica se individualiza según los factores pronósticos, principalmente la edad (infantil o de adultos), el subtipo inmunológico y la genética. La leucemia linfoblástica es sensible a varios fármacos, por lo que se usan diversas combinaciones de los mismos. Es obligatorio el tratamiento profiláctico de los santuarios, en particular del sistema nervioso central. En la LLA, a diferencia de la mieloblástica o mieloide, se ha demostrado la utilidad del tratamiento de mantenimiento.
Para el tratamiento de inducción la combinación básica consiste en vincristina, prednisona y asparaginasa, que se administran a lo largo de 4 semanas. En los grupos de alto riesgo –como los casos cromosoma Philadelphia positivo– se asocia además daunorubicina y otros fármacos, como inhibidores de la tirosina cinasa (TKI). Con este esquema, >90% de los pacientes entran rápidamente en respuesta completa con restablecimiento de la hematopoyesis, siendo la lentitud en la respuesta o la persistencia de la enfermedad mínima residual detectable por inmunofenotipo o citogenética uno de los factores pronósticos adversos más relevantes.
La meningitis leucémica es la forma de recaída de hasta el 60% de los niños con LLA si no reciben profilaxis del sistema nervioso central. La quimioterapia sistémica atraviesa mal la barrera hematoencefálica, por lo que se constituye un santuario donde los blastos leucémicos permanecen intactos, se reproducen localmente y, eventualmente, generan una recaída generalizada. Por lo tanto, la profilaxis del sistema nervioso central se debe efectuar de forma rutinaria en esta entidad y consiste en inyecciones intratecales seriadas de metotrexato o, en algunos protocolos más intensivos, con una combinación de metotrexato, citarabina e hidrocortisona (triple terapia intratecal), que comienza ya durante la inducción.
Una vez alcanzada la respuesta completa, se continúa con terapia de consolidación e intensificación(posremisión) durante los 4-6 meses siguientes, con el objetivo de prevenir la recaída y reducir la carga tumoral residual con un régimen de tratamiento distinto a la inducción. En la LLA existen multitud de protocolos distintos que combinan, en diversas formas y dosis, los fármacos útiles (ciclofosfamida, metotrexato, citarabina, antraciclinas, etopósido y corticoides) para adaptarlos al riesgo diferencial de cada situación. En casos seleccionados, de manera alternativa o complementaria a la terapia de intensificación, puede recurrirse a un trasplante alogénico de progenitores hematopoyéticos (TPH alogénico). Acabada esta fase más intensiva en pacientes no trasplantados, se pasa a un tratamiento de mantenimiento con metotrexato intramuscular semanal y 6-mercaptopurina oral, que suele durar 2-3 años. También se utilizan en esta fase vincristina, corticosteroides y otros fármacos.
En niños de riesgo estándar se pueden conseguir curaciones del 80% con una inducción y una consolidación no muy intensivas, con unos 2 años de mantenimiento suave. Por el contrario, los protocolos para los casos de mayor riesgo intensifican mucho el tratamiento de los primeros meses, aumentando el número de fármacos y sus dosis, tanto en la inducción como en las fases de consolidación e intensificación, y se siguen de un mantenimiento que periódicamente se intensifica con algún ciclo de altas dosis de quimioterapia combinada. En general, los resultados son siempre peores en adultos que en niños, incluso con factores pronósticos similares.
Además, los casos de LLA Philadelphia-positivos (Ph+) exigen protocolos específicos, en los que se combina quimioterapia intensiva con la administración continuada de imatinib o dasatinib (inhibidores de la protein cinasa Bcr-Abl) en pacientes con intolerancia o resistencia al tratamiento previo; en 2016 se autorizó en España ponatinib para el tratamiento de casos resistentes o de intolerancia a dasatinib. El pronóstico con quimioterapia es deficiente, con supervivencias prolongadas no superiores al 20%, por lo que en los casos Philadelphia-positivos (Ph+), tanto en adultos como en niños, está indicado el TPH alogénico en primera remisión, tras la inducción y la consolidación.
A pesar de un tratamiento adecuado, la leucemia puede recidivar en la médula ósea o en localizaciones extramedulares en hasta un 15-20% de los pacientes (25-30% en grupos de alto riesgo). Cabe destacar que hasta un 80% de los pacientes con recaída medular logran una segunda respuesta completa con el mismo tratamiento de inducción. Pero, sin duda, son estos casos recidivantes o refractarios al tratamiento los que presentan un peor pronóstico y comportan un mayor riesgo de muerte. El tratamiento suele consistir en quimioterapia de rescate, a ser posible con una combinación diferente de fármacos, seguida de TPH alogénico. En algunos pacientes se emplean tratamientos dirigidos (por ejemplo, frente a BCR-ABL, CD19 o CD22) o incluso tratamientos paliativos.
Hay tres fármacos actualmente autorizados con la indicación de LLA recidivante/refractaria:
En España solo está comercializado el primero, si bien ninguno de ellos parece tener capacidad curativa. La única opción potencialmente curativa en un paciente con LLA de células B recidivante es el TPH alogénico, aunque con los resultados de supervivencia global en este subgrupo de pacientes no son excesivamente prometedores (del 20-45% a los 5 años). Además, en los casos de recaída tras trasplante (o pacientes no candidatos al mismo por edad, comorbilidades, falta de donante, refractariedad) no existe ningún tratamiento estándar además de la clofarabina –con resultados muy pobres– y el tratamiento paliativo.
En el caso de la LLA de células T en recidiva hay aún menos opciones. El tratamiento posremisión debe ser intensivo y es recomendable repetir la neuroprofilaxis. La leucemia meníngea es la forma más frecuente de recaída extra-medular en la LLA. En los varones es también habitual la recidiva testicular.
La LLA recidivante/refractaria representa, por tanto, una laguna terapéutica donde las estrategias de inmunoterapia con células con receptores antigénicos quiméricos (CART-cells) pueden jugar un papel fundamental (Terwilliger, 2017).
Los linfomas2 son neoplasias del sistema linfoide que constituyen un grupo heterogéneo de enfermedades neoplásicas definidas por aspectos morfológicos, inmunofenotípicos y genéticos, que tienen su origen en los sistemas mononuclear fagocítico y linfático. Los linfomas de Hodgkin consisten en una proliferación, localizada o diseminada, de células tumorales que se originan en el sistema linforreticular y que afecta principalmente los ganglios linfáticos y la médula ósea. Los linfomas no-Hodgkin (LNH), por su parte, incluyen a todos los linfomas que no encajan dentro de la definición de linfoma de Hodgkin; por tanto, son neoplasias linfoides que pueden originarse en los linfocitos B, T y células natural killer (NK; citotóxicas naturales). Además, los linfomas se pueden clasificar por la célula maligna de origen: centro germinal o no centro germinal; también se dividen entre zona del manto y la zona marginal. También es muy importante la manera en que se infiltra el ganglio, por ejemplo: el linfoma difuso de célula grande (LDCG) es un linfoma de linfocitos B de tamaño grande (por su estado de maduración) que infiltra el ganglio de forma difusa y puede infiltrarlo tanto en el centro germinal como fuera del mismo (no centro germinal).
En los LNH, una célula linfoide inmadura, detenida en un determinado estadio madurativo, se reproduce de forma incontrolada, causando con el tiempo el aumento de tamaño del órgano en el que se producen. Dado que el tejido linfático se encuentra en todo el cuerpo, los linfomas pueden aparecer en cualquier parte del organismo y, a partir de allí, diseminarse a otros órganos y tejidos. En la mayoría de los casos empiezan con una infiltración en un ganglio linfático (formas ganglionares), pero otras veces pueden aparecer en otros órganos como el aparato digestivo, la piel, el cerebro, el bazo, el riñón u otros órganos (formas extraganglionares).
Los LNH representan el 4-5% de los nuevos casos de cáncer diagnosticados al año (con una tasa anual próxima a los 20 casos/100.000 habitantes) ocupando el quinto lugar en frecuencia. Entre los casos de LNH, los de células B representan el 80-85% y los T el 15-20%, mientras que los de NK tienen una frecuencia marginal. En conjunto, los LNH son algo más comunes entre los hombres y entre pacientes con enfermedades del sistema inmune (SIDA, inmunodeficiencias, receptores de trasplantes de órganos, enfermedades autoinmunes), infecciones (gastritis por Helicobacter, virus de Epstein Barr), y pacientes tratados con quimioterapia o radioterapia. Aunque de forma mucho menos frecuente que la leucemia linfoblástica aguda, es un tipo de cáncer que también puede afectar a los niños, si bien la mediana de presentación se sitúa en torno a los 50 años.
La etiopatogenia de los LNH varía en los distintos tipos, aunque tienen en común ciertos factores de riesgo, como tener un sistema inmune debilitado (ya sea por una enfermedad hereditaria o tras un trasplante de órganos), edad elevada, antecedentes familiares, exposición a agentes tóxicos (herbicidas) e infecciones por algunos virus (virus linfotrópico de células T del ser humano tipo 1 –HTLV-1–, virus de la inmunodeficiencia humana –VIH–, virus de Epstein-Barr –EBV–) y bacterias (Helicobacter pylori).
Los síntomas de los LNH son muy variables y dependen de cada tipo específico de linfoma y de los órganos que estén afectados. Un gran porcentaje de pacientes son diagnosticados al detectarse una adenopatía. De forma característica los pacientes sintomáticos pueden presentar fiebre, sudoración nocturna, pérdida de peso, fatiga e infecciones de repetición. También pueden producirse manifestaciones como consecuencia del crecimiento del tamaño del bazo (molestias abdominales), de la compresión de un órgano por un tumor de gran tamaño (tos, dolor lumbar o abdominal) o del mal funcionamiento de un órgano por su infiltración por las células cancerosas. Cuando se presentan al mismo tiempo fiebre, sudores nocturnos y pérdida de peso, este grupo de síntomas se denomina síntomas B. Desde un punto de vista clínico, los LNH se pueden dividir en dos grandes grupos en función de su velocidad de crecimiento (Cuéllar, 2018):
El linfomadifuso de células B grandes (LDCGB) es el tipo más común de linfoma de células B, suponiendo el 30-35% de todos los casos, por delante del linfoma folicular (20-25%; éste puede transformarse en LDCGB después de que los pacientes reciben tratamiento para el linfoma folicular), el linfoma de tejido linfoide asociado a mucosas (7-10%), el linfoma linfocítico pequeño o leucemia linfocítica crónica (6-8%), el linfoma de células del manto (5-7%), el linfoma de Burkitt (2-3%) y el linfoma mediastínico (tímico) de células B grandes (2-3%), y otros subtipos minoritarios.
Se trata de un LNH agresivo cuya incidencia media es de 50-60 nuevos casos por millón de habitantes y año (se han descrito tasas concretas de 7 casos/100.000 en EE.UU. y 3,44 casos/100.000 en la Unión Europea) y aumenta con la edad. Aunque se observa en cualquier edad, la edad mediana de los pacientes con linfomas de células grandes es de >60 años. La evolución de este tipo de linfoma es agresiva y su pronóstico depende mucho de la edad del paciente, su estado general, la extensión del tumor y la respuesta al tratamiento. Dentro de los LDCGB, se han descrito dos subtipos: el linfoma difuso de tipo centro germinal y el de célula B activada; este último tiene peor pronóstico. Es importante mencionar que hasta el 80% de los pacientes jóvenes se pueden curar, disminuyendo esta probabilidad con el aumento de edad. Sin tratamiento, la supervivencia media en pacientes es menor a un año.
El tratamiento del LDCGB se basa en la asociación dequimioterapia y radioterapia sobre áreas afectas localizadas o de gran tamaño (masa mediastínica). El esquema quimioterápico más empleado en la actualidad es el CHOP+R (ciclofosfamida, adriamicina, vincristina y prednisona, más rituximab) con una frecuencia de administración y un número de ciclos variable según cada caso; en algunos casos, se añade etopósido. La radioterapia puede ser efectiva para tratar áreas afectas localizadas. Con estas terapias, responden algo más de la mitad de los pacientes, con una supervivencia global a 5 años de, aproximadamente, el 60%.
En los casos de LDCGB recidivante/refractario, el pronóstico es desfavorable y las opciones de éxito con el tratamiento son mucho más limitadas. El tratamiento suele consistir en quimioterapia de rescate basada en platino: DHAP (dexametasona, citarabina, cisplatino), GDP (gemcitrabina, dexametasona, cisplatino), ICE (ifosfamida, carboplatino, etopósido) o IVE (ifosfamida, etopósido, epirrubicina); la adición de rituximab ha mejorado las respuestas clínicas. El trasplante de precursores hematopoyéticos, en este caso TPH autólogo, suele ser utilizado posteriormente a tales regímenes en pacientes refractarios al tratamiento de primera línea o en recaída siempre que la enfermedad permanezca quimiosensible (la mitad de pacientes no responde lo suficientemente bien a la quimioterapia de rescate como para proceder al TPH autólogo).
No obstante, la mitad de los pacientes no son candidatos al trasplante autólogo por comorbilidades o por su edad, y de aquellos que sí reciben el trasplante, más de la mitad vuelven a sufrir una nueva recaída. Ante esa situación, quedan muy pocas opciones terapéuticas. El trasplante autólogo sólo es posible en una minoría, y con resultados pobres de eficacia. El único fármaco disponible en España para estos casos de LDCGB con múltiples recaídas o refractarios al tratamiento es la pixantrona (Pixuvri®), que en monoterapia en pacientes adultos proporciona un aumento de la supervivencia libre de progresión y supervivencia global medianas de 2,7 meses y 2,6 meses, respectivamente, en comparación con otras quimioterapias mono-fármaco (Pettengell, 2012).
En casos de recaída o refractariedad de pacientes con LDCGB que han sido sometidos a dos o más líneas de tratamiento, no se dispone de ningún tratamiento estándar y la calidad de vida de los pacientes suele ser pobre. En tales casos, si no se recurre a un tratamiento experimental, solo se puede hacer tratamiento paliativo. De nuevo, esa laguna terapéutica puede ser cubierta por las novedosas terapias de células con receptores antigénicos quiméricos (CAR-T).
Tisagenlecleucel es un tratamiento de inmunoterapia consistente en células T autólogas modificadas genéticamente ex vivo mediante el empleo de un vector lentiviral para expresar un receptor de antígeno quimérico (CAR, por sus siglas en inglés) anti-CD19. El medicamento ha sido oficialmente autorizado para el tratamiento de: a) la leucemia linfoblástica aguda (LLA) de células B refractaria, en recaída postrasplante o en segunda o posterior recaída en pacientes pediátricos y adultos jóvenes de hasta 25 años de edad; y b) el linfoma B difuso de célula grande (LBDCG) en recaída o refractario tras dos o más líneas de tratamiento sistémico en pacientes adultos.
Esta terapia celular3 autóloga antineoplásica implica la reprogramación de las células T del paciente con un transgén que codifica un receptor antigénico quimérico (CAR) que les permite identificar y eliminar las células que expresan CD19. Las células que expresan CD19 –que serán eliminadas– no solo incluyen los linfocitos B malignos sino también los linfocitos B normales; de hecho, CD19 se expresa en las células del linaje B desde etapas tempranas de su desarrollo a células plasmáticas, pero no en células madre plasmáticas pluripotenciales. No obstante, la elección de CD19 como diana de la inmunoterapia se basa en sus características específicas, esto es, su expresión uniformemente distribuida (en células B de leucemias/linfomas y células B normales pero no en otros tejidos), y la aceptable toxicidad que supone la aplasia de células B, que puede manejarse clínicamente con administración parenteral de inmunoglobulinas (Vairy, 2018).
Tisagenlecleucel puede funcionar a través de múltiples mecanismos de acción. La generación de una respuesta inmunitaria antitumoral potente y sostenida requiere la activación de la producción de citocinas, la citotoxicidad y la proliferación/expansión de células T.
Tras la unión de los linfocitos T reprogramados que expresan CAR a las células que expresan CD19, la proteína quimérica transmite las señales intracelulares necesarias para activar la citotoxicidad frente a esas células CD19+, así como una señal que favorece la expansión de las células T y la persistencia de tisagenlecleucel. De hecho, tras su infusión, se observa una rápida expansión celular inicial de tisagenlecleucel, seguida de un descenso lento biexponencial. En pacientes con LLA –de forma más evidente que en los pacientes con LDCGB– se observó una correlación directa entre la expansión máxima y la respuesta al tratamiento: fue dos veces superior en los pacientes que respondieron completamente respecto a los que no respondieron al tratamiento. Sin embargo, en los casos de expansión muy rápida in vivo, se ha descrito el posible desarrollo de síndrome de liberación de citocinas (SLC) como efecto secundario.
Por último, destaca el hecho de que en los ensayos clínicos no se ha observado una asociación significativa entre la dosis (número de células perfundidas) y la respuesta o su duración.
El receptor de antígeno quimérico (CAR) que expresan los linfocitos T genéticamente modificados, también llamado CTL109, está formado por un fragmento de anticuerpo de origen murino de cadena simple (anti-CD19scFv o FMC63) –que es el que reconocerá al CD19– y que se une, mediante una bisagra CD8-α y una región transmembrana, a un dominio intracelular coestimulador 4-1BB (CD137) y a otro dominio de señalización CD3ζ (zeta) (Figura 2).
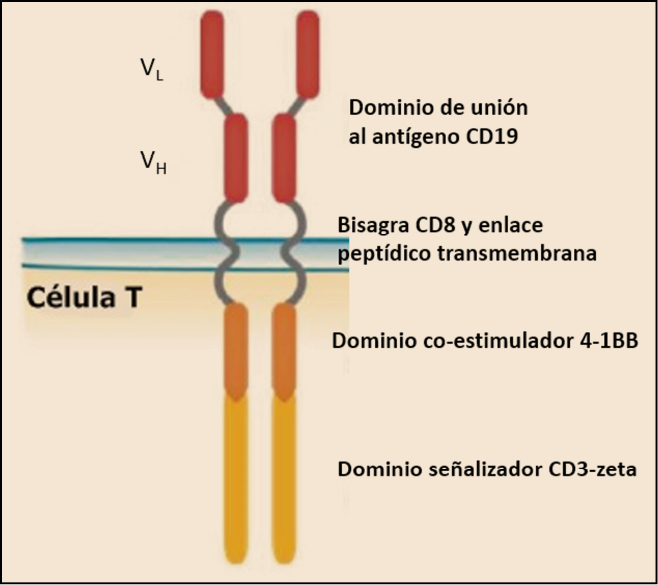
Figura 2. Estructura del receptor de antígeno quimérico CTL109 que portan las células contenidas en tisagenlecleucel. Adaptado de EMA (2018).
El vector encargado de portar el transgén que codifica para el citado CAR e introducirlo en el genoma de las células T extraídas al pacientes es un vector lentiviral autoinactivador recombinante, de tercera generación y defectuoso para la replicación, derivado del genoma lentiviral del virus de la inmunodeficiencia humana (VIH-1). La expresión del CAR anti-CD19 se controla por promotor del factor de elongación 1α (EF-1α), que también está codificado en el trasgén.
Es importante señalar que el componente CD3ζ es crítico para iniciar la activación de la célula T y la actividad antitumoral (a través de un inmunorreceptor intracelular con un motivo de activación basado en tirosina), mientras que el dominio 4-1BB es responsable de aumentar la expansión y la persistencia de tisagenlecleucel. Los receptores quiméricos CAR que portan los dominios de señalización CD3ζ son suficientes para desencadenar la activación y proliferación de las células T, pero no para impulsar la expansión in vivo y la persistencia de las células T con receptores de antígenos quiméricos (células CAR-T). Por tanto, la adición del dominio de transducción intracelular de 4-1BB (CD137) mejora la activación de las células T en comparación con los linfocitos que expresan receptores equivalentes que carecen de 4-1BB: en modelos preclínicos, la inclusión del dominio de señalización 4-1BB (CD137) incrementó significativamente la actividad antitumoral y la persistencia in vivo de los CAR en comparación con la inclusión del dominio de señalización CD3ζ solo.
Tisagenlecleucel se presenta en dispersión en una suspensión incolora o levemente amarillenta, lista para su infusión intravenosa. Es un medicamento indicado únicamente para uso autólogo, y la concentración de células CAR+ por bolsa puede ser diferente para cada paciente. Además de las células T, también pueden estar presentes células natural killer (NK) (EMA, 2018).
La eficacia y la seguridad clínicas de tisagenlecleucel han sido evaluadas en sus indicaciones autorizadas en ensayos clínicos de fase 2 (exploratorios de seguridad y eficacia): a) en un estudio pivotal multicéntrico (B2202) y dos de soporte (B2205J y B2101J), de un solo brazo y abiertos (160 pacientes en total de hasta 25 años de edad), en la indicación de leucemia linfoblástica aguda (LLA); y b) en un solo estudio pivotal multicéntrico (C2201), de un solo brazo y abierto, en la indicación de linfoma difuso de células grandes (LDCGB).
El ensayo B2202 (Maude, 2018) enroló inicialmente a un total de 92 pacientes de entre 3 y 25 años, con enfermedad refractaria o en segunda o posterior recaída o en recaída tras haber recibido un TPH alogénico o no haber sido candidatos al mismo. Además de numerosos criterios de exclusión, se definieron entre los criterios de inclusión los siguientes: enfermedad medible por ≥5% linfoblastos en médula ósea, expectativas de vida >12 semanas, estado funcional ≥50 puntos (según Karnofsky4 en mayores de 16 años y Lansky en menores de 16 años) y expresión de CD19 en células tumorales. Se podían incluir pacientes con LLA cromosoma Philadelphia positivo intolerantes o refractarios a inhibidores de tirosina cinasa.
De los 92 pacientes iniciales, 75 recibieron la perfusión de tisagenlecleucel, mientras que los otros 17 no pudieron ser tratados por imposibilidad de fabricación del medicamento, muerte o reacciones adversas mientras esperaban la fabricación. Casi todos los pacientes perfundidos (72) recibieron quimioterapia de linfodepleción previa a la perfusión. Las características basales principales de esta población eran: edad mediana de 11 años (con un 41% de los pacientes de <10 y otro 41% entre 10 y 18 años), un 57% eran varones, un 92% de pacientes había sufrido una recaída de la patología (solo el 8% eran refractarios a tratamientos previos); la mediana de tratamientos previos fue 3 (rango de 1 a 8), incluyendo 1 o 2 TPH alogénicos en el 61% de pacientes.
En un primer análisis de los datos con una mediana de seguimiento de los pacientes de 13,1 meses, la variable primaria de eficacia –tasa de remisión global (TGR) a los 3 meses, mantenida al menos 28 días– fue alcanzada por 61 pacientes (81,3%) de los tratados con el fármaco en estudio (IC95% de 70,7 a 89,4; p<0,0001). En el espectro de variables secundarias, todos ellos alcanzaron una enfermedad mínima residual (EMR) negativa en médula ósea: 45 pacientes (60%) alcanzaron una remisión completa (RC) –definida como <5% de blastos en médula ósea, <1% de blastos en sangre periférica, sin evidencia de enfermedad extramedular y recuperación completa del recuento celular en sangre periférica sin transfusiones– y 16 tuvieron una remisión completa con recuperación hematológica incompleta (RCi). Es importante subrayar que ninguno de los pacientes incluidos en el estudio que no recibieron la perfusión de tisagenlecleucel (un total de 17) alcanzó la TGR ni, por tanto, EMR negativa.
Con respecto a la duración de la respuesta (DR), ésta persistía en el 79,5% de los pacientes infundidos a los 6 meses, y en el 50,5% a los 12 meses. En términos de supervivencia global (SG) de los pacientes tratados, la mediana se situó en 19,1 meses, con una SG a los 6 meses del 90,3% y a los 12 meses del 76,4% (frente al 77,4 y el 70,3%, respectivamente, en el global de pacientes incluidos en el estudio). La supervivencia libre de eventos a los 12 meses se verificó en el 50,5% de los pacientes tratados. Además, otra variable secundaria a destacar fue el análisis de calidad de vida que se realizó mediante los cuestionarios específicos PedsQL y EQ-5D: los resultados indican una mejora clínicamente significativa desde los 3 a los 12 meses después de la infusión en los pacientes que respondieron (N=48), sin diferencias en el análisis por subgrupos.
Los dos ensayos clínicos de soporte B2205J y B2102J confirmaron la línea de los resultados de tisagenlecleucel en poblaciones similares de pacientes. En la siguiente Tabla 1 se muestran los principales resultados de eficacia de estos dos estudios, referentes a pacientes tratados con tisagenlecleucel (sin considerar los pacientes incluidos en el estudio pero no tratados), comparándolos con los previamente comentados del estudio pivotal B2202.
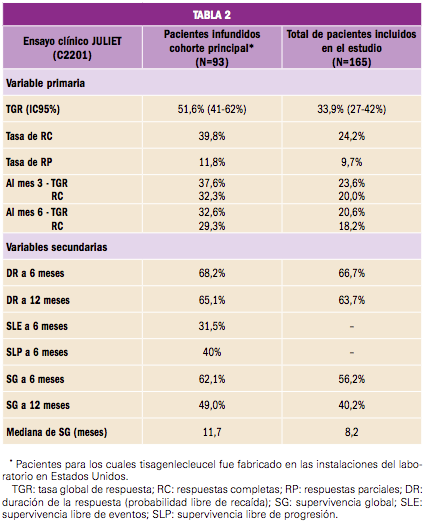
El perfil de seguridad clínica de tisagenlecleucel en pacientes con LLA se determinó en base a los datos de todos los pacientes de los estudios B2202 y B2205J (104 pacientes infundidos), en los que se aprecia que el 95% de los pacientes sufrieron algún efecto adverso atribuido al fármaco; algunos de éstos no son debidos directamente a tisagenlecleucel sino a la quimioterapia previa de linfodepleción. Cabe destacar que en torno al 88% de los pacientes presentó efectos adversos graves de grado ≥3 atribuibles al fármaco, la mayoría de los cuales aparecieron en los dos primeros meses posperfusión.
Por orden de frecuencia, las reacciones adversas no hematológicas –las anomalías hematológicas (sobre todo, disminución de glóbulos blancos, de neutrófilos y de linfocitos) acontecieron en la práctica totalidad de pacientes tratados– más notificadas fueron el síndrome de liberación de citoquinas (SLC; 77% de los pacientes), infecciones (65%), hipogammaglobulinemia (47%), pirexia (40%) y disminución del apetito (39%).
Los investigadores del ensayo B2202 consideraron de especial relevancia en la salud del paciente los siguientes efectos adversos: SLC, neurotoxicidad, infecciones, neutropenia febril, citopenias persistentes (>28 días pos-infusión), y síndrome de lisis tumoral. Estos efectos se atribuyeron al fármaco, de forma dosis-independiente, en el 86,7% de los pacientes tratados en ese estudio, siendo el SLC el más frecuente y más grave (grado ≥3 en 44% de los casos); tuvo una duración mediana de 8 días y aconteció a una mediana de 3 días tras la perfusión. Casi la mitad de los pacientes que sufrieron SLC fueron tratados con tocilizumab5.
Del resto de efectos adversos, se pueden destacar los cardíacos (47%, grado ≥3 en 21%), insuficiencia renal con necesidad de diálisis (siempre en el contexto de SLC grave), hemorragias (29%, grado ≥3 en 14%), vómitos (36%), dolor de cabeza (35%), hipotensión (31%) y náuseas (31%). De los 104 pacientes tratados, se notificaron 29 muertes, la gran mayoría de las cuales (22) se debieron a progresión de la propia LLA.
El ensayo pivotal C2201 –JULIET– (Shuster, 2019) incluyó un total de 165 pacientes adultos con LDCGB en recaída o refractario que recibieron ≥2 líneas de quimioterapia (incluido rituximab y antraciclinas) o bien que habían recaído después de un trasplante de progenitores hematopoyéticos (TPH) autólogo o no eran candidatos al mismo. También se permitió la administración de quimioterapia en los pacientes que la necesitaran para estabilizar su patología hasta la perfusión de tisagenlecleucel (terapia puente). Como requisito, se incluyeron en el estudio, por las diferencias entre sus pronósticos, al menos 25 pacientes de los subtipos de centro germinal y de fenotipo activado del LDCGB. Sin embargo, se excluyeron pacientes que presentaban algunos subtipos de tumores relacionados, entre otros, el LDCGB rico en histiocitos/células T o positivo para el virus de Epstein-Barr, linfoma cutáneo o de mediastino primario de células B grandes, o el linfoma de Burkitt.
De los 165 pacientes iniciales, el 30% interrumpieron el estudio por diversas causas (sobre todo, fallecimiento, reacciones adversas o decisión del médico) antes de la administración de tisagenlecleucel, de forma que solo 111 recibieron la perfusión con el fármaco; para 12 pacientes no se pudo fabricar el fármaco. Entre las características principales de la población de pacientes tratados destacan: edad mediana de 56 años (con un 78% de pacientes de <65 años), un 61% eran varones, un 49% habían recibido previamente un TPH autólogo y el 76% presentaba una enfermedad en fase III/IV al inicio del estudio. Con respecto a los tratamientos previos, el 51% de los pacientes habían recibido 3 o más líneas de tratamiento (21% de ellos, 4 líneas), el 55% había sido refractario a la última línea de tratamiento y el 45% había recaído tras la misma. Hasta la perfusión, el 90% de los pacientes recibieron terapia puente, siendo las más usadas rituximab (54,5%) y gemcitabina (38,4%).
Las variables primaria y secundarias del ensayo C2201 fueron muy similares a las comentadas para los estudios con pacientes de LLA. Los resultados fundamentales de eficacia se resumen en la siguiente Tabla 2. De los 111 pacientes tratados, 93 tuvieron un seguimiento superior a los 3 meses y constituyen la población del análisis por protocolo.
Por otra parte, el análisis comparativo de eficacia de tisagenlecleucel en LDCGB primario y en linfoma folicular transformado (a LDCGB) demostró que los resultados de las variables primarias y secundarias de eficacia eran significativamente superiores en el caso del linfoma folicular transformado; sin embargo, el bajo número de pacientes con este subtipo -18- impide sacar conclusiones definitivas. Además, los resultados obtenidos de los cuestionarios de calidad de vida sugieren una pequeña mejoría después de los 3 meses de desde la perfusión en aquellos pacientes con respuesta al tratamiento.
En relación a la seguridad clínica de tisagenlecleucel en LDCGB, los datos derivan de los 111 pacientes perfundidos en el ensayo C2201, con una mediana de seguimiento de 14 meses. De forma similar a lo comentado anteriormente, las reacciones adversas no hematológicas más frecuentes en los primeros dos meses postratamiento fueron: síndrome de liberación de citoquinas (SLC; 58% de los pacientes), infecciones (54%), pirexia (35%), diarrea (32%), náuseas (29%), hipotensión (26%) y fatiga (26%).
Casi un 90% de pacientes sufrió al menos una reacción adversa de grado 3 y 4, siendo las infecciones y el SLC las reacciones graves más frecuentes (en el 32 y 22% de los pacientes, respectivamente); hasta un cuarto de los pacientes con SLC fueron tratados con tocilizumab. También se observó neutropenia febril en el 15% de los pacientes y citopenias prolongadas en el 36%. Al igual que éstas, las anomalías hematológicas de laboratorio (disminución del recuento de linfocitos, neutrófilos, glóbulos blancos, hemoglobina y plaquetas) –muy frecuentes (60-95% de los pacientes)– fueron de grado ≥3. La incidencia de reacciones de neurotoxicidad fue algo menor en estos pacientes (21%, grado ≥3 en 12%). Ninguna de las tres muertes notificadas en los 30 días pos-perfusión fue atribuida al fármaco o a reacciones derivadas de su uso.
Por último, habida cuenta de que el fragmento extracelular de la proteína quimérica de tisagenlecleucel es de origen murino, existe el riesgo de inmunogenicidad, que se ha observado en el 35% de los pacientes con LLA y el 5% de los pacientes con LDCGB. Los anticuerpos antifármaco no se han asociado en los ensayos clínicos con una menor eficacia ni con menor expansión celular.
El tisagenlecleucel es un tratamiento de inmunoterapia consistente en células T autólogas modificadas genéticamente ex vivo mediante el empleo de un vector lentiviral para expresar un receptor de antígeno quimérico (CAR, por sus siglas en inglés) anti-CD19. Tras la unión de los linfocitos T reprogramados que expresan CAR a las células que expresan CD19 –células del linaje B desde etapas tempranas de su desarrollo a células plasmáticas, tanto malignas como normales–, la proteína quimérica transmite las señales intracelulares necesarias para activar la citotoxicidad frente a esas células CD19+, así como una señal que favorece la expansión de las células T y la persistencia de tisagenlecleucel.
El medicamento ha sido oficialmente autorizado como medicamentohuérfano para el tratamiento, en perfusión única, de pacientes pediátricos y adultos jóvenes de hasta 25 años de edad con leucemia linfoblástica aguda (LLA) de células B refractaria, en recaída postrasplante o en segunda o posterior recaída, y de pacientes adultos con linfoma difuso de células grandes B (LBDCG) en recaída o refractario tras dos o más líneas de tratamiento sistémico.
La eficacia y seguridad clínicas de tisagenlecleucel en LLA han sido evaluadas en un ensayo pivotal de fase 2 (B2202), abierto y de un único brazo (sin comparadores directos), en que el fármaco indujo una tasa de respuesta global del 81% (66% incluyendo pacientes tratados y no tratados), teniendo un 60% de los pacientes RC y el 21% RCi. Si se analizan estos resultados junto a los obtenidos en los ensayos de soporte B2205J y B2102J, con un total de 160 pacientes perfundidos con tisagenlecleucel, se observa la consistencia de los resultados clínicamente relevantes de la respuesta global en todos los subgrupos pronósticos y demográficos. La población tratada en estos ensayos refleja en gran medida la población clínica de pacientes pediátricos y adultos jóvenes con LLA de células B recidivante o refractaria. No obstante, las peculiaridades de la patología y del tipo de pacientes estudiados con los demás fármacos aprobados en la misma indicación (se han descrito tasas de respuesta global del 30% para clofarabina, del 44% para blinatumomab y del 81% para inotuzumab onogamicina6) hacen difícil realizar comparaciones indirectas y establecer la superioridad clínica de tisagenlecleucel.
En relación con la duración de la respuesta, la mediana de la duración de la respuesta no fue alcanzada en el momento de la autorización de tisagenlecleucel, pero en torno a la mitad de los pacientes presentaban respuesta a los 12 meses posperfusión. A pesar de los prometedores resultados, las limitaciones del periodo de seguimiento en los ensayos clínicos acota la posibilidad de establecer que tisagenlecleucel mantiene a los pacientes libres de la enfermedad a largo plazo, lo cual se esclarecerá con mayor robustez estadística con los nuevos resultados que vayan surgiendo.
Con respecto a la indicación del fármaco en LDCGB, los datos de eficacia y seguridad proceden de un único ensayo de fase 2 (C2201) –también abierto y sin comparadores directos– en que la perfusión del fármaco se realizó en un total de 111 pacientes; casi la mitad de ellos había recibido previamente un TPH autólogo y, a pesar de ello, estaban en recaída/refractariedad, situación en la cual las opciones terapéuticas son muy limitadas y el pronóstico desfavorable. En general, la población incluida en el estudio se puede asemejar a la población diana, lo que en parte valida sus resultados principales: la tasa de respuesta global alcanzó valores del 52% (34% en el total de pacientes incluidos en el estudio, tratados o no), con una mediana de supervivencia global de 13 meses (8 meses en el total de pacientes incluidos).
De forma interesante, al tratarse de un estudio de un único brazo, en el EPAR (Public Assessment Report) de la EMA –como se recoge también en el IPT de la AEMPS– se realizan comparaciones indirectas de los resultados de eficacia del estudio C2201 con datos cohortes históricas de pacientes con LDCGB que habían recibido diferentes terapias de rescate. La diferencia entre los pacientes tratados con tisagenlecleucel y aquellos del estudio SCHOLAR-1 (Crump, 2018) –que analizó datos retrospectivos de pacientes con linfoma no Hodgkin agresivo refractario a varias líneas de quimioterapia– fue del 31% en la tasa de respuesta completa (IC95% 20-42%, p<0,01), con una razón de riesgos (HR) de 0,68 para la supervivencia global (IC95% 0,48-0,96, p<0,05). Los resultados fueron parecidos al comparar con el ensayo CORAL (Thieblemont, 2011) –que evaluó el régimen quimioterapéutico R-DHAP frente al R-ICE–, demostrando, para los pacientes perfundidos, una diferencia significativa en la tasa de respuesta completa (+12%; IC95% 1-23%, p<0,01) y una mejoría de la supervivencia global (HR 0,41; IC95% 0,31-0,54, p<0,01) a favor de tisagenlecleucel.
Para la misma indicación de LDCGB, también se ha aprobado recientemente otro fármaco de terapia celular CAR-T anti-CD19, denominada axicabtagén ciloleucel7 (Yescarta®), que no ha sido aún comercializado en España. Por las notables diferencias en los criterios de inclusión y exclusión de pacientes en los ensayos clínicos, no se pueden establecer conclusiones de la comparación de eficacia de ambos fármacos, si bien los datos publicados para axicabtagén ciloleucel (Roberts, 2018) sugieren que, con un perfil de seguridad similar, puede aportar una tasa de respuesta global (y completa) y una supervivencia global a 12 meses superiores a tisagenlecleucel y a los controles históricos. No obstante, un ensayo que compare ambas terapias arrojará luz sobre la cuestión de cuál de los dos tratamientos es más eficaz.
Es importante resaltar el hecho de que el retraso en la fabricación de la terapia celular –desde que los pacientes son incluidos en el estudio hasta que son tratados– puede suponer, a pesar de la quimioterapia puente (en general, con efectos limitados en pacientes en recaída/refractarios), un avance del tumor que podría enmascarar los resultados parcialmente. Podría ser esperable que una mejora en el tiempo de producción se tradujera a su vez en una mejora de la tasa de respuestas. Por otro lado, ese retraso fue la causa de que muchos pacientes se retiraran de los ensayos clínicos (en mayor número en el caso del ensayo con pacientes con LDCGB) previamente a la infusión de tisagenlecleucel. Esto pudo implicar, no obstante, que solo fueran tratados con el fármaco aquellos pacientes con un mejor estado clínico, lo cual podría representar un factor de confusión en la tasa de respuesta global y de duración de la misma. Hasta el momento, además, no se ha establecido el impacto del tratamiento puente (ni de la quimioterapia de linfodepleción previa a la perfusión) en los resultados observados con tisagenlecleucel (IPT, 2018).
El perfil toxicológico de tisagenlecleucel es importante, con un alto porcentaje (cercano al 90%) de pacientes que sufren reacciones adversas específicas graves (grado ≥3) o potencialmente mortales en el periodo posperfusión, y principalmente en las primeras 8 semanas. Los eventos adversos, también influidos por la quimioterapia previa, fueron algo menores en los pacientes con LDCGB frente a los de LLA, lo cual se refleja, por ejemplo, en la frecuencia del síndrome de liberación de citoquinas (58% vs 77%). El manejo de este SLC –para el que se requiere la disponibilidad de tocilizumab– es quizá la cuestión clave de la seguridad de esta terapia celular. Por su relevancia clínica, también sobresalen los efectos adversos neurológicos graves (en un 12-13% de los pacientes) e infecciones (20-44%) y neutropenia febril severas (13-36%). Debe tenerse en cuenta que el uso de dosis elevadas de corticoesteroides para el tratamiento de SLC o neurotoxicidad podrían interferir en la eficacia de tisagenlecleucel por su efecto sobre el sistema inmune.
Otros efectos secundarios comunes de tisagenlecleucel son, por ejemplo, fiebre, diarrea, náuseas, cansancio, presión arterial baja, edema y dolores de cabeza. No hay datos, para ninguna de las dos indicaciones, de seguridad (ni de eficacia) en mujeres embarazadas o en fase de lactancia, ni para la producción del fármaco en pacientes positivos para el VIH o los virus de la hepatitis B o C. Además, la persistencia de tisagenlecleucel en el organismo (a día de hoy se ha confirmado su presencia en líquido cefalorraquídeo y en plasma durante 1 y 2 años posperfusión, respectivamente) hace necesario desarrollar estudios clínicos que evalúen el efecto del fármaco sobre la seguridad clínica a largo plazo, identificando el riesgo potencial de desarrollo de neoplasias secundarias, reactivaciones virales, etc.
De acuerdo con lo anterior, a fin de minimizar los riesgos derivados del empleo de estas novedosas terapias CAR-T, el Ministerio ha publicado recientemente un Plan de abordaje de las terapias avanzadas en el Sistema Nacional de Salud (MSSSI, 2018), que define una serie de requisitos y medidas que permitan atenuar y manejar la toxicidad clínica por parte de profesionales sanitarios especialmente entrenados en centros capacitados.
A modo de resumen, el diseño no comparativo de los estudios de fase 2 conducentes a la autorización de tisagenlecleucel en sus dos indicaciones (casos en recaída o refractarios de LLA-B y LDCGB) dificulta el análisis del impacto real de su utilidad clínica. Sin embargo, teniendo en consideración las escasas opciones terapéuticas y sus limitados resultados en ambas patologías, los resultados aquí comentados –sugerentes de un aumento de la tasa de respuesta global y duración de la misma (que pueden suponer un incremento de supervivencia global)– aportan la base científica que refleja un beneficio clínico para determinados pacientes. Los limitados periodos de seguimiento de los pacientes en los ensayos clínicos y la escasa experiencia posautorización hasta la fecha –incertidumbres asumibles para este tipo de medicamentos huérfanos– impiden confirmar todavía su consideración como tratamiento curativo, y se debe esperar a estudios con un seguimiento más largo (de hasta 5 años), a los que se ha comprometido el titular de autorización, para verificar si existe beneficio en términos de supervivencia libre de enfermedad a largo plazo.
En definitiva, tisagenlecleucel representa la primera terapia celular CAR-T comercial disponible en España. Por su fundamento, mecanismo de acción y resultados clínicos, supone una innovación disruptiva en el área clínica de la onco-hematología, que viene a posicionarse como una herramienta terapéutica con gran utilidad potencial en dos indicaciones consideradas necesidades médicas no cubiertas. Es probable que los criterios de inclusión tan restrictivos en los ensayos clínicos y su coste económico releguen el acceso a corto plazo a tisagenlecleucel a pacientes con relativo buen estado funcional que no dispongan de otras alternativas farmacológicas adecuadas. También se puede presumir que la mayor experiencia clínica y los resultados de futuros estudios que evalúen su eficacia cuando se administra a otro tipo de pacientes (por ejemplo, en refractariedad o recaída tras una única línea de tratamiento o incluso sin trasplante de precursores hematopoyéticos previo) permitan ampliar el espectro de pacientes en sus indicaciones.

El medicamento Biktarvy® consiste en una novedosa combinación coformulada de tres agentes antirretrovirales, que incluye dos inhibidores de la transcriptasa inversa (ITI) ampliamente utilizados (emtricitabina y tenofovir alafenamida) y un nuevo inhibidor de la integrasa (bictegravir). Ha sido oficialmente autorizado para el tratamiento de adultos infectados por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) sin resistencia viral actual o previa a los inhibidores de la integrasa, a emtricitabina o a tenofovir. Esta combinación de principios activos –que actúan a través de mecanismos de acción distintos– se utiliza con el fin de reducir drásticamente la carga viral y frenar el deterioro del sistema inmunitario de los pacientes y la incidencia de infecciones oportunistas y otras patologías asociadas al SIDA. Las tres moléculas tienen actividad antirretroviral frente a VIH-1 y VIH-2, actuando sinérgicamente frente al VIH-1.
Su eficacia y seguridad clínica han sido adecuadamente contrastadas mediante cuatro amplios ensayos pivotales de fase 3 con comparador activo realizados en las poblaciones diana: dos ensayos doblemente ciegos incluyeron pacientes no tratados previamente (naïve) y otros dos ensayos, uno abierto y otro ciego, evaluaron pacientes virológicamente suprimidos. Los datos clínicos a 48 semanas son consistentes en los distintos subgrupos de pacientes e indican que la combinación bictegravir/emtricitabina/tenofovir alafenamida es no inferior en términos de eficacia –definida como tasas de supresión viral (<50 copias de ARN viral/mL)– en pacientes naïve frente a la combinación de abacavir/dolutegravir/lamivudina (92,4% de pacientes vs 93,0%) ni frente a dolutegravir más emtricitabina/tenofovir alafenamida (89,4% vs 92,9%). También ha demostrado no inferioridad en pacientes tratados previamente –con independencia del TAR anterior– que cambiaron al nuevo tratamiento, alcanzando tasas elevadas de supresión virológica similares a las conseguidas en pacientes que siguieron en tratamiento con los comparadores activos (93,6% vs 95% de la combinación abacavir/dolutegravir/lamivudina, y 92,1% vs 88,9% de la combinación de atazanavir o darunavir –potenciados con ritonavir o cobicistat– más emtricitabina/tenofovir disoproxilo o más abacavir/lamivudina); las tasas de fracaso virológico para la nueva combinación fueron muy bajas (1,1-1,7%).
Con respecto a la seguridad, su perfil de eventos adversos está bien caracterizado en base a los datos de los 1.288 pacientes incluidos en los ensayos pivotales, y se puede considerar aceptable y similar a la del resto de regímenes de primera línea. Las reacciones adversas descritas con más frecuencia han sido cefalea, diarrea y náuseas, que afectaron a más de 4 pacientes por cada 100 tratados, siendo la frecuencia de efectos adversos graves que supusieron una interrupción del tratamiento muy baja (<1%). El perfil toxicológico se sitúa próximo al de regímenes basados en otro inhibidor de la integrasa (INI) como dolutegravir: cabe prestar especial atención al riesgo de toxicidad hepatobiliar, nefrotoxicidad o desórdenes psiquiátricos (incluyendo ideaciones suicidas). No obstante, el número de pacientes con una duración del tratamiento >72 semanas fue muy limitado, por lo que la evaluación de la seguridad a largo plazo requiere de estudios poscomercialización.
En resumen, según se recoge en las principales guías clínicas nacionales e internacionales, la combinación bictegravir/emtricitabina/tenofovir alafenamida emerge como opción preferente en el tratamiento antirretroviral (TAR) de inicio o como alternativa tras cambio de TAR (en pacientes virológicamente suprimidos), con una prometedora barrera genética a mutaciones de resistencia. Bictegravir, el nuevo fármaco inhibidor de la integrasa, presenta una alta potencia, similar a dolutegravir, con la ventaja frente a otros fármacos del grupo de que no necesita potenciador farmacocinético. Se trata de un “régimen de pastilla única” que, sin aportar una ventaja adicional en términos de adherencia terapéutica frente a otras combinaciones en primera línea, supone la primera coformulación de un INI no potenciado con los dos ITI emtricitabina y tenofovir. A falta de conocer las consideraciones derivadas del IPT y de futuros estudios de coste-efectividad, Biktarvy® se posiciona en primera línea como una alternativa eficaz más para el tratamiento de la infección por VIH-1 a los regímenes preferentes de antirretrovirales combinados, sin aportar, a priori, otros elementos que supongan una innovación disruptiva.
El virus de la inmunodeficiencia humana (VIH) es un retrovirus o virus de ARN monocatenario retrotranscrito que forma parte de los lentivirus (familia Retroviridae, género Lentivirus), una subfamilia cuyo nombre alude a la forma insidiosa (lenta e inicialmente desapercibida) en que se desarrolla su infección; sin embargo, el VIH tiene una cinética de replicación muy agresiva, y muy diferente a la que suelen presentar los lentivirus. Su genoma se compone de dos copias idénticas de cadenas de ARN que dependen para su replicación en la célula huésped de una enzima denominada transcriptasa inversa o retrotranscriptasa, que da lugar mediante el proceso de retrotranscripción a un ADN provisional que a menudo se inserta en el genoma del hospedador por la acción de una ADN polimerasa dependiente de ARN, la integrasa viral.
Los VIH son virus de geometría esférica, con un diámetro medio aproximado de 100 nm (Figura 1). La capa más externa o cubierta está formada por una membrana lipídica derivada de la célula hospedadora, con un alto contenido en lipoproteínas. En dicha membrana se insertan antígenos de histocompatibilidad (HLA) del hospedador, que le permitirán una primera unión a la célula hospedadora y varios complejos heterodiméricos de glucoproteínas, compuestos por trímeros de la glicoproteína de superficie gp120 y de la glicoproteína transmembrana gp41. Debajo de la membrana lipídica se encuentra la matriz proteica, formada por la proteína p17, que recubre a la cápsula o cápside propiamente dicha, constituida por la proteína p24. En el interior de la cápside se encuentran el material genético –que se presentará como dos hebras idénticas de ARN monocatenario–, la nucleoproteína y algunas enzimas (entre otras, la transcriptasa inversa y la integrasa). El tamaño del genoma del VIH es inferior a 10.000 nucleótidos y contiene varios genes: tres principales (gag, pol y env) que codifican, cada uno, varias proteínas (estructurales, enzimas y glicoproteínas de la cubierta viral, respectivamente) y varios genes que codifican proteínas reguladoras (tat, rev, vif, vpu, nef y vpx). Con la participación de todos esos elementos, el virus se apodera de la maquinaria celular y silencia la replicación de numerosos genes celulares en favor de la replicación de los propios.
Figura 1. Estructura del VIH-1.
A nivel mundial se describen dos tipos de VIH: VIH-1, derivado del virus de la inmunodeficiencia de los simios de los chimpancés, y el VIH-2, que se originó a partir del retrovirus que infecta a los primates denominados mangabeysgrises. La muestra de suero más antigua en la que se comprobó la presencia del VIH-1 data de 1959, aunque el estudio de la evolución de la epidemia desde su introducción en Tailandia (1986) lleva a los investigadores a estimar que la infección en humanos se inició entre 1910 y 1950, probablemente alrededor de 1930. En 1983 se identificó el primer tipo de VIH (VIH-1) y en 1986 un segundo tipo (VIH-2).
En general, los virus VIH se encuentran más localizados en individuos de diversos países de África Occidental, donde se distribuyen principalmente los primates portadores, y áreas con lazos históricos como Portugal y la India. Tanto el VIH-1, el más prevalente y distribuido a nivel mundial, como el VIH-2 son capaces de evolucionar a SIDA (síndrome de inmunodeficiencia adquirida) sin tratamiento. Dentro de cada uno de esos dos grandes grupos de VIH, se han descrito numerosos subtipos, algunos de los cuales han evolucionado mayoritariamente de forma pandémica (Figura 2). La gran variabilidad que presenta el virus VIH es uno de los mayores inconvenientes para conseguir un tratamiento eficaz.
Figura 2. Distribución mundial de los subtipos de VIH-1.
La tipificación del VIH resulta muy relevante en cuanto al pronóstico y al tratamiento de la infección, ya que los diferentes subtipos presentan características biológicas diferentes que pueden condicionar su patogenicidad, su capacidad de transmisión y su resistencia o sensibilidad a los antirretrovirales. Así, las cepas del grupo O del VIH-1 son naturalmente resistentes a inhibidores de la transcriptasa inversa no análogos de los nucleósidos; en los distintos subgrupos del grupo M, algunas cepas del subtipo G son menos sensibles in vitro a inhibidores de la proteasa; y algunas mutaciones que condicionan la resistencia a inhibidores de la transcriptasa inversa no nucleósidos son más frecuentes, por ejemplo, en el subgrupo C que en el B. El VIH-2, por su parte, no es sensible a los inhibidores de la transcriptasa inversa no análogos de los nucleósidos y, para algunos subtipos, la determinación de carga viral puede dar resultados falsamente negativos.
El VIH es un microorganismo extraordinariamente sensible al medio, no puede sobrevivir fuera del torrente sanguíneo o del tejido linfático y es muy sensible a las altas temperaturas y a la desecación (aunque el ARN es detectable en sangre seca durante 4-5 días, no es infeccioso), y a pH alcalinos o ácidos. Por ello, la transmisión del VIH entre personas ha de producirse a través de algún fluido biológico en el que pueda sobrevivir el VIH, fundamentalmente la sangre y algunas secreciones (vaginal, espermática, etc.), que entre en íntimo contacto con estructuras potencialmente receptoras, como los vasos sanguíneos, o pequeñas erosiones en la piel o las mucosas. Las principales vías de transmisión del VIH son la sexual, la parenteral y la vertical (madre-hijo), siendo la vía sexual la que origina más del 80% de los contagios a nivel mundial.
El VIH presenta tropismo por los macrófagos y los linfocitos T CD4+. Las células infectadas por el VIH pueden transferir el virus a las células del sistema inmune local, presentes en el epitelio vaginal o en la mucosa ano-rectal. En relaciones heterosexuales, el primer tejido en ser infectado es la mucosa cervical, donde pueden infectarse las células dendríticas y los linfocitos T CD4+. La proporción de linfocitos T CD4+ infectados en sangre periférica es mínima (1-10%), pero su capacidad de producir viriones es muy alta y, a partir de estos, la infección puede difundirse hacia los nódulos linfáticos regionales y, posteriormente, ser distribuida por todo el organismo a través del torrente circulatorio. Una vez en los nódulos linfáticos, se produce una intensa replicación del VIH que en unos casos provoca la lisis de las células infectadas, pero en otros queda como una infección latente en los macrófagos y los linfocitos T en reposo. Estos últimos actúan como “reservorios naturales” del VIH y dificultan notablemente la respuesta inmunológica del organismo y, en la misma medida, la adopción de medidas terapéuticas eficaces desde el mismo inicio de la infección. Solo una pequeña proporción de los linfocitos infectados (1%) replica activamente el VIH en determinado momento, permaneciendo latente en la inmensa mayoría de ellos. Aún no se conoce bien de qué forma estas células con infección latente pasan a la replicación activa, pero los factores implicados en este proceso podrían constituir nuevas dianas terapéuticas, ya que el virus latente carece de efectos patológicos.
La primoinfección por el VIH-1 es sintomática en más de la mitad de los casos, pero generalmente puede pasar desapercibida ya que sus síntomas son los de una virosis común, como un resfriado o una gripe. Los síntomas y signos más comunes son fiebre, odinofagia, adenopatías de predominio laterocervical, mialgias, exantema, sudoración nocturna y artralgias; algunos pacientes pueden tener un cuadro similar al de la mononucleosis infecciosa. Apenas 10-12 días después del contagio ya se puede detectar el ARN viral en la sangre, lo que implica que el paciente está en condiciones de contagiar a otra persona. Tras la primera replicación viral después de la infección, la carga viral puede llegar a alcanzar valores de hasta 100 millones de copias por mL de sangre. Esta elevada viremia provoca una rápida respuesta del sistema inmunológico.
La respuesta inmunitaria frente al VIH-1 se produce tanto en la vertiente humoral (con una intensa producción de anticuerpos contra las proteínas reguladoras y estructurales, así como producción de complemento e interferones) como en la celular (activación de poblaciones linfocitarias T colaboradoras, citotóxicas y de estirpe NK o natural killer). Inicialmente, se produce una intensa expansión clonal de linfocitos CD8+ con actividad citotóxica, dirigida frente a diversas proteínas del VIH-1, y que originan una cierta supresión de la replicación del VIH y una drástica disminución de la viremia –en varios órdenes de magnitud – pudiendo incluso hacerse indetectable en la sangre (generalmente, <50 copias de ARN/ mL de sangre). Entre 3 y 5 semanas después de la infección, aparecen anticuerpos, lo que determina la seroconversión, ya que a partir de este momento el paciente se transforma en seropositivo. Esta potente respuesta antiviral llega a contener la replicación vírica, pero es incapaz de erradicar el virus del organismo.
A la fase aguda de la infección le sigue una fase asintomática (fase crónica no SIDA) de duración variable pero raramente inferior a 18 meses, durante la que el virus continúa replicándose masivamente en diferentes compartimentos orgánicos, siendo solo parcialmente controlado por la activación constante de la respuesta inmunitaria natural del organismo; esto provoca un estado inflamatorio de carácter crónico que puede mantenerse por largos períodos de tiempo (Figura 3). El pronóstico y la evolución de la infección dependerán del equilibrio entre la virulencia del virus y la respuesta inmunológica del huésped.
Figura 3. Historia natural de la infección por VIH.
En la gran mayoría de pacientes infectados1, a lo largo del periodo asintomático se produce un deterioro progresivo del tejido linfoide, con reducción de los recuentos de linfocitos CD4+, fruto de la replicación viral y del estado inflamatorio crónico. Al ocupar un lugar preeminente en la organización y la activación del sistema inmune, la caída brusca de las células CD4+ origina un deterioro funcional de otras poblaciones celulares y se reduce la eficacia del sistema inmunológico: disminuyen los niveles de anticuerpos frente a diversas estructuras virales y se vuelve a elevar significativamente la carga viral, cuya difusión se generaliza por todo el organismo. En estos estadios finales de la enfermedad, la fase deSIDA propiamente dicha viene definida a partir de un contaje del número de linfocitos CD4+ inferior a 200 células por mililitro de sangre o por la asociación de cualquier manifestación grave independientemente del recuento celular.
En esta fase se magnifica el riesgo de nuevas infecciones o la reactivación de infecciones latentes, siendo frecuentes las infecciones oportunistasy también ciertas patologías malignas, como ciertos tumores, asociados a la inmunodeficiencia provocada por el VIH. Los microorganismos oportunistas más frecuentemente causantes de infecciones son varias especies de Mycobacterium (M. tuberculosis, complejo avium-intracellulare, etc.), Pneumocystis jiroveci, Candida albicans, Herpes simple y zóster, Citomegalovirus, Criptosporidium, Giardia, Isospora, etc. A día de hoy, la tuberculosis continúa siendo la principal causa de muerte entre las personas que viven con el VIH (es responsable de una de cada tres muertes relacionadas con el SIDA), seguida de neumonía y candidiasis. Las neoplasias emergentes más comúnmente relacionadas con el VIH son el sarcoma de Kaposi y ciertos linfomas. No es infrecuente el desarrollo de una encefalopatía progresiva inducida por el VIH, que consigue infectar células de la microglía.
La progresión de la infección a SIDA es más rápida en los pacientes inicialmente sintomáticos y se asocia a diversos factores de la infección, como la mayor gravedad de la sintomatología en la infección aguda, un mayor descenso inicial del número de linfocitos CD4+ (sobre todo si es < a 500 células/µL), nivel de la carga viral plasmática basal (si es > 100.000 copias/mL), la cuantía de ADN proviral inicial, la infección por más de un virus VIH-1 y el perfil genético de los individuos infectados. En ausencia de tratamiento, se estima que la mediana de supervivencia de los pacientes con infección por VIH avanzada (recuento de linfocitos CD4+ < 50 células/µL) es de 12 a 18 meses, y el tiempo medio de evolución natural transcurrido entre la primoinfección y la muerte del paciente se sitúa sobre los 11 años.
Según los datos publicados por la OMS en 2018, referentes a las cifras epidemiológicas del año 2017, en todo el mundo viven infectadas con el virus VIH un total de 36,9 millones de personas de todos los rangos de edades (incluyendo 1,8 millones de niños menores de 15 años). Casi tres cuartas partes de los casos se ubican en el continente africano (25,7 millones), especialmente en África Subsahariana, región en que la tasa media de prevalencia del VIH se aproxima al 4,7% y alcanza el 25% en algunos países, mientras que en Europa viven “solo” 2,3 millones de infectados, lo que evidencia la desigualdad en la prevalencia de la infección por zonas geográficas. El Caribe tiene la segunda tasa de prevalencia de VIH más alta del mundo, donde más del 50% de población infectada se encuentra en Haití. La prevalencia general en Asia es baja (con una tasa cercana al 0,6%), aunque en el Sudeste Asiático se estiman unos 3,5 millones de personas infectadas, de los que más de la mitad de casos se localizan en la India. El total de personas en el mundo que han contraído el virus desde el inicio de la epidemia se sitúa en 77,3 millones.
Sin embargo, la epidemia se ha estabilizado o está mostrando signos de recesión. Así, la incidencia de la infección continúa en descenso, con un significativo descenso desde los 3,4 millones de nuevos casos a nivel global en 1996 hasta las 1,8 millones de nuevas infecciones en 2017. La mayoría de nuevos casos se concentran en las poblaciones de riesgo: mujeres en África, hombres homosexuales, prostitutas o drogadictos. Es importante destacar que el número de muertes anuales debidas a enfermedades relacionadas con el SIDA también se ha reducido en más de la mitad, desde el pico de 1,9 millones en 2004 hasta las 940.000 defunciones en 2017. En el registro histórico, este dato eleva a 35,4 millones las muertes producidas por enfermedades relacionadas con el SIDA desde el comienzo de la epidemia.
El informe de Vigilancia Epidemiológica del VIH y SIDA publicado en 2017 (datos del año 2016) indica que, en España, viven entre 140/145.000 personas con VIH, de las cuales alrededor del 18% desconocen que están infectadas por el virus. En 2016, se diagnosticaron 3.353 nuevos casos (tasa de 8,6 por 100.000 habitantes); el 34% de los nuevos diagnósticos se realizó en personas nacidas fuera de España. El 83% de nuevos casos eran hombres y la mediana de edad fue de 35 años, el 46% presentaba diagnóstico tardío (cuando hay presencia de una cifra <350 células CD4+/μL); la vía de transmisión más frecuente fue la sexual en hombres homosexuales. Desde el inicio de la epidemia en España en 1981, se han notificado un total de 86.663 casos de SIDA. Tras alcanzar su cénit a mediados de la década de los 90, el número de casos notificados ha experimentado un progresivo declive hasta 2016 por la generalización de la terapia antirretroviral. Sin embargo, la proporción de casos de SIDA en personas cuyo país de origen no es España ha ido subiendo progresivamente, afectando principalmente a personas originarias de Latinoamérica y África Subsahariana.
Mientras no se disponga de fármacos con capacidad para erradicar definitivamente el VIH (como ha ocurrido ya con el virus de la hepatitis C), el objetivo fundamental del TAR es suprimir la replicación del VIH para mantener la carga viral plasmática (CVP) en niveles indetectables –el estándar terapéutico es una CVP <50 copias de ARN/mL– durante el máximo tiempo posible. Así se persigue restaurar la función inmunitaria y limitar el desarrollo de resistencias virales, a fin de reducir la morbi-mortalidad asociada y mejorar la calidad de vida del paciente para que su esperanza de vida se aproxime a la de la población general (lo cual actualmente se consigue), así como en la prevención de la transmisión del VIH.
La última versión del Documento de Consenso GeSIDA/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana (Pérez, 2019) establece, en base a estudios observacionales y grandes ensayos clínicos, que el TAR debe iniciarse en todos los pacientes con infección por VIH-1 confirmada, con o sin sintomatología, independientemente del número de linfocitos T CD4+ y del valor de la CVP, si bien estos parámetros sí deben determinarse previamente. El inicio precoz del TAR se relaciona con una menor frecuencia de transmisión del VIH y de nuevas infecciones, y de manifestaciones clínicas debidas a infecciones o procesos tumorales. Es fundamental considerar que, una vez iniciado, el TAR debe administrarse por tiempo indefinido, reevaluando periódicamente (dependiendo de comorbilidades, riesgo de interacciones y resistencias, mala adherencia, etc.) la pauta farmacológica administrada a partir de los 6 meses, cuando la infección pasa a ser crónica.
Los fármacos antirretrovirales disponibles actualmente son:
A día de hoy, el TAR de inicio de elección se basa en combinaciones de, al menos, tres fármacos (Tabla 1). El uso combinado de fármacos limita la supervivencia de cepas resistente, pero su uso inadecuado puede limitar el uso de algunas alternativas. Aunque se han estudiado combinaciones eficaces con menor número de fármacos, las autoridades sanitarias no las han autorizado aún como TAR de inicio.
Para seleccionar una u otra familia de fármacos se valorarán las ventajas que aportan: el potencial de interacciones farmacológicas (menor en los INI, seguido de ITINN y mayor en los IP), la mayor barrera genética frente a resistencias (en los IP), y el menor coste (de los ITINN). Una pauta con dos ITIAN (preferentemente tenofovir/emtricitabina) y un INI (raltegravir) puede tener la ventaja de una mayor concentración en las secreciones genitales para reducir más rápidamente la CVP durante las primeras 4-8 semanas en comparación con los IP o ITINN, facilitando la reducción de los contagios por vía sexual. Si no se dispone del resultado del estudio de mutaciones de resistencia es preferible comenzar con una pauta basada en un IP potenciado (que presentan actividad frente a la mayoría de cepas VIH-1) hasta tener los resultados. No se recomiendan como tratamientos de inicio pautas libres de ITIAN ni monoterapia con IP potenciados.
Las combinaciones de tres fármacos (2 ITIAN + 1 INI o 1 ITINN o 1 IP potenciado) previamente indicadas constituyen también el tratamiento de inicio de elección de la infección crónica por VIH, referida al período de más de seis meses tras la primoinfección. En este período de la infección, rige igualmente la recomendación principal de iniciar el tratamiento en todos los pacientes diagnosticados de VIH-1, presenten o no sintomatología, e independientemente del recuento de linfocitos T CD4+ y la determinación de CVP.
Debe tenerse presente que la aparición de resistencias es un fenómeno inevitable y su detección por métodos genotípicos es muy útil en casos de fracaso virológico, orientando a la prescripción de terapias de rescate. Diversas circunstancias, incluyendo el fracaso virológico (motivado en muchos casos por la acumulación de resistencias virales), obligan a un cambio proactivo del TAR, si bien no se recomienda suspender el TAR en pacientes con fracaso virológico avanzado y sin opciones terapéuticas de rescate (Fernández-Moriano, 2018).
Se trata de una combinación de tres agentes antirretrovirales, dos inhibidores de la transcriptasa inversa (emtricitabina y tenofovir alafenamida) y un nuevo inhibidor de la integrasa (bictegravir), coformulados en un mismo medicamento que ha sido autorizado para el tratamiento de adultos infectados por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) sin resistencia viral actual o previa a los inhibidores de la integrasa, a emtricitabina o a tenofovir. Esta combinación se utiliza con el fin de reducir drásticamente la carga viral y frenar el deterioro del sistema inmunitario de los pacientes y la incidencia de infecciones oportunistas y otras patologías asociadas al SIDA. Tanto bictegravir como emtricitabina y tenofovir tienen actividad antirretroviral frente a VIH-1 y VIH-2, actuando sinérgicamente frente al VIH-1.
Tenofovir y emtricitabina son dos inhibidores de la transcriptasa inversa análogos de nucleósidos o de nucleótidos (ITIAN), con capacidad de impedir la síntesis de ADN viral a partir de la cadena de ARN infectante. Actúan como finalizadores de cadena: la transcriptasa inversa los incorpora como eslabones en la cadena de ADN en formación, pero es incapaz de unir a ellos el eslabón siguiente; obstaculizan así la incorporación del ADN viral a la dotación genética de la célula infectada. El Documento de Consenso GeSIDA 2019 considera como combinaciones de ITIAN de elección en regímenes de inicio las formadas por emtricitabina/tenofovir alafenamida y por abacavir/lamivudina, que deberían administrase siempre que sea posible en medicamentos coformulados que incluyan varios principios activos.
El tenofovir pertenece a subclase de derivados nucleotídicos (nucleósidos fosforilados) –es un análogo de la adenosina monofosfato (AMP)– y presenta la peculiaridad de acortar el proceso bioquímico intracelular de fosforilación, un paso imprescindible para la activación farmacológica de estos fármacos, además de facilitar su paso a través de las membranas celulares. Tenofovir intracelular se fosforila al metabolito farmacológicamente activo tenofovir difosfato, el cual inhibe la replicación del VIH mediante su incorporación en el ADN viral por la transcriptasa inversa, produciendo la interrupción de la cadena de ADN. Tenofovir muestra actividad frente al VIH-1, VIH-2 y también frente al virus de la hepatitis B (VHB), con un mejor perfil de seguridad que otros ITIAN2.
Más concretamente, el tenofovir alafenamida es la formulación de tenofovir de más moderno desarrollo clínico y requiere dosis menores que tenofovir disoproxilo fumarato porque se concentra selectivamente como fármaco activo (es un profármaco de tenofovir difosfato) en las células diana. Presenta una mayor estabilidad plasmática, permeabilidad celular y activación intracelular rápida mediante hidrólisis por la catepsina A, aportando una mayor carga de tenofovir en las células mononucleares de sangre periférica (incluyendo linfocitos y otras células diana del VIH) y en macrófagos. Ha demostrado eficacia superior a tenofovir disoproxilo en tratamiento combinado tras 3 años de seguimiento, con menor riesgo de nefrotoxicidad y afectación de la densidad mineral ósea, por lo que es preferible en pacientes con alteración renal u osteopenia (Arribas, 2017).
La emtricitabina pertenece a subclase de derivados nucleosídicos de los ITIAN y presenta un marcado parecido estructural, farmacológico y clínico con la lamivudina (aunque solo requiere una única administración diaria, frente a las dos que supone normalmente lamivudina). Emtricitabina se fosforila por enzimas intracelulares para formar emtricitabina trifosfato, que es el compuesto que se incorpora al ADN viral (compitiendo con la desoxicitidina-5’-trifosfato celular) e inhibe la replicación del virus mediante la transcriptasa inversa, ocasionando la finalización de la cadena de ADN. Emtricitabina también muestra actividad frente al VIH-1, VIH-2 y VHB. Es un fármaco de elección entre los fármacos ITIAN, por poseer una toxicidad comparativamente baja, siendo, en cambio, muy susceptible a la resistencia viral. Se han observado resistencias a la emtricitabina y al tenofovir debido al desarrollo de mutaciones que expresan sustituciones de la transcriptasa inversa en las posiciones 184 (M184V/I), o al tenofovir por sustitución en la 65 (K65R).
Por último, bictegravir es un nuevo fármaco inhibidor la integrasa del VIH (INI), que se une al sitio activo del enzima y bloquea la transferencia de hebras3 para la integración del ADN retroviral, que es esencial para el ciclo de replicación del VIH. Bictegravir muestra actividad frente al VIH-1 y al VIH-2. En ensayos bioquímicos bictegravir ha demostrado su capacidad de inhibir la actividad de transferencia de hebras con una concentración inhibitoria para el 50% (IC50) de 7,5 ± 0,3 nM, comparable a dolutegravir (IC50: 7,4 nM) y con mayor potencia que elvitegravir (IC50: 8,8 nM); bictegravir también es capaz de inhibir el paso de procesamiento en 3´, aunque de forma mucho menos potente (IC50: 241 ± 51 nM). En estudios in vitro en modelos celulares, se demostró por técnicas de PCR que los niveles de las formas circulares de ADN viral no integrado eran aproximadamente 5 veces mayores en presencia de bictegravir (en comparación con células no tratadas); correspondientemente, la cantidad del ADN del VIH-1 integrada se vio reducida unas 100 veces.
De manera interesante, cabe destacar que los resultados de ensayos in vivo e in vitro apuntan a que bictegravir tiene un perfil de resistencias mejorado respecto a otros inhibidores de la integrasa. En un screening frente a un conjunto de 8 virus resistentes a INI, que incluyen mutantes simples y dobles de las cuatro vías principales de resistencia a raltegravir y elvitegravir (E92, Y143, Q148 y N155) –mutaciones genéticas que conducen a cambios en la secuencia peptídica de la integrasa principalmente por sustitución de un aminoácido–, demostró que bictegravir era más activo frente a ellos. Por ejemplo, el mutante Q148H/G140S, que se ha documentado en numerosos casos de fallos de regímenes basados en INI (tanto en primera línea como en segunda línea), fue al menos dos veces más susceptible a bictegravir que a dolutegravir (un cambio de 4,8 veces en la EC50). Los ensayos clínicos que se comentarán más adelante no hicieron sino confirmar la ausencia de resistencias a la coformulación de antirretrovirales que incluye el bictegravir, independientemente del subtipo de VIH (Pham, 2019).
Figura 4. Fórmula química de los tres principios activos incluidos en el medicamento Biktarvy®.
El tenofovir alafenamida (TAF) fumarato es un inhibidor de la transcriptasa inversa análogo de nuleótido, que se presenta en forma de su sal de fumarato. TAF es un profármaco tipo éster de tenofovir, siendo un análogo de 2’-desoxiadenosina monofosfato (Figura 4). El nombre químico es {9-[(R)-2-[[(S)[[(S)-1-isoproproxicarbonil-etil]-amino]-fenoxifosfinil]-metoxi]-propil]-adenina}, y tiene una fórmula empírica de C21H29O5N6P · C4H4O4 y un peso molecular de 534,5 g/mol (el peso molecular de tenofovir alafenamida sin la molécula de fumarato es de 476,46 g/mol). Su solubilidad en agua es de 4,7 mg/mL.
La emtricitabina es un inhibidor de la transcriptasa inversa análogo nucleósido de la 2’-desoxicitidina que presenta un átomo de flúor en la posición 5’ (Figura 4). Su nombre químico es 5-fluoro-1-(2R, 5S)-[2-(hidroximetil)-1,3-oxatiolan-5-il] citosina y su fórmula molecular es C8H10FN3O3S con un peso molecular de 247,2 g/mol. Se presenta como un polvo blanquecino con una solubilidad de aproximadamente 112 mg/mL en agua a 25 °C.
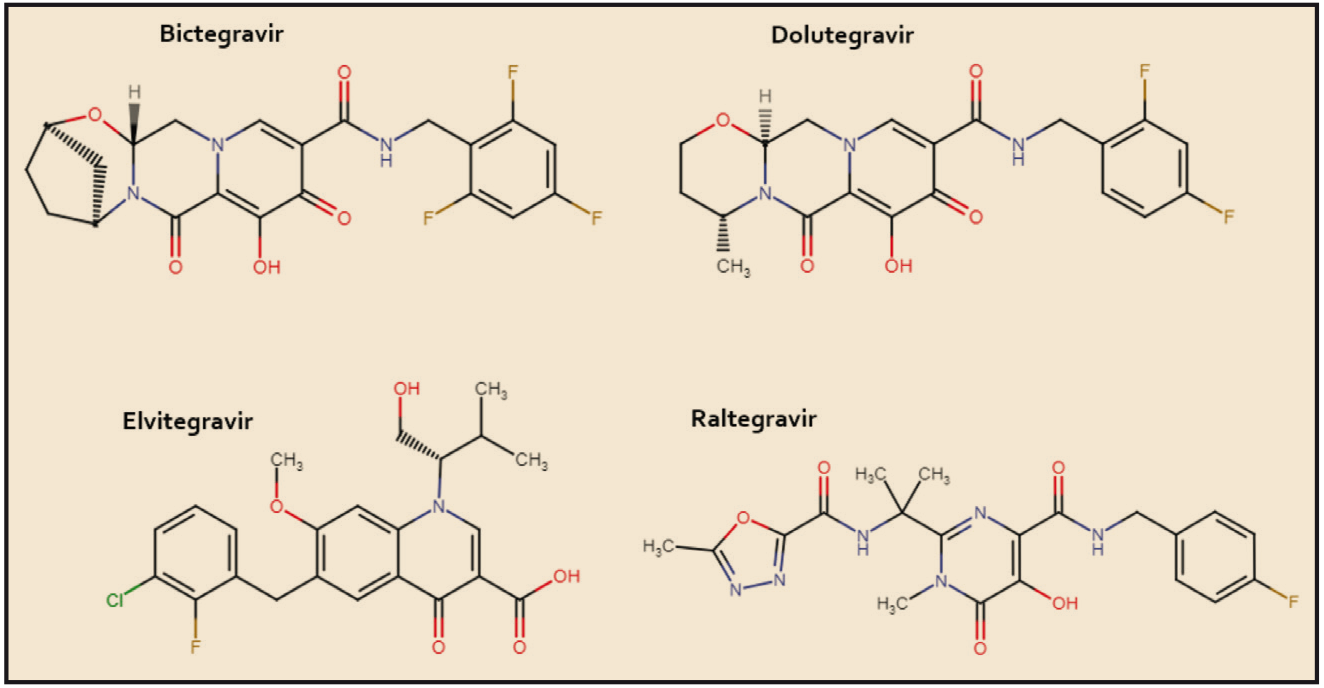
Figura 5. Fórmula química de los cuatro inhibidores de la integrasa del VIH disponibles en
España.
Por último, el bictegravir representa la principal novedad de la combinación de principios activos aquí evaluada. Se trata de un inhibidor de la integrasa de segunda generación, estrechamente relacionado con su predecesor farmacológico directo, el dolutegravir (Figura 5). Como este, presenta una estructura pseudo-peptídica que emula químicamente una fracción proteica y así es capaz de actuar de señuelo bioquímico para bloquear la integrasa del VIH, conduciendo a su bloqueo. Su estructura química sí difiere notablemente con respecto a los otros dos inhibidores de la integrasa disponibles (elvitegravir y raltegravir). Con un nombre químico complejo, bictegravir tiene una fórmula empírica de C21H18F3N3O5, con un peso molecular de 449,38 g/mol; es un polvo blanco-amarillo con una solubilidad de 0,1 g/mL en agua a 20 ºC.
La eficacia y seguridad clínicas de la combinación bictegravir, emtricitabina y tenofovir alafenamida han sido adecuadamente contrastadas en la indicación autorizada en base a datos de seguimiento de 48 semanas de dos amplios ensayos clínicos pivotales de fase 3 (confirmatorios de seguridad y eficacia) aleatorizados y doblemente ciegos con comparador activo en pacientes infectados por el VIH-1 no tratados previamente (naïve), y mediante dos ensayos aleatorizados con comparador activo, uno abierto y otro doblemente ciego, en pacientes virológicamente suprimidos (tratados previamente). Además, en su desarrollo clínico se desarrollaron varios ensayos de fase 1 y otros de soporte de fase 2 que apoyan la evidencia derivada de los ensayos fase 3 para su autorización.
Los dos ensayos pivotales realizados en pacientes sin tratamiento previo fueron multicéntricos, multinacionales, aleatorizados (1:1) y doblemente ciegos. Ambos evaluaron la eficacia y seguridad de bictegravir, emtricitabina y tenofovir alafenamida mediante el empleo de comparador activo: la combinación abacavir/dolutegravir/lamivudina coformulada en una única dosis en el ensayo GS-US-380-1489 (n=629) y la combinación de dolutegravir con emtricitabina/tenofovir alafenamida en el ensayo GS-US-380-1490 (n=645).
Ambos estudios enrolaron pacientes naïve4 para el TAR, infectados con VIH-1 con niveles plasmáticos de ARN viral ≥ 500 copias/mL, libres de infección crónica por el virus de la hepatitis B, negativos para el alelo HLA-B*5701, con una tasa de filtración glomerular ≥50 mL/min y sometidos a un estudio de genotipado para evidenciar sensibilidad a emtricitabina, lamivudina, tenofovir y abacavir. La variable primaria de eficacia fue la consecución de una concentración de ARN viral < 50 copias/mL a la semana 48, mientras que como variables secundarias se fijaron la evaluación de la eficacia (mediante el recuento de copias de ARN/mL y el recuento de células CD4+), seguridad y tolerabilidad de los dos tratamientos en las semanas 48, 96 y 144; el ensayo GS-US-380-1489 también investigó como variable secundaria la densidad mineral ósea de la columna vertebral en esos puntos temporales.
Entre las características basales de la población de pacientes incluida en ambos estudios, bastante equilibradas entre los distintos grupos de tratamiento, destaca: una edad media de 35 años, un 89% hombres, 58% de raza blanca (33% negros y 3% asiáticos), una media del ARN basal del VIH-1 en plasma de 4,4 log10 copias/mL (18% de pacientes tenía una carga viral basal superior a 100.000 copias/mL), predominancia del subtipo B del VIH-1 (en el 11% de pacientes fueron subtipos no-B), media de recuentos basales de células CD4+ de 460 células/mm3 (el 11 % tuvo recuentos basales de células CD4+ <200 células/mm3). En ambos estudios los pacientes se estratificaron en función del ARN viral basal (≤100.000 / >100.000 copias/mL; o ≤400.000 / >400.000 copias/mL), por recuento de células CD4+ (<50, 50-199, o ≥200 células/μl), y por región geográfica (EE.UU. o fuera de EEUU).
Los principales resultados del análisis primario por intención de tratar5 de los datos a la semana 48 se muestran en la siguiente tabla (Tabla 2).
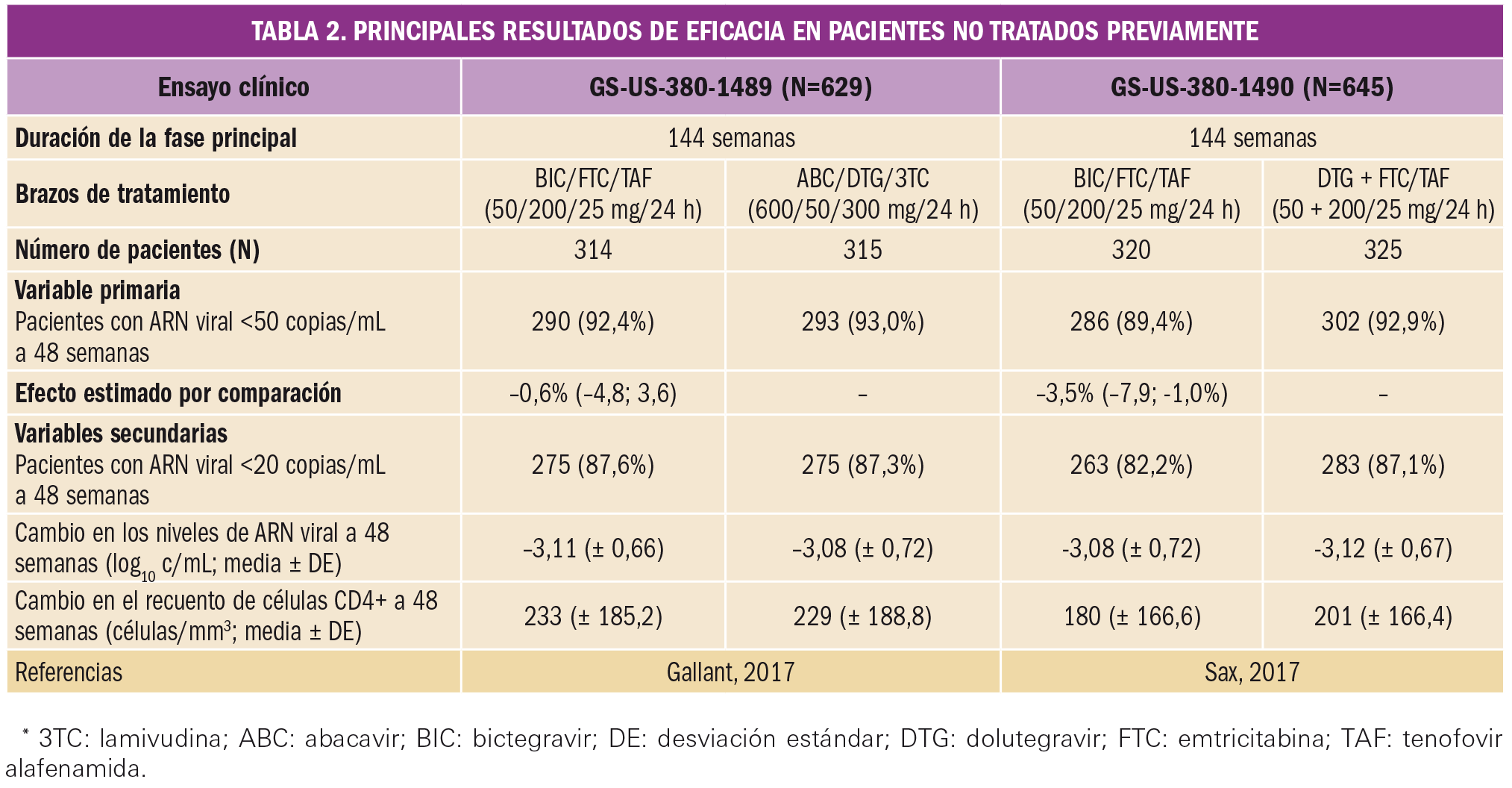
Tales resultados fueron confirmados por el análisis por protocolo, con tasas virológicas de éxito para la asociación bictegravir/emtricitabina/tenofovir alafenamida del 99,3% (vs 98,6% del comparador activo en el ensayo GS-US-380-1489) y del 98,9% (vs 99,7% en el ensayo GS-US-380-1490). Ningún paciente desarrolló, en el periodo de duración del estudio, resistencias a ninguno de los fármacos estudiados. Además, el análisis por subgrupos de pacientes confirmó que los resultados de los distintos brazos de tratamiento eran consistentes e independientes de factores como la edad, el sexo, la raza, la región geográfica, el nivel basal del ARN viral, el recuento basal de células CD4+ y la adherencia terapéutica durante el estudio.
Los dos ensayos pivotales de fase 3 realizados en pacientes con tratamiento previo fueron también multicéntricos, multinacionales y aleatorizados (1:1). Uno de ellos (ensayo GS-US-380-1844; n: 563) evaluó, con un diseño doblemente ciego, la eficacia y seguridad de bictegravir, emtricitabina y tenofovir alafenamida empleando como comparador activo la combinación de abacavir con dolutegravir y lamivudina –coformulados o no–, mientras que el otro ensayo pivotal (ensayo GS-US-380-1878; n=577) fue abierto, e investigó los efectos de la combinación bictegravir, emtricitabina y tenofovir frente a la combinación de atazanavir o darunavir –potenciados con ritonavir o cobicistat– más emtricitabina/tenofovir disoproxilo o más abacavir/lamivudina.
El objetivo primario de estos estudios consistía en evaluar la eficacia a la semana 48 del cambio del tratamiento retroviral previo a una dosis única diaria coformulada de la combinación bictegravir, emtricitabina y tenofovir alafenamida en comparación con la continuación en el tratamiento previo, que posteriormente se emplea como comparador activo en cada uno de los ensayos. Como variable secundaria, ambos estudios investigaron la seguridad y tolerabilidad de los tratamientos a la semana 48; el ensayo GS-US-380-1844 adicionalmente evaluó la densidad mineral ósea de la columna vertebral en ese punto temporal. La principal diferencia entre las poblaciones incluidas en ambos estudios radica en que en el estudio GS-US-380-1844 se excluyeron pacientes con infección crónica con el virus de la hepatitis B, que sí fueron admitidos (igual que aquellos con infección crónica por hepatitis C) en el ensayo GS-US-380-1878.
En la siguiente tabla (Tabla 3) se muestran las características basales más importantes de los dos ensayos clínicos, así como los resultados de eficacia más relevantes surgidos del análisis primario de los datos por intención de tratar.
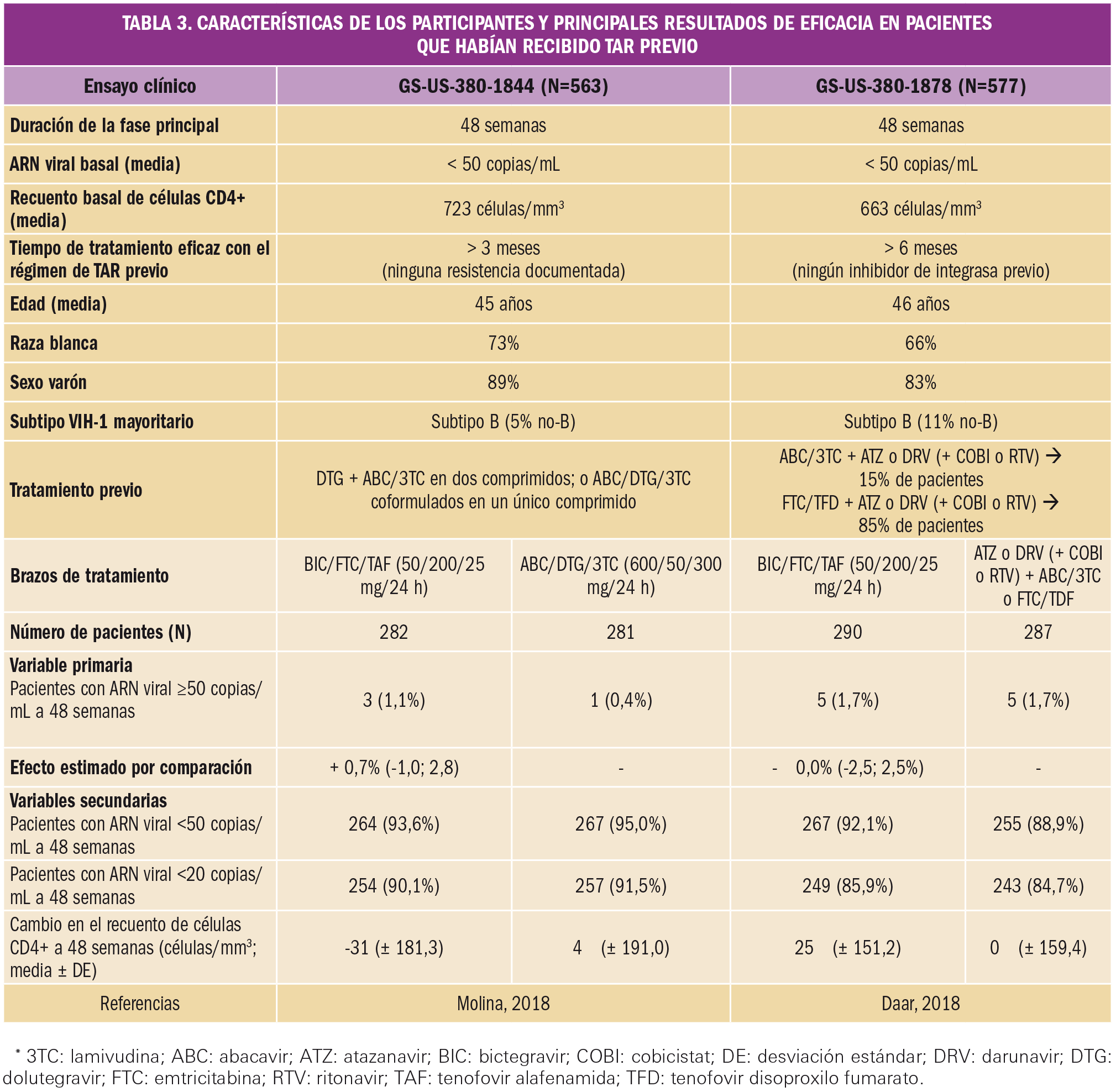
Estos resultados fueron también confirmados por el análisis por protocolo, en que se demostraron tasas de fracaso virológico mínimas para la combinación bictegravir/emtricitabina/tenofovir alafenamida: del 0,4% y 1,1% en los dos ensayos. Cabe destacar que ningún paciente desarrolló resistencias emergentes a tal combinación de fármacos. Los resultados de supresión virológica fueron consistentes en todos los subgrupos de pacientes analizados, sin detectarse diferencias por factores como la edad, el sexo, la raza, la región geográfica y la adherencia terapéutica durante el estudio. El estudio GS US-380-1878 concluyó, de forma interesante, que las tasas de respuesta a la nueva combinación de antirretrovirales eran elevadas y similares independientemente de los inhibidores de la transcriptasa inversa incluidos en el tratamiento previo.
Por otro lado, la seguridad clínica de la combinación bictegravir/emtricitabina/tenofovir alafenamida parece suficientemente definida, en base a un total de 1.511 pacientes que han recibido al menos una dosis de la misma en los estudios de fase 2 y 3, entre los que se incluyen los 1.288 pacientes estudiados en los ensayos pivotales aleatorizados de fase 3, una gran mayoría de los cuales han sido tratados durante al menos 48 semanas. En general, el perfil de eventos adversos relacionados con la administración de la combinación fue muy similar en ambos tipos de pacientes en relación a cualquier evento adverso (tasa de incidencia del 21,9% y del 8,2-18,6% en pacientes naïve y tratados con anterioridad, respectivamente) y eventos adversos graves de grado ≥3 (0,8% y 0,7%, respectivamente).
La incidencia de eventos adversos graves fue muy similar en todos los grupos de tratamiento de los ensayos pivotales, lo que indica que el perfil toxicológico no difiere significativamente del de las otras combinaciones de antirretrovirales incluidas como comparadores activos en los mismos. Es importante reseñar que la nueva combinación de antirretrovirales demostró un perfil de seguridad ósea similar a la combinación abacavir/dolutegravir/lamivudina, típicamente no asociada con toxicidad a nivel óseo; así, tanto en pacientes naïve como virológicamente suprimidos, el cambio desde el estado basal en cuanto a densidad ósea de la cadera o de la columna vertebral fue muy similar entre ambos grupos de tratamiento. El régimen basado en bictegravir demostró, en comparación con lo conocido para regímenes basados en dolutegravir, mayores tasas de elevación de transaminasas e hiperbilirrubinemia, si bien éstas fueron moderadas y transitorias.
En los brazos del tratamiento en desarrollo clínico, las interrupciones prematuras del estudio debidas a eventos adversos fueron en todo caso inferiores al 1%, y solo se describieron 4 muertes en los ensayos pivotales, ninguna de las cuales fue relacionada con el tratamiento por el investigador. Cabe destacar, además, que en los ensayos clínicos de fase 1 en que se evaluó el bictegravir como agente único, así como regímenes formados por bictegravir y emtricitabina/tenofovir alafenamida no se describió ningún caso de muerte emergente por el tratamiento.
Los efectos adversos más comúnmente descritos en pacientes naïve (ensayos clínicos GS-US-380-1489 y GS-US-380-1490; N=634), fueron, en orden descendiente de frecuencia, los siguientes: cefalea (4,6%), diarrea (4,6%), náuseas (4,1%), fatiga (2,5%), mareo (2,1%), insomnio (1,7%), alteraciones del sueño (1,4%), distensión abdominal (1,1%) y estreñimiento (1,1%). Otras reacciones adversas, como flatulencia, vómitos o dispepsia, se reportaron de forma minoritaria (<1%). Un total de 139 pacientes (21,9%) reportó cualquier efecto adverso (EMA, 2018).
Se trata de una combinación de tres agentes antirretrovirales, que incluye dos inhibidores de la transcriptasa inversa (emtricitabina y tenofovir alafenamida) y un nuevo inhibidor de la integrasa (bictegravir), coformulados en un mismo medicamento que ha sido oficialmente autorizado para el tratamiento de adultos infectados por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) sin resistencia viral actual o previa a los inhibidores de la integrasa, a emtricitabina o a tenofovir. Esta combinación se utiliza con el fin de reducir drásticamente la carga viral y frenar el deterioro del sistema inmunitario de los pacientes y la incidencia de infecciones oportunistas y otras patologías asociadas al SIDA. Tanto bictegravir como emtricitabina y tenofovir tienen actividad antirretroviral frente a VIH-1 y VIH-2, actuando sinérgicamente frente al VIH-1.
La eficacia y seguridad clínica de esta asociación de principios activos han sido adecuadamente contrastadas en la indicación autorizada mediante cuatro amplios ensayos pivotales de fase 3 con comparador activo realizados en las poblaciones diana: dos ensayos doblemente ciegos incluyeron pacientes no tratados previamente (naïve) y otros dos ensayos, uno abierto y otro ciego, evaluaron pacientes virológicamente suprimidos por un tratamiento previo. El diseño de los estudios, así como el análisis estadístico, las variables de medida y los criterios de inclusión/exclusión parecen aceptables para aportar una evidencia sólida. De igual modo, la elección como comparador activo de regímenes de dos inhibidores de la transcriptasa inversa más el inhibidor de la integrasa dolutegravir en tres ensayos pivotales es adecuada, pues dolutegravir es el INI de referencia –frente a raltegravir y elvitegravir– y son asociaciones consideradas de elección por las guías clínicas (Tabla 1). La comparación de la nueva combinación frente a un régimen basado en inhibidores de la proteasa potenciados –que suelen ser pautas alternativas– en el restante ensayo pivotal refuerza la robustez de los resultados obtenidos.
Los datos clínicos a 48 semanas son consistentes en los distintos subgrupos de pacientes e indican, por la ausencia de diferencias estadísticamente significativas, que la combinación bictegravir/emtricitabina/tenofovir alafenamida administrada a pacientes naïve es no inferior en términos de eficacia –definida como tasas de supresión viral (<50 copias de ARN viral/mL)– frente a la combinación de abacavir/dolutegravir/lamivudina (92,4% de pacientes vs 93,0%) ni frente a dolutegravir más emtricitabina/tenofovir alafenamida (89,4% vs 92,9%).
Igualmente, ha demostrado esa condición de no inferioridad en pacientes tratados previamente –con independencia del TAR anterior– que cambiaron al nuevo tratamiento, alcanzando tasas de supresión virológica elevadas y muy similares tanto a las conseguidas en pacientes que se mantuvieron en tratamiento con los comparadores activos (93,6% vs 95% de la combinación abacavir/dolutegravir/lamivudina, y 92,1% vs 88,9% de la combinación de atazanavir o darunavir –potenciados con ritonavir o cobicistat– más emtricitabina/tenofovir disoproxilo o más abacavir/lamivudina) como a las obtenidas en pacientes naïve. Además, en estos pacientes, las tasas de fracaso virológico para la combinación incluida en Biktarvy® fueron muy bajas (1,1-1,7%).
Desde el punto de vista toxicológico, el perfil de eventos adversos de la combinación bictegravir/emtricitabina/tenofovir alafenamida ha sido bien caracterizado en base a los datos de los 1.288 pacientes incluidos en los ensayos pivotales. Su seguridad clínica se puede considerar aceptable y similar a la del resto de regímenes de antirretrovirales de primera línea. Las reacciones adversas descritas con más frecuencia han sido cefalea, diarrea y náuseas, que afectaron a más de 4 pacientes por cada 100 tratados. Además, la frecuencia de efectos adversos graves que supusieron una interrupción del tratamiento fue muy baja (<1%). No obstante, estos datos proceden de seguimiento a 48 semanas de tratamiento y, teniendo en cuenta que el número de pacientes con una duración del tratamiento >72 semanas fue muy limitado, la evaluación a largo plazo de la seguridad de esta combinación que contiene el nuevo principio activo bictegravir deberá realizarse es estudios poscomercialización.
En general, se puede considerar que el perfil de seguridad se sitúa muy próximo al de aquellos regímenes que contienen otro inhibidor de la integrasa como dolutegravir, si bien el EPAR de la EMA sugiere que el riesgo de toxicidad hepatobiliar podría ser mayor con bictegravir, sin conclusiones claras al respecto. Por tanto, no habiéndose identificado nuevos riesgos específicos para el régimen con bictegravir, cabe prestar especial atención a los efectos adversos de especial riesgo descritos, en términos cualitativos, para otros inhibidores de la integrasa, como la nefrotoxicidad o los desórdenes psiquiátricos (incluyendo ideaciones suicidas).
Con un perfil farmacocinético y un potencial de interacciones farmacológicas (las más probables a nivel de metabolismo por CYP3A4 y UGT1A1) similar al del resto de regímenes basados en inhibidores de la integrasa e inhibidores de la transcriptasa inversa, entre los aspectos innovadores de mayor interés de la nueva combinación, se puede subrayar el hecho de que ninguno de los pacientes incluidos en alguno de estos estudios desarrolló mutaciones de resistencias a los fármacos utilizados, por lo que se prevé que el perfil favorable de barrera genética de bictegravir sea similar o superior al de dolutegravir. Sin embargo, hay que mencionar que todos los pacientes incluidos en los ensayos debían demostrar ausencia de historial de resistencias a inhibidores de la integrasa, por lo que se plantean incertidumbres sobre la eficacia del fármaco en pacientes que hayan sufrido alguna mutación de resistencia conducente a fracaso virológico con regímenes basados en ese tipo de antirretrovirales; en tal caso, podrían aparecer resistencias cruzadas a bictegravir que hicieran necesario dosis más elevadas del principio activo en la combinación. Se requiere una mayor experiencia clínica que permita aclarar esta cuestión y que podría situarlo también como un régimen preferente en segunda línea como alternativa eficaz en pacientes cuyo virus ha sufrido mutaciones de resistencia a otros inhibidores de la integrasa.
En definitiva, la combinación bictegravir/emtricitabina/tenofovir alafenamida incluida en el medicamento Biktarvy® presenta un favorable balance beneficio/riesgo y emerge como régimen preferente en el tratamiento antirretroviral de inicio o como alternativa tras cambio de TAR (en pacientes virológicamente suprimidos), como así ha sido recogido en las recomendaciones de las principales guías clínicas nacionales (GeSIDA) e internacionales (IAS, DHHS, etc.). Bictegravir, el nuevo fármaco inhibidor de la integrasa, presenta una alta potencia, similar a dolutegravir, con la ventaja frente a otros fármacos del grupo (como elvitegravir) de que no necesita potenciador farmacocinético. Se trata de un régimen de pastilla única, de una administración diaria por vía oral, que, si bien por este hecho aislado no aporta una ventaja adicional en términos de adherencia terapéutica respecto a otras combinaciones coformuladas de primera línea, sí que supone la primera formulación conjunta de un inhibidor de la integrasa no potenciado con los dos inhibidores de la transcriptasa inversa emtricitabina y tenofovir6.
A falta de conocer las consideraciones derivadas del IPT, así como de futuros estudios de coste-efectividad, la combinación bictegravir/emtricitabina/tenofovir alafenamida se posiciona en primera línea como una alternativa más para el tratamiento de personas infectadas con el VIH-1 a los regímenes preferentes de antirretrovirales combinados, con una eficacia similar a éstos, y sin aportar, a priori, otros elementos que supongan una innovación disruptiva.

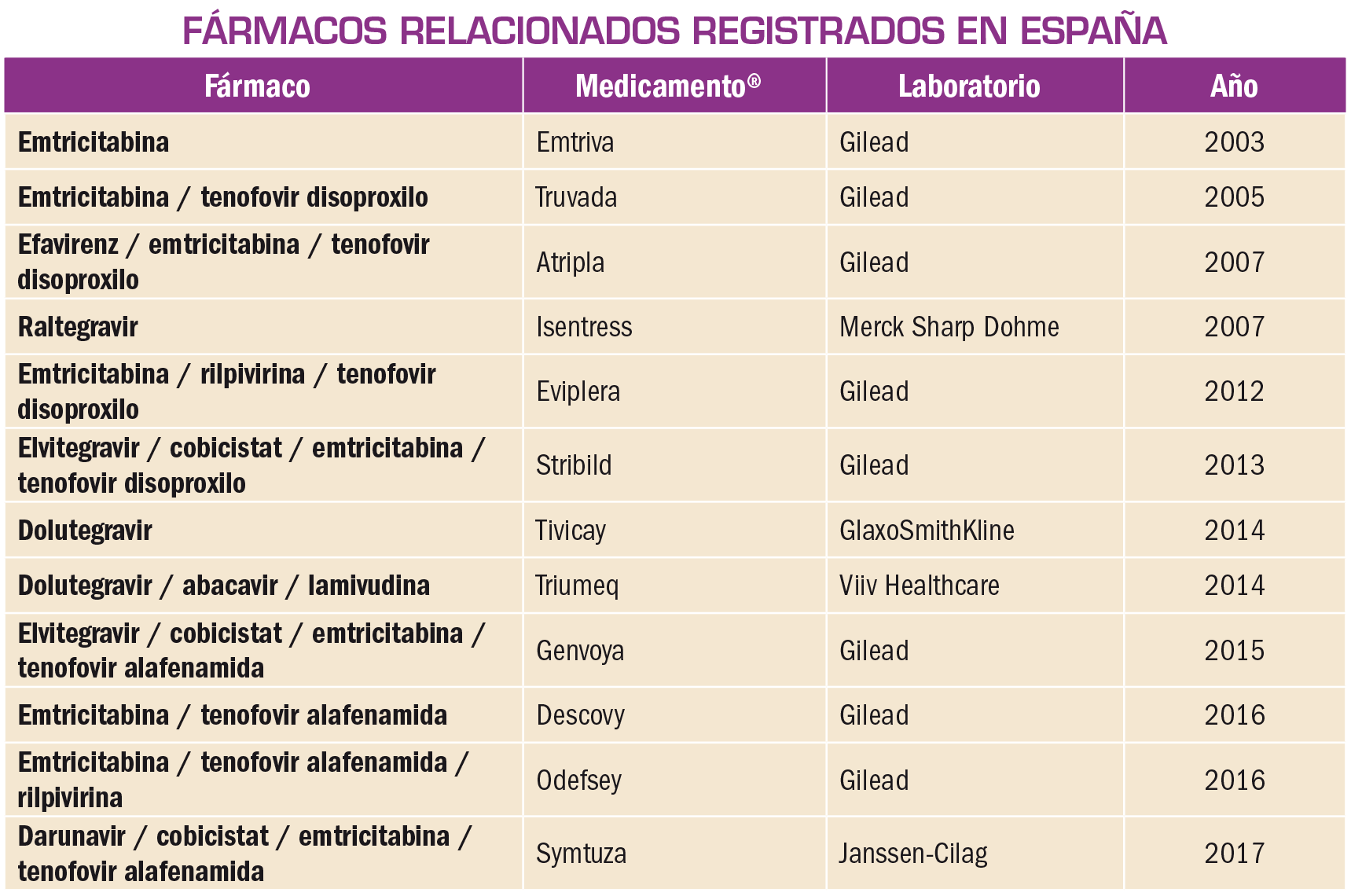
BIBLIOGRAFÍA
Miopatía significa “enfermedad del tejido muscular”. Específicamente, las miopatías constituyen un numeroso grupo de enfermedades que afectan el tejido muscular, tanto a su estructura como a su función, y cuyo síntoma clásico es la debilidad muscular. Las miopatías son enfermedades originadas por distintas causas, lo que determina que cada una se desarrolla con sus peculiaridades y, por lo tanto, se deben tratar de forma diferente.
Pueden aparecer de forma aislada, como una enfermedad primaria, o, pueden constituir una manifestación producto de una enfermedad multisistémica, o sea, que afecte varios órganos. Ocasionan problemas con el tono y la contracción de los músculos del esqueleto (músculos que controlan los movimientos voluntarios). Estos problemas van desde la rigidez (llamada miotonía) hasta la debilidad, con diferentes grados de severidad.
En el campo de las miopatías, el problema se plantea desde un punto de vista práctico para llegar a etiquetar al paciente con debilidad muscular, síntoma fundamental entre los enfermos con esta patología. Obtener un diagnóstico correcto se ve dificultado dado la extensa lista de posibles causas y la escasa especificidad de las pruebas diagnósticas. Una rigurosa historia clínica, la determinación de enzimas musculares, electromiograma y biopsia muscular son fundamentales para establecer el diagnóstico y un tratamiento óptimo.
Las miopatías son trastornos estructurales y funcionales del músculo esquelético, secundarios a una deficiencia o anomalía en los distintos grupos musculares, incluyendo el corazón, que no es sino un músculo muy peculiar. Sus orígenes y, por ende, sus tratamientos son muy diversos, pudiendo ser secundarios a mecanismos como inflamación, alteración genética, metabólica o exposición a tóxicos.
Para que se produzca un correcto funcionamiento de un grupo muscular deben intervenir de manera secuencial y ordenada:
Entre los síntomas cabe destacar la existencia de debilidad muscular, debiéndose diferenciar ésta de la debilidad subjetiva, no propiamente por afectación del músculo, sino secundaria a otras causas como dolor, disfunción articular, alteración sistémica grave. Otros síntomas frecuentes son: fatiga, intolerancia al ejercicio (frecuente en miopatías metabólicas), mialgias, calambres musculares, dificultad para la relajación muscular (miotonía), y orinas oscuras en caso de rabdomiolisis con mioglobinuria.
En este artículo nos centraremos en el primer grupo, es decir en aquellas miopatías con afectación del impulso nervioso y, por tanto, que presentan características de patología neurológica.
Es una enfermedad que podría catalogarse de poco frecuente, afectando en torno a 10.000 personas en total en España. Cada año se diagnostican unos 700 casos nuevos aproximadamente, según la Sociedad Española de Neurología. La prevalencia de la enfermedad ha ido en aumento debido a los avances en su diagnóstico y a la expectativa de una mayor esperanza de vida.
Puede manifestarse a cualquier edad, aunque suele tener dos picos de incidencia: uno temprano en la segunda-tercera década (de predominio femenino) y otro tardío en la octava década (de predominio masculino).
Actualmente se sabe que puede asociarse con otras enfermedades autoinmunes como la neuromielitis óptica, la enfermedad tiroidea autoinmune, la artritis reumatoide o el lupus eritematoso sistémico.
Es la enfermedad autoinmune mejor caracterizada. Está mediada por autoanticuerpos en un 80-90% de los casos, cuya presencia causa debilidad de los músculos esqueléticos al alterar la unión neuromuscular. Los síntomas aparecen principalmente en la adolescencia y la edad adulta, siendo mayoritariamente un trastorno adquirido. La aparición en recién nacidos y en la infancia se conoce como miastenia congénita.
El principal autoanticuerpo causante de la MG es el dirigido contra el receptor de acetilcolina (AChR), aunque recientemente se ha conocido que los autoanticuerpos dirigidos contra la tirosina quinasa del receptor específico de músculo (MuSK) tienen un papel destacado en su desarrollo. Existen diferencias entre las manifestaciones clínicas en función del autoanticuerpo causante.
Los linfocitos T también juegan un papel relevante en la miastenia gravis, ya que pueden atacar al receptor de acetilcolina al estimular la secreción de anticuerpos por parte de las células B. Los autoanticuerpos están presentes en el 80% de los pacientes afectos de MG, pero se conoce una forma seronegativa para ambos anticuerpos (AChR y MuSK), que tiene prácticamente las mismas características clínicas que las formas seropositivas.
A día de hoy se propone que el timo es el origen de los antígenos causantes de la autoinmunidad en la miastenia, al presentar la mayoría de los pacientes seropositivos anormalidades en este órgano. Además, la enfermedad a menudo remite después de la timectomía.
Hay dos formas clínicas de miastenia gravis:
Pese a que más de la mitad de los pacientes presentan inicialmente síntomas oculares como ptosis y/o diplopía, alrededor del 50% de estos desarrollarán la forma generalizada. De manera inversa, muchos pacientes que se presentan sin manifestaciones oculares la desarrollarán con los años.
Respecto a la debilidad o fatiga, esta se caracteriza por el empeoramiento de la fuerza contráctil de los músculos, no por una sensación de cansancio (la presencia de cansancio muscular sin debilidad muscular no es característica de la MG). La característica principal de los pacientes que la sufren es la debilidad fluctuante de los músculos afectados, pudiendo aparecer a lo largo del día, siendo más común al final del día o la noche o después del ejercicio. Al inicio de la enfermedad pueden encontrarse asintomáticos a lo largo del día.
Actualmente se reconocen tres etapas en la enfermedad marcadas por la respuesta al tratamiento, aunque estas se han alterado por la inmunoterapia actual.
Se debe hacer una especial mención a la crisis miasténica, causada por una debilidad bulbar grave que produce disfagia intensa e insuficiencia respiratoria neuromuscular, siendo una condición que amenaza la vida del paciente. Ésta puede aparecer en cualquiera de las tres fases. En ocasiones, se puede presentar como un aumento de la debilidad generalizada, siendo los pródromos de una rápida instauración de insuficiencia respiratoria.
Como se sugirió anteriormente, el timo está íntimamente relacionado con la patogénesis de la MG, pudiendo asociarse la enfermedad con tumores tímicos y otros trastornos autoinmunes. En aquellos pacientes con patología tímica o hiperplasia tímica, el tumor predominante es el timoma, presente hasta en un 15%. Se han descrito formas de tumores tímicos invasivos, así como la asociación de estos con otras neoplasias fuera del órgano (cáncer de pulmón o linfomas). Por tanto, las pruebas de imagen son un componente importante en la evaluación de cualquier paciente afectado de miastenia.
Se basa tanto en las manifestaciones clínicas como en las pruebas serológicas para autoanticuerpos y en los estudios electrofisiológicos.
Otras pruebas incluyen, por ejemplo, la prueba del Tensilon (Edrofronio), que es un fármaco que inhibe la acetilcolina y cuya administración produce una mejoría inmediata y transitoria. Pese a ser fácil de realizar y tener una elevada sensibilidad, presenta limitaciones debido al exceso de resultados falsos positivos. Actualmente se considera un elemento complementario a la exploración neurológica más que una prueba diagnóstica.
La demostración de los anticuerpos AChR y/o MuSK, presentes en aproximadamente el 90% de los pacientes con enfermedad generalizada, proporciona la confirmación de laboratorio para el diagnóstico de miastenia gravis. No obstante, su presencia lo confirma pero su ausencia no excluye el diagnóstico, ya que no son patognomónicos de esta enfermedad. Como se ha mencionado anteriormente, existe una variante seronegativa de la MG, presente en el 6-12% de los pacientes. En esos casos, el diagnóstico se alcanza gracias a la clínica junto con las pruebas electrofisiológicas. Éstos son más propensos a presentar la enfermedad puramente ocular y responden mejor al tratamiento.
En aquellos pacientes con enfermedad puramente ocular, la sensibilidad de las pruebas para determinar los autoanticuerpos es considerablemente menor, ya que solo aparecen aproximadamente en la mitad de los pacientes.
Las pruebas electrofisiológicas se basan en la estimulación nerviosa repetitiva y la electromiografía de fibra única, siendo un complemento importante de las pruebas serológicas para confirmar el diagnóstico. En esta enfermedad, las velocidades de conducción nerviosa son normales, la amplitud del potencial de acción ante un estímulo único es normal, sin embargo la estimulación nerviosa repetitiva produce una respuesta decremental.
Actualmente existen cuatro líneas de tratamiento básicas para la MG:
El tratamiento con inhibidores de la acetilcolinesterasa se considera la primera línea de tratamiento de la MG sintomática, siendo la piridostigmina (Mestinon®) el fármaco de elección. Algunos de los pacientes logran el control de la enfermedad con esta única línea de tratamiento.
Sin embargo, los inhibidores de la acetilcolinesterasa solo proporcionan un alivio sintomático y, por lo general, no son suficientes para el control de la MG generalizada. Cuando éstos no logran su objetivo de manera adecuada o sola aportan una respuesta temporal, se recomienda agregar inmunoterapia al tratamiento. La mayoría de los pacientes necesitarán algún tipo de inmunoterapia.
Los fármacos inmunomoduladores que se utilizan más comúnmente en la miastenia gravis son los glucocorticoides, la azatioprina, la ciclosporina o el micofenolato de mofetilo. En otras circunstancias como la MG de mal control se pueden emplear otros fármacos o técnicas como el rituximab, el tacrolimus, la administración periódica de inmunoglobulinas o la plasmaféresis.
Los pacientes con MG son considerados como pacientes crónicos e inmunodeprimidos. Por ello tienen mayor riesgo de exacerbaciones miasténicas y compromiso vital durante cualquier tipo de infección.
Se debe recomendar la vacunación antigripal anual y la vacuna antineumocócica para todos los pacientes. Las vacunas vivas atenuadas deben evitarse. De la misma forma, se debe pautar un tratamiento antibiótico precoz ante cualquier sospecha de infección.
Hay que tener especial cuidado con la administración de otros medicamentos en estos pacientes, ya que algunos tratamientos de uso habitual pueden exacerbar la enfermedad, pudiendo causar una crisis miasténica. Algunos ejemplos de éstos son: antibióticos (como fluoroquinolonas, clindamicina, vancomicina, telitromicina o los aminoglucósidos), depresores del centro respiratorio (como las benzodiacepinas), o anticonvulsivantes (como la fenitoína o la gabapentina). Una especial mención merece el tratamiento con estatinas, ya que su uso puede enmascarar una miastenia latente o exacerbar una miastenia preexistente debido a la conocida miotoxicidad de las estatinas.
El síndrome miasténico de Lambert-Eaton (LEMS) es una de las enfermedades de la unión neuromuscular probablemente más infradiagnósticada, con una incidencia estimada en 0,5 casos por millón/año y una prevalencia global de 3,4 casos por millón. Se caracteriza por la presencia clínica de debilidad muscular espinal y una evolución fluctuante. Como hecho clínico característico, no compromete la musculatura craneal en su inicio. Se acompaña de síntomas autonómicos, síntomas sensitivos y arreflexia. Es considerado un síndrome neurológico paraneoplásico clásico, pero puede ser idiopático. El tumor más frecuente al que se asocia es la neoplasia pulmonar de células pequeñas, aunque se han registrado casos secundarios a síndromes linfoproliferativos.
Se trata de un defecto de la transmisión neuromuscular sináptica, de tipo presináptico, autoinmune y adquirido, donde la existencia de anticuerpos anti canales voltaje-dependientes de calcio (VGCC) impiden la entrada de este ión a la terminal presináptica axonal. La acción normal del calcio ocurre cuando el potencial de acción de membrana llega a la terminal axónica y genera la apertura de estos canales voltaje-dependientes. El calcio ingresa al axón distal y promueve, con su unión a una serie de proteínas citoplasmáticas, la liberación de acetilcolina. La acetilcolina asegura así la despolarización de la membrana postsináptica muscular (neurotransmisión química). Debemos destacar que la salida de acetilcolina se realiza de dos formas:
La estimulación a alta frecuencia o la contracción voluntaria y sostenida de breve duración permiten una mayor entrada de calcio en la terminal presináptica resolviendo transitoriamente el defecto mencionado.
La debilidad muscular afecta típicamente al sector espinal, inicialmente de predominio en miembros inferiores (proximal y de forma simétrica). No comienza con una afectación craneal, pero estos grupos musculares pueden verse afectados en la evolución y en casos severos. Pacientes con Eaton-Lambert pueden presentar hipo o arreflexia. Una manifestación típica pero presente tan solo en un 40% de los pacientes es la aparición de los reflejos osteotendinosos exaltados y la mejoría de la fuerza muscular postejercicio.
El síndrome no cursa exclusivamente con afectación motora, sino que también pueden aparecer síntomas a nivel autonómico. Los más frecuentes son la sequedad de boca y la disfunción eréctil en el hombre. Otros síntomas menos comunes descritos son: hipotensión ortostática, sequedad ocular, alteraciones en la sudoración, disfunción en la micción y estreñimiento. La exploración clínica y electrofisiológica sensitiva somática es normal. El diagnóstico es difícil, debido a su forma de presentación y evolución lentamente progresiva. Los diagnósticos erróneos incluyen desde miopatías hasta patología degenerativa de columna como la estenosis del canal lumbar.
Cuando la afectación ya compromete el sector craneal, el diagnóstico de Miastenia Gravis suele ser otro de los errores más comunes. Cabe destacar en este último caso, que la estimulación repetitiva a baja frecuencia en ambas patologías demuestra un decremento de potenciales motores, por lo cual su mala interpretación puede colaborar al diagnóstico erróneo si se analiza este hecho exclusivamente. La diferencia radica en que, en la miastenia gravis, los potenciales motores no son de baja amplitud y el perfil de descenso en la transmisión repetitiva a baja frecuencia es diferente, sin anormalidades en la transmisión repetitiva a altas frecuencias, como sí ocurre en el LEMS. Para llegar al diagnóstico de Eaton-Lambert, es útil –pero no imprescindible– la dosificación de anticuerpos VGCC tipo P/Q, si bien éstos pueden encontrarse en otras patologías paraneoplásicas, aunque paradójicamente su presencia no siempre indica un riesgo de neoplasia asociada.
El riesgo de un LEMS paraneoplásico de origen pulmonar aumenta en mayores de 50 años y en hábito tabáquico. El “Delta p-score” es una escala de valoración en pacientes con LEMS que permite determinar la probabilidad de presentar una neoplasia pulmonar asociado al momento del diagnóstico. La prueba de imagen inicial de elección es una tomografía de cuerpo entero y si esta es normal la realización de un PET-FDG. Si la primera evaluación de búsqueda de neoplasia es negativa, deberá realizarse un seguimiento durante los siguientes 5 años, siendo los dos primeros años donde se registra la mayor incidencia de estudios positivos. La frecuencia con que se repetirán dichos estudios podrá sistematizarse mediante el Delta p-score.
Es sintomático y etiopatogénico. Para el control de los síntomas se utiliza la piridostigmina vía oral que promueve la inhibición de la enzima acetilcolinesterasa, o la 3,4-diaminopiridina vía oral que promueve la inhibición de canales de potasio presinápticos, lo cual provoca una mayor liberación de vesículas de acetilcolina. Ambos tratamientos incrementan la disponibilidad de acetilcolina en la terminal postsináptica.
En cuanto al tratamiento etiopatogénico, éste se basa es la eliminación de los anticuerpos a través de inmunoglobulinas o plasmaféresis. También pueden utilizarse inmunosupresores para inhibir su producción. El tratamiento oncoespecífico se realizará una vez detectada la neoplasia en las formas de Eaton-Lambert paraneoplásicas.
La evolución y pronóstico dependerán de si se trata de un síndrome idiopático o paraneoplásico. Las formas severas requieren un tratamiento con inmunosupresores e inmunoglobulinas o plasmaféresis y, en general, se relacionan con formas paraneoplásicas, siendo habitualmente suficiente el tratamiento sintomático en los casos idiopáticos.
Es una intoxicación causada generalmente por la ingesta de alimentos contaminados por las bacteria Clostridium botulinum, la cual produce espora termorresistentes y ampliamente difundidas en el medio ambiente, que en ausencia de oxígeno germinan, crecen y excretan toxinas. Existen siete formas diferentes de toxina botulínica identificadas con las letras de la A a la G. Cuatro de ellas (tipos A, B, E y ocasionalmente el F) pueden causar botulismo humano. Los tipos C, D y E provocan enfermedades en otros mamíferos, aves y peces. La toxina botulínica se ingiere con alimentos elaborados inapropiadamente. Aunque es principalmente una intoxicación de transmisión alimentaria, también puede deberse a infección intestinal por C. botulinum en los lactantes, heridas infectadas e inhalación.
El crecimiento de la bacteria y la formación de toxinas tienen lugar en productos con bajo contenido de oxígeno y no se generan en alimentos cocinados (aunque un pH bajo no degrada ninguna toxina ya existente). Por ello, las combinaciones de baja temperatura de almacenamiento y contenidos de sal, y/o el pH, se utilizan para prevenir el crecimiento de la bacteria o la formación de la toxina.
A pesar de que las esporas de C. botulinum son termorresistentes, la toxina producida por la bacteria – que crece a partir de las esporas en condiciones anaeróbicas – se destruye mediante el hervor (por ejemplo, a una temperatura superior a los 85 ºC durante al menos cinco minutos). Por consiguiente, los casos de botulismo de transmisión alimentaria frecuentemente guardan relación con alimentos listos para el consumo y empaquetados con poco oxígeno.
Esto ocurre mayoritariamente en conservas de alimentos hechas sin las debidas precauciones y en alimentos inapropiadamente procesados, enlatados o embotellados en casa, aunque en ocasiones se ven implicados alimentos elaborados con fines comerciales, tales como: las conservas vegetales con bajo grado de acidez (judías verdes, espinacas, setas y remolachas), pescados (incluido el atún en lata y los pescados fermentados, salados y ahumados), y productos cárnicos (por ejemplo, jamón y salchichas). Las muestras de alimentos vinculados a casos sospechosos se deben obtener inmediatamente, guardar en envases herméticos y enviar al laboratorio para identificar la causa y prevenir otros casos.
Los síntomas iniciales incluyen fatiga intensa, debilidad y vértigo, seguidos generalmente por visión borrosa, sequedad de boca y dificultad para tragar y hablar. También pueden concurrir vómitos, diarrea, e inflamación abdominal. La enfermedad puede dar lugar a debilidad en el cuello y los brazos, y afectar posteriormente a los músculos respiratorios y los músculos de la parte inferior del cuerpo (parálisis flácida descendente). No se presentan síntomas febriles o pérdida de consciencia.
Este cuadro por lo general se manifiesta entre 12 y 36 horas después de la ingesta (con un plazo mínimo de cuatro horas y un máximo de ocho días). La incidencia del botulismo es baja, pero la tasa de mortalidad es alta si no se realiza un diagnóstico precoz y se dispensa el tratamiento adecuado de manera inmediata (pronta administración de antitoxina y atención respiratoria intensiva). La enfermedad puede ser mortal en el 5 a 10% de los casos.
Suele afectar a niños menores de seis meses, cuando ingieren esporas de C. botulinum que germinan como bacterias, colonizan el intestino y liberan toxinas. En la mayoría de los adultos y los niños mayores de seis meses esto no ocurre, porque las defensas naturales del intestino que el propio cuerpo desarrolla con el tiempo impiden dicha germinación y, con ello, el crecimiento bacteriano.
En los lactantes los síntomas clínicos incluyen, pérdida de apetito, debilidad y llanto alterado y una apreciable pérdida del control de la cabeza. Aunque son varias las fuentes posibles de infección de lactantes con botulismo, la miel contaminada con esporas se ha asociado a algunos casos. Por lo tanto, se aconseja a los padres y cuidadores de niños que no alimenten con miel a los lactantes menores de un año.
Es poco usual y se produce cuando las esporas entran en una herida y comienzan a reproducirse en un medio anaeróbico. Los síntomas son similares al botulismo de trasmisión alimentaria, pero pueden tardar hasta dos semanas en aparecer. Esta forma de la enfermedad se ha relacionado con el abuso de sustancias y, especialmente, con la inyección de heroína.
Es muy infrecuente y no se produce naturalmente. Está asociado a sucesos accidentales o intencionales (como el bioterrorismo) que dan lugar a la liberación de las toxinas en aerosoles. El botulismo por inhalación presenta manifestaciones clínicas similares a las del botulismo de trasmisión alimentaria. La dosis letal media para el ser humano se ha estimado en dos nanogramos de toxina por kilo de peso corporal, o sea, aproximadamente, el triple que en los casos de transmisión alimentaria.
Tras la inhalación de la toxina, los síntomas aparecen después de uno a tres días, y ese tiempo es mayor cuando los niveles de intoxicación son más bajos.
En teoría, el botulismo transmitido por el agua puede producirse mediante la ingestión de la toxina. Sin embargo, dado que los procesos habituales de tratamiento del agua (por ejemplo, cocción hasta la ebullición, desinfección con una solución al 0,1% de hipoclorito) destruyen la toxina, el riesgo es considerablemente bajo.
El botulismo de origen desconocido suele afectar a adultos, y en esos casos no es posible determinar si el origen es alimentario o por heridas. Estos casos son comparables al botulismo en los lactantes, y pueden ocurrir cuando la flora intestinal se altera debido a procedimientos quirúrgicos o terapia antibiótica.
Por lo general, el diagnóstico se basa en la historia clínica y el examen físico, seguidos de la confirmación de laboratorio, especialmente para demostrar la presencia de la toxina botulínica en el suero, las heces o los alimentos, o un cultivo de C. botulinum en heces, heridas o alimentos. En ocasiones el botulismo se diagnostica equivocadamente, ya que suele confundirse con accidente cerebrovascular, síndrome de Guillain-Barrè o miastenia gravis.
La antitoxina se debe administrar lo antes posible tras el diagnóstico clínico. La pronta administración es eficaz para reducir las tasas de mortalidad. Algunos casos de botulismo requieren un tratamiento de apoyo, especialmente ventilación mecánica, que puede ser imprescindible durante semanas e incluso meses. Los antibióticos no son necesarios (excepto en casos de botulismo por heridas).
Existe una vacuna contra el botulismo, pero se utiliza en muy pocas ocasiones, dado que su eficacia no se ha evaluado totalmente y se han demostrado efectos secundarios negativos.
La prevención se basa en las buenas prácticas de preparación de los alimentos, en particular el calentamiento/esterilización, y la higiene. La ebullición puede destruir las formas vegetativas de la bacteria, pero las esporas pueden seguir siendo viables tras horas de ebullición, aunque es posible su destrucción con tratamientos a muy altas temperaturas, como el enlatado comercial.
La pasteurización comercial (incluidos los productos pasteurizados envasados al vacío y ahumados en caliente) no siempre es suficiente para acabar con todas las esporas y, por consiguiente, la inocuidad de esos productos se deberá basar en la prevención del crecimiento bacteriano y la producción de toxinas. Las temperaturas de refrigeración combinadas con el contenido de sal y/o las condiciones de acidez impedirán el crecimiento de la bacteria y la formación de toxinas.
Las cinco claves de la OMS para asegurar la inocuidad de los alimentos sirven de base a los programas de formación y capacitación de manipuladores de alimentos e informan a los consumidores. Son particularmente importantes para prevenir esta intoxicación alimentaria las siguientes:
Bibliografía
La hemofilia –más correctamente, las hemofilias– es un trastorno hemorrágico congénito vinculado al cromosoma X, provocado por la deficiencia de la actividad del factor VIII o del factor IX de la coagulación sanguínea, como resultado de mutaciones de los respectivos genes que los codifican. Aunque considerados ambos tipos de hemofilia como enfermedades raras, la ausencia de actividad del factor VIII o hemofilia A provoca una alteración de la hemostasia que afecta a 1-3 varones de cada 10.000 personas a nivel mundial, siendo el déficit de factor IX o hemorragia B menos prevalente (afecta a 1 de cada 30.000 varones). Se trata de una enfermedad de herencia autosómica recesiva ligada al cromosoma X, por lo que en muy raras ocasiones se manifiesta en mujeres (que son portadoras sanas). En España, hay unas 3.000 personas diagnosticadas de hemofilia, si bien se cree que hay un importante infradiagnóstico. En general, en casi un tercio de los casos –mayoritariamente en casos de hemofilia A– aparecen mutaciones de novo que determinan la aparición de autoanticuerpos inhibidores, que van a suponer un hándicap para el tratamiento estándar; es la denominada hemofilia adquirida.
La principal manifestación clínica de la hemofilia es la hemorragia, que puede tener localizaciones muy diversas y una gravedad dependiente del grado de actividad residual del factor de la coagulación afectado. Se manifiesta de forma crónica, debutando generalmente en la infancia, cuando los niños aprenden a caminar. El sello distintivo de la hemofilia es la hemorragia a nivel de articulaciones (hemartrosis), que suele desembocar en la artropatía hemofílica, de gran morbilidad. También puede implicar hemorragias potencialmente mortales, como aquellas a nivel del sistema nervioso central, gastrointestinales o en músculos profundos. El diagnóstico definitivo de hemofilia depende de la cuantificación del nivel de actividad del factor mediante técnicas de biología molecular. Se recomienda también un análisis genético que permita identificar el tipo de mutaciones presente, a fin de cuantificar el riesgo de desarrollo de inhibidores y de identificar posibles portadoras dentro de los grupos familiares.
El tratamiento se ha centrado clásicamente en el reemplazo del factor sanguíneo deficitario, inicialmente con concentrados plasmáticos liofilizados y, en la actualidad, con una mayor tendencia al empleo de factores de coagulación recombinantes, de mayor vida media plasmática y menor riesgo de transmisión de patógenos virales. Con el objetivo de prevenir y tratar las complicaciones derivadas de hemorragias recurrentes, muchas personas reciben una profilaxis a largo plazo mediante la administración intravenosa frecuente de esos concentrados exógenos de factores de coagulación, que permite que los niños hemofílicos tratados desde la infancia puedan albergar una expectativa de vida normal. No obstante, se estima que solo un 25% de los pacientes a nivel mundial tienen acceso a un tratamiento adecuado; en caso contrario, muchos niños mueren antes de convertirse en adultos.
En el presente artículo se abordan aspectos relativos a la fisiopatología de la hemofilia y se revisa en profundidad la farmacoterapia de reemplazo actualmente empleada en la práctica clínica, así como las más modernas opciones de tratamiento planteadas y otros aspectos del abordaje sanitario al paciente hemofílico. El profesional farmacéutico puede realizar una labor asistencial relevante, que permita, entre otros beneficios, promover la adherencia al tratamiento, prevenir e identificar reacciones adversas o interacciones farmacológicas derivadas del mismo, e impulsar una serie de hábitos favorables en la prevención de hemorragias.
Entre los trastornos hereditarios de la coagulación, sin duda alguna es la hemofilia (palabra derivada del griego hemo –sangre–, philos –amado o amigo–, y el sufijo ia –cualidad–) –y más correctamente, las hemofilias– el más conocido popularmente, si bien no es el más prevalente de entre ellos. De hecho, actualmente recibe la calificación de enfermedad rara. A grandes rasgos, se puede definir la hemofilia como un trastorno hemorrágico congénito vinculado al cromosoma X, provocado por la deficiencia de la actividad del factor VIII de coagulación (en el caso de la hemofilia A) o del factor IX (en el caso de la hemofilia B), como resultado de mutaciones de los respectivos genes de los factores de la coagulación.
La hemofilia es conocida –al menos desde el punto de vista descriptivo clínico– desde al menos el siglo II, estando descrita en el Talmud1; no obstante, su conocimiento científico y, desde luego, su tratamiento son mucho más recientes.
Sin duda alguna, fue el caso de la reina Victoria de Gran Bretaña el factor que más contribuyó al crecimiento de la popularidad de la hemofilia. Además de un largo reinado (durante 64 años, entre 1837 y 1901), Victoria tuvo una prolífica descendencia –9 hijos– con su primo el príncipe Alberto de Sajonia-Coburgo-Gotha. Conocida con el apelativo de abuela de Europa, tuvo 42 nietos, la mitad de los cuales siguieron el ejemplo de sus padres y se casaron con miembros de la realeza europea, dando forma a un parentesco que aún hoy conecta a muchas de las monarquías europeas. A pesar de que la reina Victoria no sufrió nunca la enfermedad, ni ésta se había descrito en ninguno de sus antepasados, se cree que en ella se produjo de novo (de forma espontánea) la mutación del gen del factor IX de la coagulación –causa de la hemofilia B– en uno de sus cromosomas X.
Por tanto, su relación principal con la hemofilia se debe a la propagación de su alelo a través de la realeza europea, que tuvo efectos nefastos sobre el destino de algunas monarquías. Uno de sus hijos, Leopoldo, se vio afectado por la enfermedad y murió con apenas 30 años; posteriormente, se comprobó que dos de sus cinco hijas, Alicia y Beatriz, eran también portadoras del gen defectuoso. Los tres contribuyeron a propagar la enfermedad entre los descendientes reales, muchos de los cuales la padecieron clínicamente y murieron por su causa. Entre ellos se encuentran los bisnietos de la reina, Alejo Nikoláyevich de Rusia2, heredero del último zar –y asesinado, junto con el resto de la familia real rusa a manos de los bolcheviques en la Revolución Rusa de julio de 1918–, el Infante de España Alfonso de Borbón y Battenberg y Gonzalo de Borbón y Battenberg (ambos tío-abuelos del Rey Juan Carlos I de España, y afectados a través del linaje de Beatriz).
Según estas consideraciones históricas, puede comprenderse que la hemofilia se haya denominado clásicamente como la “enfermedad de los reyes” (Ingran, 1997).
Para comprender la fisiopatología de la hemofilia, se debe hacer una mención previa al proceso de hemostasia. Grosso modo, se trata de un mecanismo de defensa del organismo para prevenir la hemorragia cuando se produce una lesión vascular, con participación de las plaquetas y de la coagulación. El estímulo que desencadena la activación de la hemostasia es la lesión a nivel del endotelio (barrera natural entre la circulación y el tejido a irrigar) provocando el contacto de la sangre con el tejido conectivo subendotelial. Inicialmente se produce la adhesión y agregación de plaquetas al subendotelio lesionado y, posteriormente, se pone en marcha el mecanismo de la coagulación para formar un coágulo de fibrina.
La respuesta hemostática incluye tres procesos –la hemostasia primaria, la hemostasia secundaria y la fibrinólisis– de forma que siempre existe una interacción entre la pared vascular y la sangre. El proceso se resume en la Figura 1.
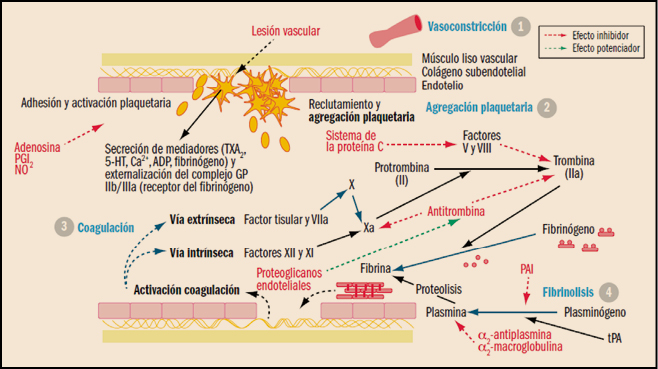
Figura 1. Sistema hemostático. La lesión vascular desencadena una respuesta vasoconstrictora en el músculo liso (1). La exposición de las plaquetas al colágeno subendotelial favorece su adhesión y, junto con factores de coagulación, su activación, que atraerá a nuevas plaquetas (2). La agregación plaquetaria favorece también la fase de coagulación (3) que dará lugar a la formación de la red de fibrina que atrapará elementos celulares formando el coágulo. En rojo, los factores antihemostáticos endógenos. Debe existir un equilibrio entre los factores prohemostáticos y antihemostáticos. 5-HT: serotonina; ADP: adenosín difosfato; NO: óxido nítrico; PAI: inhibidores del factor activador del plasminógeno; PGI2: prostaciclina; tPA: activador tisular del plasminógeno; TXA2: tromboxano A2.
La hemostasia primaria se inicia a los pocos segundos de producirse la lesión, mediante la interacción de las plaquetas y la pared vascular, y tiene una importancia enorme para detener la salida de sangre en los capilares, arteriolas pequeñas y vénulas. Se produce una vasoconstricción derivando la sangre fuera del área lesionada. Las plaquetas se adhieren al vaso lesionado y se agrupan formando el tapón plaquetario. Así se “sella” la lesión de la pared y cede temporalmente la hemorragia. La adhesión plaquetaria a la pared vascular está controlada por el equilibrio entre las prostaglandinas (tromboxano A2 y prostaciclina) y se ve favorecida por diversas sustancias, como el factor de von Willebrand.
La coagulación o hemostasia secundaria resulta de la interacción de proteínas plasmáticas –factores de coagulación– entre sí, que se activan (normalmente están inactivos pero pueden activarse en cuestión de pocos segundos) en una serie de reacciones en cascada conduciendo a la formación de fibrina. Intervienen en el proceso tanto una serie de proteínas procoagulantes (los 12 factores de la coagulación, de síntesis hepática) como otras proteínas anticoagulantes que regulan y controlan la coagulación, evitando que los factores activados en un punto concreto se dispersen y produzcan una coagulación generalizada; los más importantes agentes anti-hemostáticos son: antitrombina III, proteína C y proteína S.
La coagulación se inicia por la exposición al factor tisular debido a la lesión de los tejidos, formándose el complejo factor tisular-factor VII activado (VIIa). Se cree que la activación de los factores IX y X por parte del factor tisular-VIIa desempeña un papel importante en la inducción de la hemostasia. A través de esta interacción, el inhibidor de la vía del factor tisular bloquea el proceso y diversos elementos de la vía intrínseca en particular el factor VIII y IX se convierten en reguladores principales de la formación de trombina (dando lugar previamente al factor Xa que promueve la generación de ésta), enzima activa que convierte el fibrinógeno circulante en fibrina. La fibrina es la responsable última de la formación de la malla en que quedan atrapadas las plaquetas para formar un tapón hemostático (trombo plaquetario), construyéndose finalmente el coágulo o trombo final que detendrá la hemorragia.
Los diferentes pasos de la coagulación se propagan en la superficie celular en presencia de los cofactores plasmáticos unidos a las células. Además, los monocitos y los neutrófilos circulantes interaccionan con las plaquetas y las células endoteliales, iniciándose una serie de uniones que producirán una interacción estable entre leucocitos y plaquetas en el coágulo. Los neutrófilos y los monocitos participan en la reacción inflamatoria local, y los monocitos son también inducidos a expresar el factor tisular, por lo que también contribuyen en la trombogénesis y en el primer nivel de curación de la herida.
Aunque la descripción del mecanismo de la coagulación se pueda dividir en diferentes fases, todas están estrechamente relacionadas entre sí: las plaquetas activadas aceleran la coagulación plasmática y los productos de activación de la coagulación, como la trombina, inducen la activación plaquetaria.
La fibrinolisises el último proceso en el que se elimina la fibrina no necesaria para la hemostasia con la finalidad de la reparación del vaso y el restablecimiento del flujo vascular. Los principales activadores fisiológicos de la fibrinolisis son el activador tisular del plasminógeno (t-AP) y el activador urinario del plasminógeno (u-AP) que difunden desde las células endoteliales y convierten el plasminógeno, absorbido en el coágulo de fibrina, en plasmina. La plasmina es la enzima activa que degrada el polímero de fibrina en pequeños fragmentos o productos de degradación que son eliminados por el sistema de limpieza monocito-macrófago.
Aunque la plasmina también puede degradar el fibrinógeno, la reacción es localizada, debido, en primer lugar, a que el t-AP (y algunas formas del u-AP) activa el plasminógeno de forma más efectiva cuando está absorbido por el coágulo de fibrina; en segundo lugar, porque cualquier molécula de plasmina que pase a la circulación es rápidamente neutralizada por la α2-antiplasmina (el principal inhibidor de la plasmina); y en tercer y último lugar, debido a que las células endoteliales liberan el inhibidor del activador del plasminógeno (IAP) que bloquea directamente la acción del t-AP.
Todo este sistema hemostático se encuentra habitualmente en una situación “de reposo” en el organismo, normalmente inactivo, con un perfecto y delicado equilibrio que favorece la fluidez circulatoria y la oxigenación tisular y en que los fenómenos de coagulación-descoagulación se producen constantemente, siendo capaz de activarse rápidamente tras la aparición de una agresión.
Sin embargo, una alteración de este equilibrio por activación excesiva del sistema hemostático –con formación de fibrina que ocluye la luz vascular– favorece el desarrollo de la trombosis. Por otra parte, una activación deficiente del sistema hemostático consecuencia de una lesión vascular excesiva o del déficit de uno o varios de sus componentes desencadenará un cuadro hemorrágico persistente que puede requerir la administración de hemoderivados. Para prevenir o tratar estas alteraciones se emplean agentes que modulan diversas facetas de la hemostasia. Ante el riesgo de pérdida excesiva de sangre del torrente circulatorio, se suelen administrar concentrados de factores de coagulación –que de forma normal están presentes en sangre como precursores inactivos de enzimas proteolíticas y cofactores– para garantizar el aporte de cantidades funcionalmente suficientes.
La cirugía3 (como cualquier otro procedimiento invasivo que conlleve lesión vascular secundaria), por ejemplo, supone un importante reto para el sistema hemostático, ante el cual la respuesta normal del sistema de la coagulación será pro-coagulante, con la puesta en marcha de los mecanismos antihemorrágicos, inicialmente la hemostasia primaria. Es por ello que en el periodo peri-operatorio se suele pautar de forma protocolizada una profilaxis antitrombótica (basada principalmente en medidas farmacológicas, mecánicas y de movilización precoz) dependiente del tipo de cirugía realizada, la patología del paciente y el período de inmovilización para evitar las complicaciones trombóticas.
Como se ha sugerido anteriormente, la hemofilia se enmarca en el grupo de trastornos de la coagulación o coagulopatías y, dentro de ellas, en el grupo de las coagulopatías hereditarias. Además de éstos, se pueden identificar defectos de la coagulación debidos a deficiencias adquiridas (por la patología del paciente o incluso por fármacos) en la síntesis de factores de la coagulación o debidos a un aumento del consumo de estos factores en la dieta, que no son el objetivo de la presente revisión.
Centrando el foco en los trastornos asociados a una deficiencia congénita en la producción de factores de la coagulación sanguínea, éstos constituyen uno de los ejemplos típicos de las metabolopatías congénitas de mayor impacto, aunque se trata de patologías poco frecuentes. Se estima que cerca de 7 millones de personas viven en el mundo con algún trastorno hereditario de la coagulación, si bien hasta un 75% de los casos permanece sin diagnosticar y no recibe un tratamiento adecuado o incluso no recibe ningún tratamiento. En general, todos ellos tienen una herencia autosómica recesiva (salvo algunos subtipos de la enfermedad de von Willebrand) y clínicamente se expresan con hemorragias de intensidad variable, de mayor gravedad en los casos homocigóticos en los que existe una muy baja concentración de factor de coagulación. Su tratamiento suele implicar la administración de plasma fresco congelado, concentrados del factor deficiente o formas recombinantes de dicho factor.
La más común de las coagulopatías hereditarias –en realidad, la menos infrecuente– es la enfermedad de von Willebrand, un trastorno congénito transmitido autosómicamente, caracterizado por el déficit cualitativo y/o cuantitativo del factor de von Willebrand (factor vW)4. Se ha calculado que en determinadas áreas puede llegar a tener una prevalencia del 1-3% en la población, aunque los pacientes con problemas hemorrágicos graves no son numerosos y casi hasta el 90% de los afectados pueden estar sin diagnosticar. Algunos autores hablan de unos 125 casos graves por cada millón de habitantes.
El resto de las coagulopatías congénitas son mucho más infrecuentes. Además de las hemofilias (que se tratarán en detalle más adelante), cabe destacar el déficit de protrombina (factor II), que ocurre en 1-2 casos por millón de habitantes. La deficiencia completa del factor II parece ser incompatible con la vida, no obstante, en la mayoría de los casos se trata de deficiencias tipo I (disminución del factor II antigénico y funcional), aunque hay descritos casos de disprotrombinemia. Se caracteriza por sangrado después de maniobras invasivas (incluyendo la caída del cordón umbilical), hemartrosis, hematomas musculares y sangrado mucoso.
El déficit de factor VII tiene una incidencia estimada en 1 caso por cada 500.000 habitantes y sus manifestaciones clínicas son similares al déficit de factor II. También el déficit de factor X tiene una frecuencia similar a la deficiencia de factor II, con una clínica parecida al déficit de los otros factores del complejo protrombínico (II o VII).
Por su parte, el déficit de factor XI (también llamado hemofilia C) tiene una prevalencia de 1:500.000. La clínica hemorrágica suele ser moderada o severa (mayor severidad a menores niveles del factor) y el lugar más frecuente son las mucosas, de tal forma que las mujeres con este déficit presentan menorragias abundantes. La deficiencia de factor V tiene una incidencia de un caso por millón de habitantes y se caracteriza por sangrado relativamente leve tras maniobras invasivas y sangrado mucoso.
La incidencia del déficit de factor XIII es también muy baja (1:2.000.000), siendo característica la aparición tardía de las hemorragias en el recién nacido, como por ejemplo, la hemorragia umbilical varias horas o días después de la caída del cordón. También hay que añadir el sangrado intracraneal como un lugar característico de la expresión hemorrágica. La deficiencia de factor XIII se asocia a abortos y/o pérdidas fetales y retraso en la cicatrización de las heridas. Por último, las deficiencias de factor XII o de otras proteínas de la fase de contacto (precalicreína o cininógeno de alto peso molecular) son asintomáticas (Franchini, 2018).
La hemofilia A (hemofilia clásica o común) representa el tipo más prevalente de hemofilia, caracterizada por el déficit del factor VIII de la coagulación, que está implicado en la fase de amplificación de la coagulación (Figura 2). Afecta a entre 1 y 3 varones de cada 10.000 personas a nivel mundial y, por tanto, se puede considerar como una enfermedad rara (definidas así por una prevalencia de < 5 casos por 10.000 habitantes en la Unión Europea), al igual que el resto de la coagulopatías hereditarias, aún menos frecuentes que la hemofilia A salvo la mencionada enfermedad de von Willebrand. Cabe destacar, además, que la enfermedad de von Willebrand implica también un descenso proporcional de factor VIII asociado al de factor von Willebrand, de ahí que contribuya a la expresión y gravedad del cuadro hemorrágico.
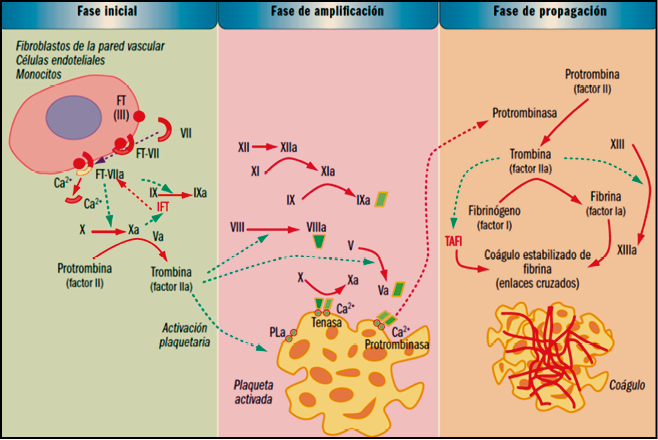
Figura 2. Cascada de la coagulación sanguínea.
Por su parte, la hemofilia B (o enfermedad de Christmas) implica una deficiencia de factor IX y afecta a 1 de cada 30.000 varones. En ambos casos, la deficiencia de estos factores determina un retraso en el proceso de coagulación, con mayor riesgo de hemorragias graves.
Según último informe anual de la Federación Mundial de Hemofilia (FMH)5, en todo el mundo viven con hemofilia confirmada casi 200.000 personas, aunque estos datos proceden únicamente de 116 países, entre los que no se incluye España. Las estimaciones globales de la FMH apuntan a que el total de afectados puede llegar a alcanzar las 400.000 personas en todo el mundo, de las cuales solo 1 de cada cuatro afectados reciben la el tratamiento y la atención sanitaria adecuada (Stonebraker, 2010). En global, el 80-85% de pacientes tiene hemofilia A y el otro 15-20% tiene hemofilia B; la hemofilia puede darse en todos los grupos raciales y sociales.
El último sondeo mundial de la FMH que incluye datos de España corresponde al año 2014. En ese año, había en nuestro país 3.050 pacientes diagnosticados de hemofilia (independientemente del tipo), lo que supone una tasa de prevalencia de 6 pacientes por cada 100.000 habitantes. En España se diagnostican cada año entre 20 y 25 nuevos casos, si bien la tasa de prevalencia se estima estable en torno a 3.000 casos durante los últimos años. El ratio de hemofilia A y hemofilia B es de 6,5:1.
En general, la hemofilia es un trastorno de herencia autosómica recesiva6 ligada al cromosoma X (y, por ello, al sexo de la persona), en cuyo brazo largo se encuentran los genes que codifican para los factores VIII y IX –concretamente, situados en el par 23–. Ello supone que, como norma habitual, la transmiten las mujeres (portadoras) y la padecen los hombres, debido a la dotación de dos cromosomas X de la mujer (XX) y de un único cromosoma X en el hombre (XY). Así pues, el 100% de las hijas de hombres hemofílicos son portadoras y el 50% de los hijos de mujeres portadoras manifiestan la forma clínica de la hemofilia (Figura 3). Aunque la probabilidad es más remota, las mujeres también pueden padecer la hemofilia hereditaria, normalmente con una clínica más leve. Se estima que alrededor del 70% de pacientes hemofílicos tienen un historial familiar positivo para la enfermedad y por cada varón afectado de hemofilia hay una media de 4 portadoras en la familia.
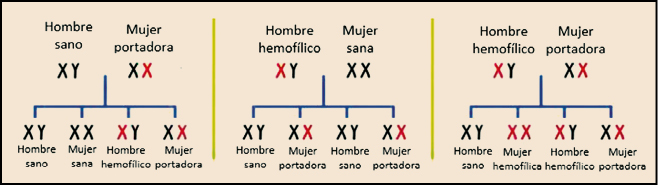
Figura 3. Herencia de la hemofilia. En rojo se representa el cromosoma que porta el alelo mutado de un determinado gen de la hemofilia. Los genes del factor VIII y IX se encuentran en el cromosoma X, de los cuales las mujeres tienen dos alelos y los hombres solo uno. Por tanto, lo más común es que las mujeres lo transmitan siendo portadoras sanas (porque conservan un alelo sano procedente de uno de sus progenitores que las protege de la enfermedad) y los hombres la padezcan (porque cuando aparece el alelo de la enfermedad, es la única copia que tienen para ese gen). Es una enfermedad genética de herencia mendeliana ligada al sexo.
A pesar de ser poco prevalente, la hemofilia (y, especialmente, la hemofilia A) es una de las ocho enfermedades más costosas económicamente, con un consumo de recursos económicos 30 veces superior que un paciente medio. El coste anualizado medio por paciente puede incluso superar los 200.000 euros, solo para la terapia de reemplazamiento de factores. No obstante, son diversos los costes indirectos derivados de la enfermedad, debidos a hospitalizaciones prolongadas, situaciones de emergencia en que no hay disponibilidad de tratamiento adecuado inmediato, cirugía de reemplazo de articulaciones y terapia física para mantener funcionales las articulaciones (O’Hara, 2017).
La causa primaria de las hemofilias A y B hereditarias reside en mutaciones de los genes que codifican para los factores VIII (F8) y IX (F9), respectivamente. Se ha demostrado que el tipo de mutación en los genes influye en la tendencia al sangrado en hemofilia severa: los defectos genéticos menos severos determinarán un fenotipo más leve.
El gen humano del F8 se ubica en la banda más distal (Xq28) del brazo largo del cromosoma X, y es muy largo y complejo: está formado por una secuencia de 186.000 pares de bases (186 kb) –que dan lugar a un RNAm de prácticamente 9 Kb, incluyendo 7.053 nucleótidos codificantes– dividida en 26 exones y 25 intrones. El factor VIII se expresa principalmente en el hígado, en las células endoteliales sinusoidales. La proteína precursora de dicho factor contiene dominios de homología interna y su estructura desde el extremo amino (–NH2) al carboxilo (–COOH) sigue una secuencia A1-A2-B-A3-C1-C2, en la que el dominio B es escindido para formar el factor VIII, que es activado por la trombina.
La hemofilia A puede aparecer como consecuencia de muy diversas mutaciones en el gen F8, de las que aproximadamente un tercio son de novo, es decir, en pacientes sin historial familiar, como puede ocurrir también en la hemofilia B. Entre estas mutaciones, destacan las inversiones (cambio estructural por el cual un segmento cromosómico cambia de sentido dentro del propio cromosoma), las supresionesgrandes –que se asocian con formas clínicas graves de hemofilia y un mayor riesgo de anticuerpos inhibidores–, inserciones, duplicaciones y reordenamientos cromosómicos. Las mutaciones puntuales, deleciones o inserciones pequeñas y mutaciones sin sentido están vinculados a formas clínicas específicas, de menor severidad.
La inversión en la secuencia de ADN que se encuentra en el intrón 22 del gen F8 es la anomalía molecular más frecuentemente detectada en pacientes con hemofilia A grave, siendo responsable de la enfermedad en el 45-50% de los casos; en el 3-5% de casos se halla una inversión en el intrón 1. El otro 50% de hemofílicos con afección grave y la práctica totalidad de hemofílicos con diátesis hemorrágica moderada o leve presentan mutaciones puntuales, como, por ejemplo, los polimorfismos bialélicos HindIII/BclI. Se han descrito más de 2.000 defectos moleculares únicos, tal como se recoge en la base de datos Worldwide Factor VIII Variant Database (Jayandharan, 2012).
Por su parte, el gen humano F9 responsable de la producción de factor IX, está situado en la región subtelomérica (Xq27) del brazo largo del cromosoma X; tiene una extensión de 34 kb y se divide en 8 exones y 7 intrones. El factor IX se sintetiza exclusivamente en el hígado por los hepatocitos y se procesa durante su secreción en el torrente sanguíneo. El polipéptido maduro del factor contiene tres dominios: Gla / péptido de activación / dominio catalítico. El péptido de activación es liberado durante la conversión del factor IX activado, que es modificado mediante hidroxilación y por un proceso de carboxilación dependiente de vitamina K.
A diferencia de lo comentado para la hemofilia A, los defectos genéticos severos se describen solo en un 3% de pacientes con hemofilia B, siendo las mutaciones sin sentido y de empalme (normalmente mutaciones de nucleótido único) –descritas en número superior a 1.000 y distribuidos por toda la longitud del gen– las que representan el principal defecto molecular en esta enfermedad, en más del 70% de los casos. Las deleciones parciales o completas del gen F9 también pueden estar presentes, aunque con mucha menor frecuencia. Por lo general, estos defectos son fáciles de identificar con enzimas de restricción.
La existencia de mutaciones puntuales es, también, responsable de las formas variantes de enfermedad (alteraciones funcionales). Muchos de los pacientes con hemofilia B se clasifican como positivos para el material de reacción cruzada (CRM), caracterizados por presentar una concentración normal de antígeno de factor IX pero una actividad reducida del mismo. Estos pacientes CRM-positivos generalmente tienen mutaciones sin sentido en regiones codificantes o mutaciones de empalme o de transcripción que dan lugar a actividades reducidas de factor IX. No obstante, también hay pacientes CRM negativos: con niveles reducidos de antígeno de factor IX y de actividad; los pacientes CRM-negativos suelen tener mutaciones de diversa tipología. En la hemofilia B –y no tanto en la hemofilia A– hay una intensa correlación entre la presencia de deleciones parciales o completas de genes y el desarrollo de inhibidores (Santagostino, 2013).
Si bien se trata de patologías comúnmente hereditarias, los genes del F8 y del F9 son proclives a nuevas mutaciones espontáneas. Así, por causas que aún se desconocen, algunos pacientes sin antecedentes familiares pueden desarrollar autoanticuerpos circulantes –consecuencia de mutaciones genéticas de novo (no heredadas)– que actúan como inhibidores de los factores de la coagulación en el torrente sanguíneo, suponiendo un gran hándicap para el tratamiento.
La concordancia global sobre la existencia o no de inhibidor en hermanos con hemofilia es del 70%, y hasta de un 90% si son monocigotos. La tasa de inhibidor es de un 50% en los pacientes con historia previa de inhibidor frente a un 15% en los que no la referían. El riesgo de desarrollo de inhibidor es tres veces mayor en pacientes con historia familiar (54% vs 23%). Sin embargo, la concordancia en familiares ya de 2º y 3º grado es mucho más baja. Los pacientes tratados durante al menos cinco días consecutivos en el primer episodio de sangrado o por cirugía, presentan un riesgo de aparición de inhibidor 3,3 veces más alto que los que reciben tratamiento solo uno o dos días consecutivos.
Existe una gran variabilidad en la literatura respecto a la frecuencia de inhibidores. Se ha descrito que los autoanticuerpos aparecen tras las primeras exposiciones al factor (por término medio al cabo de 10-12 días de exposición) y que la prevalencia de inhibidores puede variar del 21 al 88% en pacientes de hemofilia A y del 6 a 60% en pacientes con hemofilia B con defectos severos. Sin embargo, en la estratificación del riesgo de desarrollo de inhibidor, el tratamiento intensivo en la primera exposición resulta el factor de riesgo más importante. En pacientes con hemofilia A en los que la dosificación de factor VIII es < 2 UI/dL, existe correlación entre la edad de la primera exposición al factor y el desarrollo de inhibidor (41% cuando el tratamiento se inició antes de los 6 meses, 29% entre los 6-12 meses y un 12% si se trataron después del año) (Cuéllar, 2013).
De forma mayoritaria, es el factor VIII el que se ve afectado, resultando en una hemofilia A adquirida que puede generar cuadros hemorrágicos graves y, en ocasiones, potencialmente fatales. La incidencia global de aparición de inhibidores en pacientes con hemofilia A grave se encuentra entre un 25% y un 30%, si bien la prevalencia es del 12% dado el carácter transitorio de los anticuerpos en algunos casos. En cuanto a la hemofilia B grave, la incidencia es menor, y solo un 3-5% de los casos se explican por la presencia de inhibidores del factor IX.
Considerada también como una enfermedad rara, algunos autores sugieren que la incidencia de esta enfermedad autoinmune puede estar infraestimada debido a la carencia de registros, a su desconocimiento por parte de muchos especialistas, a la complejidad de su diagnóstico en el laboratorio y, en ocasiones, a una presentación clínica tan fulminante –la mortalidad oscila entre el 9 y el 33%– que impide su confirmación.
Este trastorno hemorrágico adquirido, que supone un problema terapéutico importante (como se verá más adelante), se produce con igual frecuencia en ambos sexos. La prevalencia global de la hemofilia (A) adquirida se estima de 1 a 9 casos por millón de habitantes, aumentando su incidencia con la edad7, desde 0,045 a 14,7 casos/millón/año en niños < 16 años y ancianos > 85 años, respectivamente. La muerte es más frecuente en las primeras semanas después de la manifestación sintomática, por lo que el reconocimiento y el tratamiento inmediato son de vital importancia. Aproximadamente, la mitad de los casos de hemofilia A adquirida son atribuibles a una condición médica subyacente, normalmente otro trastorno autoinmune, cáncer, síndromes linfoproliferativos o una reacción adversa a un fármaco; el resto son de origen idiopático.
Los anticuerpos inhibidores son de tipo inmunoglobulina IgG policlonal –la más común es la de tipo IgG4– que se une a los dominios funcionales del factor VIII e impide la interacción con los factores de la coagulación relacionados. Los inhibidores que reconocen epítopos propios del dominio A2 (aminoácidos 484-508) o A3 (aminoácidos 1811-1818) suelen bloquear de forma directa los lugares de unión de alta afinidad del factor VIII activado con el factor IX activado y el factor X, impidiendo la formación del complejo tenasa. Los inhibidores dirigidos contra el dominio C2 (aminoácidos 2181 y 2243) suelen interferir en la unión del factor VIII a fosfolípidos y al factor von Willebrand.
La propensión al desarrollo de inhibidores involucra elementos del sistema inmunitario, pero también tiene un componente genético importante, relacionado con mutaciones en los genes de los factores de coagulación. Los pacientes con hemofilia hereditaria que tienen un defecto molecular grave (grandes deleciones, inversiones y mutaciones sin sentido) –y ausencia completa de la proteína pro-coagulante– parecen tener una mayor propensión a desarrollar inhibidores en comparación con aquellos con defectos moleculares leves (mutaciones de sentido erróneo o de empalme) en que sí hay cierta cantidad y actividad residual de factor VIII o factor IX. Se considera por ello necesario que a todo paciente diagnosticado de hemofilia se le realice un estudio genético con el fin de identificar la alteración genética responsable de la enfermedad, y poder así determinar el riesgo de desarrollar inhibidor (Mingot-Castellano, 2017).
La técnica más extendida a día de hoy para la cuantificación del título de inhibidor es la llamada técnica Bethesda modificada de Nijmejen. Esta implica la incubación durante 2 horas (37 ºC) de un lote de plasma normal (como fuente de factor VIII) con plasma del paciente sin diluir, y seguidamente se somete a pruebas de factor VIII residual. Una unidad de inhibidor (Unidad Bethesda o UB) se define como la cantidad que destruye la mitad del factor en esa mezcla. El resultado debe ser corregido teniendo en cuenta el deterioro espontáneo del factor VIII, mediante un control consistente en plasma normal incubado con tampón. La división de los pacientes con inhibidores en “respondedor bajo” (cuando la exposición al factor produce poco o ningún aumento del título del inhibidor, siempre < 10 UB) y en “respondedor alto” (cuando la exposición al factor es seguida por un rápido incremento en el título de inhibidor, de > 10 UB), determina el tipo de tratamiento que deben recibir.
La expresión clínica más característica de todas las formas de hemofilia es la hemorragia, que se manifiesta de forma crónica (durante toda la vida del paciente) en múltiples niveles y localizaciones orgánicas: muscular, sistema nervioso central, partes blandas y, muy especialmente, en las articulaciones. En este punto, es importante mencionar que una persona con hemofilia no sangra más rápido que una persona con un sistema de la coagulación normal, pero la duración de sus hemorragias sí es significativamente superior. Generalmente, el “debut” de la enfermedad suele ocurrir cuando los niños afectados aprenden a andar.
Aunque las hemofilia A y B tradicionalmente se consideran indistinguibles con respecto a los síntomas clínicos y, a menudo, se las considera como un trastorno hemorrágico único con presentación clínica similar, algunas evidencias antiguas y más recientes sugieren que la tendencia a la hemorragia asociada con la deficiencia de factor IX (hemofilia B) puede ser menos grave y, por consiguiente, con mejores resultados a largo plazo (Lowe, 2008; Preijers, 2019).
En el pasado, los indicadores comunes y factores pronóstico del fenotipo clínico de los pacientes con hemofilia incluían la edad a la primera hemartrosis, la frecuencia de sangrado, el nivel de consumo de factores de coagulación y las puntuaciones en escalas ortopédicas. Sin embargo, la introducción de la profilaxis continua ha modificado drásticamente la historia natural de la enfermedad.
Actualmente se acepta una clasificación de las hemofilias A y B en tres niveles de severidad, atendiendo al grado de actividad plasmática del factor de la coagulación implicado (Tabla 1). El rango normal de actividad es 50 a 150 UI/dL, es decir, del 50% a 150% de la actividad normal del factor endógeno.

Por lo general, el nivel de severidad –basado en la actividad del factor sanguíneo– de la hemofilia en una persona afectada no cambiará a lo largo de su vida. Sin embargo, el número y tipo de hemorragias sí que podría variar según el estado de salud y la actividad física de la persona, que determinará el riesgo de sufrir heridas por traumatismos o de requerir cirugías. La excepción que confirma esta regla es la llamada hemofilia B Leyden, un subtipo de deficiencia de factor IX debida a una mutación genética específica que sí puede tener una gravedad moderada-severa al nacimiento y normalizarse tras la adolescencia. Además, todos los miembros de una misma familia que padezcan la enfermedad suelen compartir el mismo nivel de actividad del factor sanguíneo, y por tanto, igual grado de severidad de la patología (Srivastava, 2013).
Se estima que en torno al 35-45% de las personas con hemofilia presentan la forma clínica grave o severa, un 10-20% son casos de hemofilia moderada y el resto (aproximadamente otro 40-45% de los casos) presentan la forma leve de la enfermedad. Dado que las mujeres son portadoras, objetivan una actividad del factor VIII o IX de alrededor del 50% y, por ello, no presentan sintomatología o solo síntomas leves.
El problema principal de la hemofilia, más allá del sangrado externo desde una herida o rasguño, son las hemorragias “invisibles” que acontecen en el interior del organismo. La sangre se puede acumularse dentro de espacios en articulaciones, músculos y órganos, pudiendo generar gran daño si no se trata y se repite varias veces.
La hemartrosis o hemorragia articular es la forma de sangrado más frecuente (65-90%), hasta el punto de que constituye el sello distintivo de las hemofilias. En pacientes con hemofilia A o B grave, más del 90% de todos los episodios hemorrágicos se producen en las articulaciones, y el 80% de estos viene representados por hemartrosis de los tobillos, rodillas y codos; es menos habitual en las articulaciones esféricas, como el hombro, la muñeca o la cadera. En cualquier caso, la hemartrosis produce dolor agudo (que puede cronificarse), tumefacción e impotencia funcional.
Por detrás de la hemartrosis, en orden de frecuencia, los hematomas musculares suponen, en algunos pacientes, hasta el 20% de las complicaciones hemorrágicas. Pueden complicarse con síndromes compartimentales (especialmente las hemorragias localizadas en antebrazos o gemelos) e incluso shock hemorrágico; a la larga pueden producir atrofia muscular. Una localización de especial gravedad es el hematoma del músculo psoas-Ilíaco, que puede suponer un riesgo vital. La complicación más grave de la hemofilia es la hemorragia intracraneal; si bien apenas constituye menos del 5% de las complicaciones hemorrágicas, sin un tratamiento rápido puede causar la muerte. Finalmente, los hemofílicos también pueden presentar complicaciones hemorrágicas en otras localizaciones, destacando la hematuria (sangre en orina) y la hemorragia gastrointestinal y orofaríngea.
A priori, las hemorragias pueden acontecer en cualquier localización tras un traumatismo o una lesión y en cualquier momento de la vida de un paciente hemofílico. La incidencia de sangrado en los pacientes con hemofilia durante el período neonatal oscila entre un 20% y un 44%, mientras que la hemorragia intracraneal (HIC) aparece en un 3,5-4% de los neonatos con hemofilia, aunque la cifra podría ser mayor si se incluyen las asintomáticas (Cuéllar, 2013).
Las hemartrosis recurrentes –consideradas como tal cuando se produce sangrado en la misma articulación 4 o más veces en un período de 6 meses– pueden desencadenar un daño progresivo de la articulación y el desarrollo de lo que se conoce como artropatía hemofílica crónica, con gran morbilidad en estos pacientes. Se caracteriza por hipertrofia sinovial, daño en el cartílago, pérdida de espacio articular y modificaciones óseas relevantes (Figura 4). Asimismo, conduce a atrofia muscular, anquilosis, osteoporosis, quistes óseos y, finalmente, la artritis paralizante. Todo ello provoca un notable deterioro de la salud y de la calidad de vida, así como altos costes derivados de la hospitalización y las necesarias intervenciones ortopédicas para aliviar el dolor y mejorar la función articular (en los casos más graves, implican implantación de prótesis).
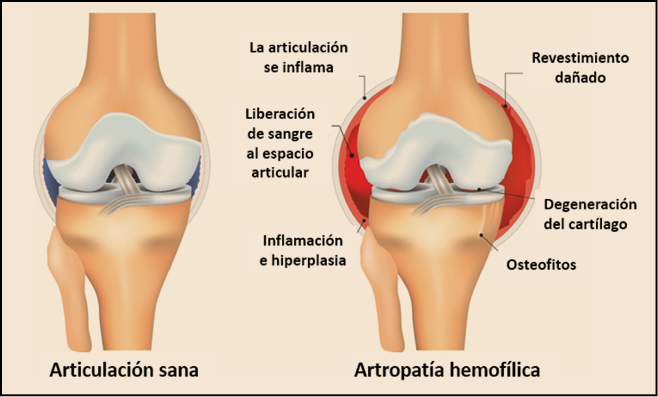
Figura 4. Artropatía hemofílica.
Tras un episodio aislado de hemorragia articular, la sangre en la cavidad de la articulación es reabsorbida gradualmente por el tejido sinovial durante 3-4 semanas, sin que ello deje secuelas. Sin embargo, cuando el sangrado articular es recurrente se produce un deterioro de la membrana sinovial debido a la acumulación de hierro ligado a la degradación de los glóbulos rojos. Este deterioro se manifiesta como hipertrofia e hiperplasia sinovial, así como por la proliferación y persistencia de células inflamatorias; en definitiva, la inflamación crónica provoca la liberación de enzimas líticas destructoras de tejidos y citocinas que contribuyen al daño progresivo de la articulación.
Por otro lado, la angiogénesis y el desarrollo neovascular son un componente esencial de la hemartrosis recurrente. De forma similar a lo que ocurre con el desarrollo tumoral, la angiogénesis parece ser necesaria para la expansión sinovial en la hemartrosis, y la neovascularización hace que la membrana sinovial gane espesor y desarrolle proyecciones vellosas. En respuesta a la irritación articular, con el fin de aumentar el flujo de sangre para eliminar los productos de degradación, se forma una red de capilares bajo de la membrana sinovial hipertrofiada. Por este motivo, cualquier intento no controlado de rehabilitación de la articulación podría provocar que las vellosidades sinoviales quedasen atrapadas en el espacio articular, pudiendo romperse con el movimiento de las articulaciones –con la consiguiente ruptura de los vasos subsinoviales– y provocar hemorragia.
La aparición de una nueva hemorragia aumentaría la irritación en la membrana sinovial, lo que incrementaría la probabilidad de atrapamiento sinovial posterior y sangrado, dando lugar a un círculo vicioso de sangrado, sinovitis y hemorragias articulares. A medida que la enfermedad articular progresa, se produce la remodelación ósea, y aumenta la actividad de los osteoclastos, lo que resulta en una pérdida de densidad mineral ósea que culmina en la osteoporosis (van Vulpen, 2018).
Mientras que estas hemorragias articulares son una característica muy notable de la hemofilia congénita, cabe destacar que la forma adquirida de la enfermedad se manifiesta comúnmente con púrpura cutánea extensa y otras hemorragias internas.
Se considera fundamental contar con un diagnóstico preciso de hemofilia para poder elaborar un plan de tratamiento adecuado. Se debe sospechar de posible hemofilia en casos de pacientes con antecedentes de: familiares con hemofilia, aparición de hematomas durante la infancia, hemorragias espontaneas sin razón aparente (en especial en las articulaciones, músculos y tejidos blandos) o hemorragia excesiva posterior a un traumatismo o una cirugía.
Ante una hemorragia, lo normal es realizar un screening bioquímico para identificar su causa potencial. Las pruebas de coagulación sanguínea en un paciente con hemofilia –bien sea congénita o adquirida– típicamente revelan un tiempo de tromboplastina parcial activada (TTPA) prolongado (que se corrige administrando plasma normal), mientras que los tiempos de trombina y protrombina son normales, al igual que el recuento y la función plaquetaria. El diagnóstico definitivo dependerá de la cuantificación del factor (y de su actividad) en una muestra sanguínea del paciente –procesada previamente a plasma pobre en plaquetas– para demostrar la deficiencia de factor VIII o factor IX. La cuantificación de estos déficits permitirá calcular el grado de severidad de la hemofilia así como el tratamiento sustitutivo adecuado en caso de traumatismo o intervención quirúrgica.
En la actualidad, las técnicas de biología molecular son esenciales para el diagnóstico del paciente y la detección de familiares enfermos o portadoras. El nivel de actividad plasmática del factor VIII se puede evaluar mediante ensayos de una etapa (basados en el TTPA) y de dos etapas o cromogénicos. Sin embargo, estos ensayos no miden con precisión niveles de factor VIII por debajo de 1 UI/dL, lo cual impide concluir sobre el fenotipo clínico. También pueden emplearse ensayos globales más modernos, como el ensayo de generación de trombina o la tromboelastografía, para medir la actividad del factor VIII en el rango de 0 a 2 UI/dL.
Es importante mencionar que en el 30% de los pacientes con hemofilia A leve o moderada suele producirse una discrepancia entre el ensayo de una etapa y el ensayo de dos etapas o cromogénico, que se ha asociado a varios defectos genéticos. Por ejemplo, los defectos agrupados en los dominios A1-A2-A3 del factor VIII se asocian con una mayor actividad cuando se mide mediante el ensayo de una etapa respecto al uso del ensayo de dos etapas o cromogénico (Croteau, 2018).
En el caso de la hemofilia B, el ensayo de coagulación en una etapa es el método estándar para medir el nivel de actividad del factor IX en plasma. También se pueden usar ensayos cromogénicos, pero aún no se han aprobado procedimientos estandarizados para su uso en laboratorios de hemostasia clínica.
Adicionalmente, se recomienda el análisis genético en todos los pacientes con hemofilia para identificar la mutación causante de la actividad reducida del factor, pues se cree que un defecto genético está implicado en, aproximadamente, el 40% de los casos de formación de inhibidores. De hecho, el mayor riesgo de esa complicación del tratamiento lo comportan las deleciones génicas grandes y las mutaciones sin sentido, mientras que las mutaciones de nucleótido único (que generan un aminoácido diferente y potencial alteración de la función y estructura proteica) y de empalme muestran el riesgo más bajo. Normalmente, se investigan las alteraciones genéticas más comunes en ambos tipos de hemofilia –comentadas anteriormente– mediante técnicas de secuenciación directa tipo Sanger o de secuenciación de próxima generación (next-generation sequencing). Otro posible método para identificar la presencia de deleciones y duplicaciones en los genes del factor VIII o del IX es el ensayo de amplificación de sondas dependiente de ligandos múltiples, una técnica modificada de la reacción en cadena de la polimerasa (Peyvandi, 2016).
Teniendo en consideración que casi el 70% de los pacientes tienen una historia familiar positiva de hemofilia, uno de los objetivos claves en el diagnóstico de la hemofilia es identificar, dentro de los grupos familiares, las probables portadoras. Las pruebas genotípicas son también de elección para el diagnóstico de portadoras que permita identificar la mutación exacta en cada familia. El conocimiento preciso del estado del portador puede llevar a la identificación de las personas afectadas en el momento del nacimiento o antes del diagnóstico prenatal. En aquellos pacientes en las que aparece la enfermedad de novo (hemofilia adquirida) se debe verificar si la madre es portadora o no; si así fuera, se estudiará a otras mujeres, en edad fértil, de dicho grupo familiar para descartar otras posibles portadoras.
El diagnóstico prenatal de la hemofilia A es posible mediante los análisis moleculares de muestras de vellosidades coriónicas, tomadas entre las semanas 11 y 14 de embarazo, o mediante amniocentesis tras la semana 15; también puede realizarse de forma temprana tras el nacimiento con un análisis de sangre del cordón umbilical. Sin embargo, todos los factores dependientes de vitamina K, incluyendo el factor IX, están reducidos en el momento del nacimiento (de forma más prominente en nacidos pre-término), por lo que el diagnóstico de la hemofilia B en el momento del nacimiento puede ser impreciso, y debe confirmarse en torno a los 6-12 meses de vida.
Por último, con respecto a la evaluación objetiva del estado estructural de las articulaciones y la detección de cambios tempranos indicativos de artropatía hemofílica, las técnicas de imagen por ultrasonido y por resonancia magnética nuclear (RMN) han demostrado una utilidad similar. Las imágenes por RMN permiten visualizar la deposición de hemosiderina y son más completas en la descripción de los cambios osteoarticulars, mientras que el análisis por ultrasonido aporta ventajas en cuanto a costes, duración y facilidad de uso, y exime de la necesidad de sedación o inyección de contrastes intraarticulares (Charlebois, 2018).
Hace ya más de 150 años que se conocen los beneficios de la transfusión de sangre para controlar la hemorragia relacionada con la hemofilia, y hace casi un siglo (1923) que Feissly demostró la superioridad de la administración de plasma sobre la de sangre total para este fin. Así, en la década de 1950 y gran parte de la década de 1960, los episodios de sangrado se trataban con plasma fresco congelado.
El punto de origen del tratamiento moderno podría situarse en 1965, cuando Judith Pool identificó la fracción del crioprecipitado en el plasma fresco. A partir de ese momento, se desarrollaron técnicas para separar los factores VIII y IX en amplios grupos de muestras congeladas de plasma de pacientes, que dieron como resultado la comercialización de concentrados liofilizados de los factores VIII y IX. La introducción en la práctica clínica en la década de 1970 de esos concentrados con factores sanguíneos (de administración intravenosa) permitió un tratamiento mucho más eficaz y funcional –pues las trasfusiones sanguíneas no aportaban cantidades suficientes de factores VIII o IX–, así como una notable reducción de la morbilidad y mortalidad asociadas a la hemorragia al iniciarse el tratamiento profiláctico.
Desafortunadamente, esa favorable evolución del tratamiento de restauración o de reemplazo de los factores sanguíneos implicados en las coagulopatías hereditarias sufrió, a principios de la década de los 80 del pasado siglo, un grave revés. Nada menos que tres de cada cuatro pacientes con hemofilia grave acabaron infectados por el virus de la inmunodeficiencia humana (VIH) y prácticamente todos con el de la hepatitis C (VHC), como consecuencia de la transfusión de concentrados de plasma contaminados.
En las décadas siguientes la seguridad de estos productos se convirtió en un objetivo crucial de la investigación farmacológica y derivó en la producción de concentrados no contaminados biológicamente, gracias a la incorporación de dobles sistemas de inactivación viral. Aunque han surgido productos más seguros, el riesgo teórico de contaminación de los hemoderivados con agentes infecciosos conocidos y desconocidos (por ejemplo, con parvovirus B19 y enfermedad asociada a priones, como la variante de Creutzfeldt-Jakob) aún permanece.
Para evitar tal riesgo, se ha desarrollado en las últimas décadas la producción de análogos recombinantes de los factores naturales, carentes por completo de contaminación por virus humanos. El primer producto de proteína recombinante disponible comercialmente apareció en 1992 para la hemofilia A y en 1997 para la hemofilia B.
Desde entonces han surgido diversos fármacos tanto recombinantes como derivados del plasma, pero todos ellos han presentado una importante limitación: su corta vida media (de 8-12 horas para el factor VIII y de 18-24 horas para el factor IX), que implica una elevada frecuencia de administración. Además, la complicación más grave del tratamiento (tanto con medicamentos derivados de plasma humano, como con fármacos recombinantes) se asociaba, y aún hoy se asocia, con el desarrollo de anticuerpos inhibidores que se dirigen contra los factores infundidos. Para salvar ambos obstáculos, se han desarrollado nuevos medicamentos que, o bien han ampliado esa vida media plasmática, o bien actúan por rutas diferentes para evitar o reducir la inmunogenicidad (por ejemplo, aumentando la producción de trombina mediante la inhibición de la actividad anticoagulante natural).
A día de hoy, asumiendo que no se dispone de un tratamiento curativo, los objetivos principales de la terapéutica de la hemofilia son: a) prevenir la hemorragia y, en su caso, tratarla; b) prevenir y tratar sus complicaciones y secuelas, incluyendo la restauración y el mantenimiento de la función articular; y c) integrar a los pacientes en la vida social normal. Aunque aún en la actualidad algunas personas con hemofilia –que no reciben el tratamiento adecuado– mueren antes de convertirse en adultas, si los pacientes hemofílicos adultos reciben un tratamiento apropiado, su expectativa de vida es aproximadamente 10 años menos que la de personas sanas; los niños hemofílicos que son tratados desde la infancia pueden albergar una expectativa de vida normal.
La farmacoterapia estándar de pacientes con hemofilia severa consiste en la administración intravenosa de concentrados exógenos del factor de coagulación sanguínea cuya actividad se encuentra reducida. El origen de estos fármacos puede ser extractivo, si proceden de concentrado plasmático humano purificado, o recombinante, cuando son diseñados por la técnica del ADN recombinante y producidos en sistemas biológicos de líneas celulares diversas (tales como células de riñón de crías de hámster –BHK–, células de ovario de hámster chino –CHO–, o línea celular embrionaria de riñón humano –HEK–). Estos últimos son los de desarrollo más moderno y, quizá, los más empleados y preferidos en la práctica clínica actual, por su mayor disponibilidad y la reducción del riesgo de contaminación biológica.
Los fármacos anti-hemofílicos actualmente disponibles en España se recogen en la siguiente Tabla 2. El mecanismo de acción de todos ellos es sencillo de comprender, pues vienen a sustituir al factor de la coagulación que se encuentra deficitario, ejerciendo las mismas acciones biológicas que éstos desarrollan en la cascada de la coagulación (Figura 2). Así, cuando se perfunde en un paciente hemofílico, el factor VIII se enlaza con el factor de von Willebrand en la circulación del paciente; el factor VIII activado funciona como cofactor para el factor IX activado, acelerando la reacción de activación del factor X. Al administrar por vía intravenosa el factor IX, éste es activado por el factor XIa para posteriormente interactuar con el factor VIII y, a su vez, activar el factor X. Esa activación final del factor X es la que transforma la protrombina a trombina, que convierte el fibrinógeno en fibrina (responsable de la formación del coágulo).
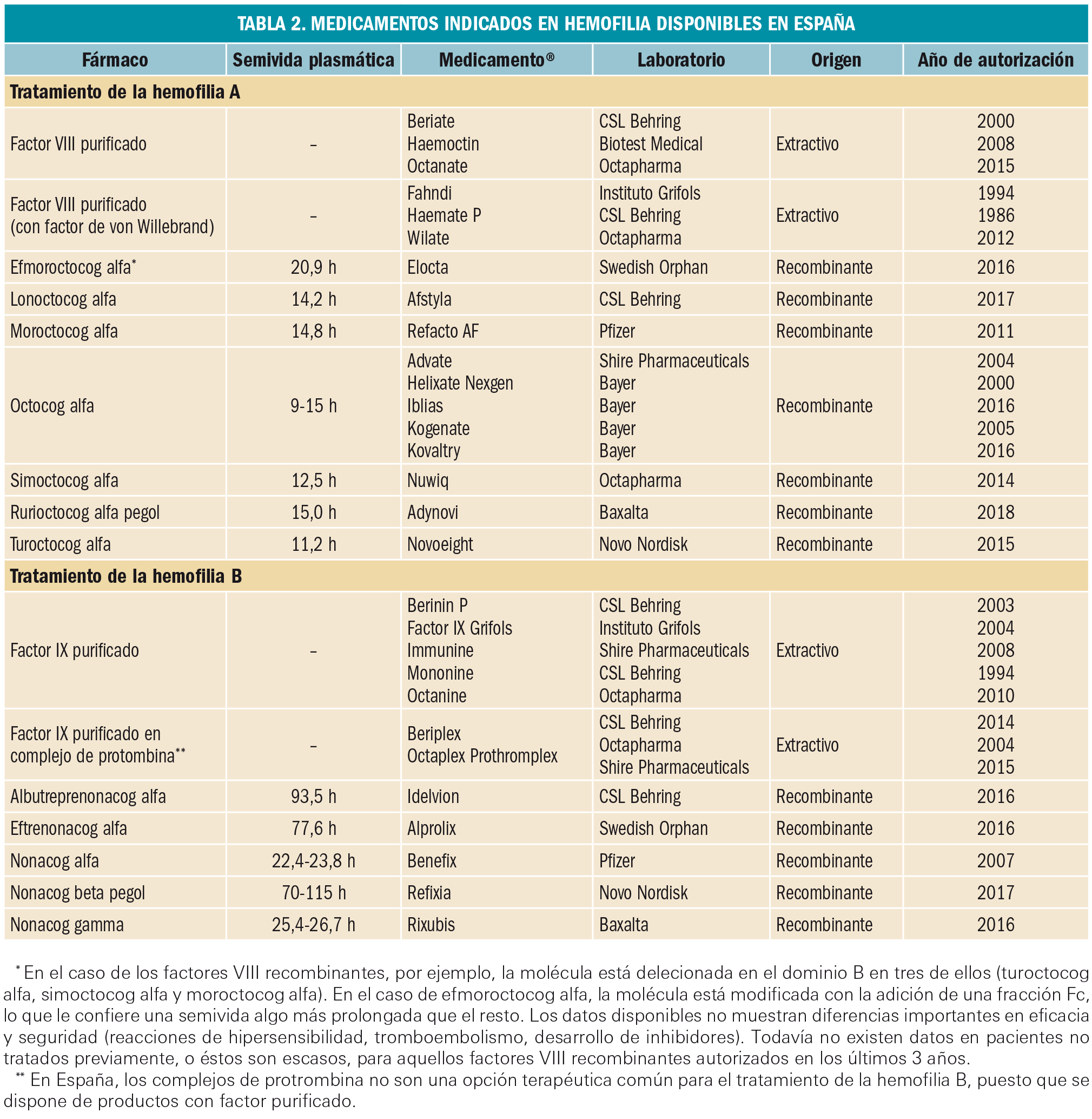
Las pautas de tratamiento (y de profilaxis) deben ser totalmente individualizadas. El cálculo de la cantidad de factor a perfundir debe tener en cuenta el peso del paciente, la intensidad de la gravedad de la hemorragia o clínica hemofílica (dependiente del grado de déficit del factor de coagulación que presente el paciente) y la sensibilidad individual ante una cantidad determinada de estos concentrados. También se debe tener en cuenta que los factores VIII y IX tienen propiedades farmacocinéticas distintas, que se observan incluso al comparar entre las formas de origen extractivo y las formas recombinantes del factor; por ejemplo, el factor IX recombinante muestra in vivo una recuperación de la actividad del factor un 30% menor que aquél factor derivado de plasma.
En episodios hemorrágicos agudos graves que podrían poner en peligro la vida del paciente hemofílico –en especial, en cabeza, cuello, tórax y tracto gastrointestinal–, la terapia con el factor correspondiente es la primera línea de tratamiento. Se recomienda iniciar el tratamiento con factor de inmediato, aun antes de completar la evaluación del diagnóstico e independientemente de que puedan ser espontáneos o debidos a un traumatismo o lesión. Para combatir hemorragias de riesgo vital se estima que se debe alcanzar inicialmente hasta un 100% de factor circulante hasta que el sangrado se detenga. Como niveles de mantenimiento, se suelen buscar unos niveles en torno al 50% de la actividad normal del factor de coagulación.
Ante una hemartrosis, el tratamiento también debe ser lo más precoz posible (preferiblemente antes de 4 horas) y, ante la duda, siempre se debe tratar. En estos casos, así como con el objetivo de profilaxis, el objetivo de factor a conseguir es de un 30-50% (habitualmente 20-40 UI/kg de factor VIII8 y 30-60 UI/kg de factor IX). Los objetivos terapéuticos en cuanto a actividad del factor se recogen en la siguiente Tabla 3.
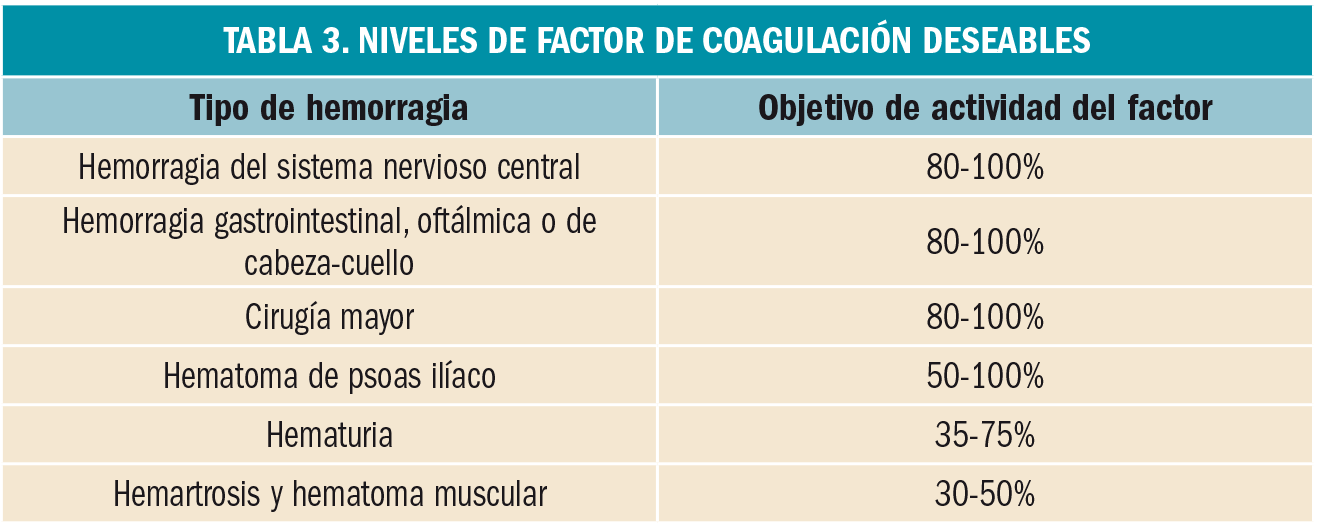
En ocasiones, la dosis requerida se puede determinar mediante la siguiente fórmula: número de UI9 de factor VIII o IX = peso corporal (kg) × incremento deseado de factor (%) × el recíproco de la recuperación observada (1,2 UI/kg en adultos; 1,4 UI/kg en niños <15 años).
Por otro lado, en casos leves de hemofilia A (a veces, incluso en casos moderados), una de las primeras opciones terapéuticas es la administración, por vía endovenosa o intranasal, de desmopresina o 1-desamino-8-D-arginina-vasopresina (Minurin®, Ostostim®, EFG) (Figura 5). Se trata de un análogo sintético de la vasopresina capaz de liberar del endotelio vascular tanto factor de von Willebrand como factor VIII al torrente circulatorio, con aumentos de 3-5 veces su valor basal y una duración de acción de 6-10 horas. Ese aumento de factor VIII puede ser suficiente para normalizar la coagulación y controlar hemorragias leves (o moderadas), como las debidas a procedimientos invasivos menores. La respuesta de cada paciente a desmopresina puede ser muy variable, por lo que se recomienda realizar la prueba de valoración de la respuesta al fármaco. Sus efectos adversos son ligeros, habiéndose descrito enrojecimiento facial, cefalea, hipotensión y taquicardia.
Figura 5. Estructuras químicas de la desmopresina y el ácido tranexámico.
Para las hemorragias mucocutáneas (como hemorragias orales, epistaxis o menorragia) se ha mostrado especialmente útil la administración adicional de ácido tranexámico (Amchafibrin®, EFG). Es un compuesto antifibrinolítico (Figura 5) con un potente efecto inhibidor competitivo sobre la activación de la fibrolisina, que está indicado, por ejemplo, en el tratamiento y profilaxis de hemorragias –asociadas a una fibrinólisis excesiva– en pacientes hemofílicos sometidos a cirugía dental. Como efectos secundarios, destaca el riesgo de complicaciones tromboembólicas o convulsiones.
Existe un caso especial de deficiencia combinada de factor V y factor VIII, que es un trastorno hemorrágico hereditario totalmente independiente de la deficiencia de factor V y de la deficiencia de factor VIII (hemofilia A) y que no parece provocar más hemorragias que las que se presentarían si solo uno u otro de los factores estuviera afectado. En esos casos, los tratamientos disponibles son también los concentrados de factor VIII o el plasma fresco congelado.
Además, en recién nacidos con sospecha de hemofilia se recomienda administrar vitamina K por vía intravenosa a través de los vasos umbilicales. En caso de que no sea posible, otra alternativa es la vía oral, con una dosis de carga de 1 mg, seguida de 25 µg durante varios días.
Grosso modo, en la terapia sustitutiva de los factores de coagulación en hemofilias, la utilización de los medicamentos puede ser a demanda para parar un episodio de sangrado manifiesto y en profilaxis previa a una cirugía, o como profilaxis general con administraciones regulares para prevenir hemorragias. La evolución será mejor en aquellos pacientes que reciban una terapia sustitutiva temprana y adaptada a su evolución clínica (Croteau, 2018; Morfini, 2019).
En estas patologías el tratamiento está especialmente unido a la prevención. De hecho, el régimen de administración más recomendado por expertos a nivel nacional e internacional en la actualidad es el tratamiento profiláctico pautado. Se han demostrado los beneficios de la profilaxis en la prevención del desarrollo de artropatías (que, una vez establecidas, son irreversibles) y en la reducción de sangrados. Los pacientes deben ser tratados con el objetivo de alcanzar y mantener constante un umbral de actividad de factor VIII o de factor IX por encima del 1% (≥1 UI/dL).
Dado que la semivida plasmática de la mayoría de los concentrados de factor VIII es de alrededor de 12 horas, el régimen profiláctico recomendado en pacientes con hemofilia A supone administraciones intravenosas frecuentes: 3 o 4 veces por semana. En el caso de los concentrados de factor IX, sus semividas son más prolongadas (>24 horas) y las administraciones se pueden espaciar cada 3-4 días: aproximadamente dos veces por semana.
Existen diversos protocolos de profilaxis. A nivel mundial, los más aceptados y empleados incluyen el protocolo sueco o de Malmö, el holandés o de Utrecht y el canadiense. El de Malmö, por ejemplo, recomienda que cada una de esas administraciones aporte 25-40 UI/kg, mientras que el de Utrecht establece ese rango entre 15 y 30 UI/kg. En todos los casos, la profilaxis implica una administración frecuente por vía intravenosa, lo cual afecta negativamente a la calidad de vida de los pacientes y podría afectar al grado de adherencia al tratamiento (Moreno, 2019).
De cualquier modo, la profilaxis, definida como la administración regular y a largo plazo –durante meses o años– de los correspondientes factores implicados para prevenir hemorragias articulares, constituye el eje central del manejo de los niños con hemofilia severa. La controversia que surgió en torno a la posibilidad de suspender la profilaxis al llegar el paciente a la edad adulta, fue mitigada por la declaración conjunta de la OMS y la Federación Mundial de la Hemofilia a finales del siglo pasado (Berntorp, 1995): recomiendan que, si es posible, la profilaxis se continúe durante toda la vida.
La profilaxis primaria consiste en la infusión regular de concentrados del factor deficitario, que se mantiene durante más de 46 semanas al año, y es iniciada antes de la aparición de alteraciones articulares. Según las definiciones aceptadas a nivel internacional (Blanchette, 2014), la profilaxis primaria es una terapia continua que comienza después de la primera hemorragia articular y antes de los 3 años. Alternativamente, la profilaxis primaria puede ser un tratamiento continuo iniciado antes de los 3 años en un paciente sin sangrado articular previo, es decir, iniciado en base únicamente a la edad. La profilaxis secundaria y terciaria se iniciaría más tarde en la vida y con diversos grados de artropatía.
Existe una sólida evidencia científica que demuestra que la profilaxis primaria desde una edad temprana protege contra el daño de las articulaciones, permite un desarrollo músculo-esquelético normal y disminuye la frecuencia de hemartrosis y otras hemorragias potencialmente mortales. A pesar de ello, suele generarse debate en torno la utilización sistemática de la profilaxis primaria en niños pequeños debido a su elevado coste económico y otros problemas como la apertura de vías venosas para la infusión de los factores. Así, una encuesta realizada en 2012 en Estados Unidos demostró que solo el 25% de los centros de tratamiento de hemofilia encuestados (62) comenzaron la profilaxis tras el primer episodio de sangrado (de cualquier tipo), y únicamente el 16% comenzó después de la segunda hemorragia; además, cerca de un tercio implantó la profilaxis al principio con infusiones una vez a la semana para evitar la necesidad de establecer una vía venosa central (Simpson, 2012).
A pesar de sus claras ventajas, la profilaxis primaria sigue siendo infrautilizada por los motivos mencionados. No obstante, una parte considerable de la población con hemofilia puede beneficiarse de la profilaxis periódica cuando se inicia después de varios episodios de sangrado comunes. Es lo que se denomina profilaxis secundaria, similar a la primaria, pero que comienza tras dos o más hemorragias en articulaciones mayores, con aparición de cierto grado de lesión articular (se considera que la artropatía no está completamente instaurada).
La implantación general de la profilaxis secundaria en adolescentes o en adultos, además de en niños pequeños, va en aumento. A largo plazo (mantenida durante más de 45 semanas por año), se asocia con una menor incidencia de episodios de hemorragias articulares y retraso de la progresión de daños existentes, facilita la práctica de ejercicio físico y la rehabilitación, y reduce el número de ingresos hospitalarios y el absentismo escolar.
Si el objetivo es simplemente evitar las hemorragias que amenazan la vida y la artropatía incapacitante, en la mayoría de los casos se puede lograr mediante la profilaxis con dosis bajas; varios estudios recientes han mostrado su buen resultado en el número de hemorragias articulares en pacientes hemofílicos en comparación con el tratamiento a demanda. Por ejemplo, un estudio realizado en la India con profilaxis secundaria –se administraron 10 UI/kg dos veces por semana durante casi un año– demostró que se reducía significativamente el número de hemorragias articulares mensuales de 0,48 (±0,37) a 0,08 (±0,13), sin un aumento significativo en el consumo global de concentrados de factor sanguíneo en comparación con el grupo de tratamiento a demanda (Verma, 2016). Sin embargo, si el objetivo es prevenir la artropatía hemofílica y las hemorragias graves, para que los niños o adultos afectados puedan llevar una vida razonablemente normal, se requieren las dosis y frecuencia de infusiones previamente comentadas.
La profilaxis terciaria se definiría como aquella que se inicia después de la aparición de la enfermedad articular para evitar la progresión del daño. En este sentido, nunca es demasiado tarde para comenzar la profilaxis. Numerosos estudios retrospectivos y prospectivos demuestran claramente que el tratamiento profiláctico, aunque mucho más caro, es clínicamente superior al tratamiento a demanda. El modelo óptimo consiste en individualizar la profilaxis teniendo en cuenta el fenotipo de sangrado y de la susceptibilidad individual a la artropatía (Ljung, 2018).
Finalmente, la profilaxis limitada o episódica se define como un período corto de terapia de restauración del factor, generalmente para prevenir los episodios hemorrágicos en situaciones específicas, tales como deportes u otras actividades con intensa actividad física, o incluso en el periodo previo y posterior a una cirugía. El objetivo de esta profilaxis es evitar completamente el sangrado y, por ello, la dosificación debe ser intensiva, para lograr un 100% de corrección de los niveles del factor afectado.
Mención aparte merece el sistema Port-a-cath. Se trata de una alternativa a la profilaxis clásica en pacientes hemofílicos, que consiste en un reservorio subcutáneo –implantado bajo la piel mediante una pequeña intervención quirúrgica– conectado a una vena profunda de la zona subclavia (bajo la clavícula); algunos modelos se insertan en el brazo. Está indicado sobre todo en niños con un difícil acceso para la administración intravenosa del factor, así como en edades muy tempranas para facilitar la adherencia. Su implantación debe valorare detenidamente, pues comporta varios riesgos entre los que destaca la incidencia de infecciones; algunos estudios han referido una tasa de infección hasta del 50% que, si bien pueden tratarse con antibioterapia, en casos graves obligan a retirar el dispositivo. Otros estudios han hecho referencia al riesgo de que se formen coágulos en la punta del catéter.
Como ya se ha indicado, el desarrollo de anticuerpos inhibidores (dirigidos contra el factor de coagulación administrado) es la complicación más importante del tratamiento restaurador crónico en el paciente hemofílico. Es recomendable realizar una detección precoz mediante determinaciones analíticas cada 5 dosis administradas, hasta la vigésima dosis y, después, cada 10 dosis hasta los 50 días. Superadas las 150 dosis, el riesgo de que aparezcan los inhibidores baja notablemente, por lo que es suficiente un control anual. También es común realizar una determinación antes de una intervención quirúrgica, cuando se cambie el tipo de concentrado administrado, y si se observa ineficacia en el tratamiento de un episodio hemorrágico.
El objetivo del tratamiento de la hemofilia A adquirida –muy mayoritaria respecto a los casos de hemofilia B adquirida– es el control de la hemorragia aguda y la erradicación a largo plazo de la producción de anticuerpos frente al factor VIII.
La elección del tratamiento hemostático está condicionada por el nivel o título del inhibidor, y la respuesta después de una nueva estimulación con administración de factor VIII. Se considera como inhibidor de título bajo aquél por debajo de 5 UB por mililitro, que se mantiene a pesar de la estimulación sucesiva. Se considera, además, como de bajo título y alta respuesta cuando se observa una producción de inhibidor rápida (alrededor de 7 días) después de la administración de una dosis de factor VIII. Los pacientes con inhibidor de bajo título y poco respondedores serán tratados con dosis más elevadas de factor VIII, y un control estrecho de sus niveles. Para el cálculo de las dosis necesarias se debe tener en cuenta que se precisa una cantidad para neutralizar el efecto del inhibidor más la cantidad de factor necesario para alcanzar los niveles terapéuticos deseados, de ahí la necesidad de un control estricto y personalizado.
Por otra parte, en pacientes con título alto puede disminuir el título en ausencia de administración de factor VIII durante periodos prolongados de tiempo. En esos pacientes, en que el tratamiento de sustitución del factor a dosis altas será ineficaz, el tratamiento actual para el control de la hemorragia aguda incluye como primera línea el uso de fármacos que aumenten la generación de trombina sin necesidad de que exista factor VIII o IX, también denominados agentes baipás (bypass). Se basan en la capacidad de los factores de coagulación activados como el factor Xa o el VIIa para poner en marcha el proceso de la coagulación en ausencia de factor VIII o IX, e incluso en presencia de un inhibidor contra éstos.
Se dispone de dos fármacos para el tratamiento o prevención de las hemorragias en pacientes con hemofilia adquirida. El primero de ellos es el concentrado de complejo protrombínico activado (CCPa; Feiba®), también conocido como complejo anticoagulante antiinhibidor, un derivado plasmático que contiene múltiples zimógenos del complejo protrombínico junto con sus productos de activación; por lo tanto, su mecanismo de acción es multifactorial. La concentración de protrombina junto con la presencia de factores activados VIIa y Xa contribuye a potenciar la generación de trombina en la superficie de las plaquetas.
Este fármaco tiene una actividad estandarizada: una solución que contiene 1 U/mL acorta el tiempo de tromboplastina parcial activada (TTPA) de un plasma con inhibidor del factor VIII en un 50% del valor obtenido con solución tampón, cuando se utilizan volúmenes iguales de solución de CCPa y de plasma con inhibidor. Su eficacia clínica se sitúa entre un 64% y un 93%; sin embargo, alrededor del 30% de los pacientes con hemofilia A e inhibidor presentan un aumento del título de inhibidor tras la administración del CCPa, aunque sin repercusión sobre la eficacia clínica.
La otra opción terapéutica, que es la más frecuentemente empleada, es el eptacog alfa activado (rFVIIa, NovoSeven®). Se trata de una forma recombinante del factor VII activado –obtenido en células BHK– que fue inicialmente desarrollado para tratar pacientes hemofílicos con inhibidores del factor VIII, pero que después ha demostrado también su utilidad en situaciones que cursan con sangrado incoercible que no responden a las medidas quirúrgicas y médicas convencionales. La unión del factor VII activado al factor tisular expuesto en la superficie de fibroblastos o células endoteliales desencadena la cascada de coagulación mediante la activación secuencial de los diferentes factores de coagulación (IXa, Xa y trombina).
El medicamento está indicado en el tratamiento de episodios hemorrágicos y su prevención en intervenciones quirúrgicas en pacientes con hemofilia adquirida o heredada con inhibidores de los factores de coagulación VIII o IX > 5 UB, o en pacientes con títulos de anticuerpos menores pero que se espera que tengan una respuesta alta de inhibidores tras la estimulación. Sus efectos adversos se relacionan con la capacidad trombogénica del fármaco; se han reportado casos agudos de síndrome coronario, accidentes cerebrovasculares y trombosis tanto arterial como venosa.
En pacientes con hemofilia B e inhibidor, se ha descrito un mayor riesgo de reacciones anafilácticas graves con CCPa, por lo que se recomienda utilizar el factor VIIa recombinante.
Por otra parte, para reducir la producción de autoanticuerpos y lograr un incremento sostenido en la concentración de factor VIII, a día de hoy se recomienda en primera línea la inmunosupresión con corticoesteroides en monoterapia o en combinación con ciclofosfamida. Un tratamiento usual incluye la administración de prednisolona (o prednisona) en dosis de 1-2 mg/kg, combinada con ciclofosfamida oral en dosis de 50-100 mg/día (1-2 mg/kg/día) durante un máximo de 6 semanas. Se recomienda el inicio de la terapia inmunosupresora tan pronto como se establezca el diagnóstico de hemofilia adquirida (Giangrande, 2012).
El anticuerpo monoclonal rituximab, otros fármacos citotóxicos (azatioprina, 6-mercaptopurina y vincristina), ciclosporina A o micofenolato pueden ser utilizados en segunda línea, y cuando el tratamiento preferente de esteroides con ciclofosfamida esté contraindicado.
La inducción de tolerancia inmunológica también ha sido el objetivo de algunos grupos, mediante la combinación de infusiones diarias de altas dosis de concentrados de factor VIII con distintos regímenes inmunosupresores (que incluyen esteroides, ciclofosfamida y vincristina), inmunoglobulinas intravenosas e inmunoadsorción. Se recomienda para pacientes con títulos altos de anticuerpos (> 5 UB) y se han descrito altas tasas de reducción de inhibidores (70-80%) pero, por su alto coste, se ha postulado su uso en ensayos clínicos o en segunda línea en casos refractarios (Charlebois, 2018).
Finalmente, merece una reseña la reciente aprobación en 2016 de susoctocog alfa (Obizur®), que no está aún comercializado en España. Se trata de una proteína purificada del factor VIII de la coagulación de origen porcino, obtenida por la tecnología del ADN recombinante (en células BHK) y que carece del dominio B. Aporta la ventaja teórica de que es un factor VIII sin reactividad cruzada contra los anticuerpos inhibidores anti-factor VIII humano, permitiendo ajustar la posología únicamente en función de los niveles del factor, sin necesidad de medir el título de anticuerpos. Si bien los datos clínicos son limitados, ha demostrado ser eficaz en el tratamiento de eventos hemorrágicos graves en pacientes con hemofilia A adquirida con títulos de inhibidores anti-susoctocog inferiores a 20 UB. Estudios posautorización deben elucidar el potencial riesgo de inducción de inmunogenicidad (reacciones de hipersensibilidad y desarrollo de inhibidores anti-susoctocog alfa) (Knöbl, 2018).
No menos importante que el tratamiento de reemplazo o terapia de restauración de los factores deficitarios es el tratamiento adyuvante destinado a controlar el dolor y la inflamación y recuperar la funcionalidad de las articulaciones afectadas por la artropatía hemofílica. Los AINE, como el ibuprofeno, son efectivos pero su potencial de hemorragia gástrica es problemático, y los opioides no se han evaluado sistemáticamente en hemartrosis, por lo que se utilizan poco en la población con hemofilia.
Por otro lado, hay pocos datos disponibles sobre la eficacia de las inyecciones intraarticulares de corticosteroides para controlar el dolor hemofílico. El descanso, con o sin colocación de una férula, limita el riesgo de una lesión mayor y reduce el dolor, pero el descanso prolongado representa un riesgo para la atrofia muscular y la formación de contracturas.
Mientras que la aplicación local de hielo disminuye el dolor, la inflamación y el daño a los tejidos, también reduce el flujo de sangre, la función de las plaquetas y las reacciones enzimáticas, incluyendo la conversión de protrombina a trombina; en consecuencia, el hielo se debe aplicar solo después de administrar el correspondiente factor de coagulación implicado. Un vendaje de compresión puede ayudar a reducir la hinchazón de los tejidos blandos, pero tiene poco impacto sobre la hemorragia. La elevación de la extremidad afectada para limitar la inflamación es teóricamente aceptable, pero difícilmente viable en el caso de niños y jóvenes. Por su parte, la hidroterapia reduce el dolor, el sangrado y la inestabilidad de las articulaciones afectadas y mejora la movilidad y la recuperación de la masa muscular.
La artrocentesis implica la extracción de la sangre presente en el interior de la articulación, con el fin limitar los daños a largo plazo. Sin embargo, para ser eficaz, la aspiración articular se debe realizar dentro de los 2 días tras el inicio de la hemorragia, administrando previamente el correspondiente factor, que debe alcanzar 100% de corrección. En general, la aplicación de este procedimiento se debe limitar a los episodios extremos de hemorragia, salvo en hemartrosis de cadera en niños pequeños, donde existe el riesgo de necrosis avascular. Cuando la hemorragia articular y la sinovitis no llegan a controlarse adecuadamente, la realización de una escisión de la membrana sinovial o sinovectomía puede ser eficaz para la eliminación de inflamación sinovial hipertrófica, disminuyendo el riesgo de hemorragia recurrente entre un 70% y un 100%.
Por último, se ha sugerido que los agentes anti-TNF (infliximab, adalimumab, certolizumab, etc.) utilizados en artritis reumatoide y otras patologías inflamatorias crónicas podrían ayudar a controlar la inflamación de los tejidos sinoviales en pacientes con hemofilia, pero aún no se dispone de estudios controlados que avalen su uso. Asimismo, considerando que la neovascularización de la membrana sinovial es una característica prominente de la hemartrosis, se ha especulado sobre el potencial terapéutico de los agentes antiangiogénicos (bevacizumab, ranibizumab, etc.).
Según lo sugerido anteriormente, en los últimos años se han desarrollado, por diversas técnicas, derivados de los factores de coagulación que han conseguido ampliar la vida media plasmática generalmente limitada de éstos. El primero de ellos fue quizá el caso del efmoroctocog alfa (Elocta®), autorizado en 2016 para el tratamiento de la hemofilia A congénita. Se trata de una proteína recombinante producido en células HEK y basada en la fusión del factor VIII (al que se le ha eliminado el dominio B) con la porción Fc de una IgG1. Su capacidad de unirse al receptor de Fc nenonatal –que se expresa a lo largo de toda la vida en células endoteliales y monocitos circulantes– promueve su estabilidad y prolonga su vida media plasmática (20,9 h) respecto de otras formas recombinantes del factor VIII (< 15 horas).
Otra vía que ha permitido prolongar la vida media de los factores recombinantes es la pegilación, sin duda una de las técnicas de bioingeniería de mayor impacto en este campo. Consiste en la unión covalente de polímeros de polietilenglicol (PEG) a la molécula terapéutica (factor VIII o factor IX) a través de una unión sitio-específica a residuos de cisteína libres o mediante ingeniería de proteínas (pegilación o glicopegilación sitio-específica). Esta técnica, igual que la anterior, se aprovecha del reciclaje mediado por el receptor Fc neonatal, lo que protege al factor de coagulación recombinante de la degradación proteolítica lisosomal e incrementa su vida media.
Entre 2018 y 2019 se han autorizado en nuestro país algunos fármacos basados en la pegilación del factor VIII recombinante para el tratamiento de la hemofilia A congénita en mayores de 12 años. Es el caso del ruricoctocog alfa pegol (Adynovi®, comercializado en 2018), que es un factor VIII recombinante de cadena completa – producido en células CHO – conjugado con una o más moléculas de PEG (aportando un peso molecular de 20 KDa) de forma no sitio-específica, pero frecuentemente en el dominio B. Comparte todas las funciones fisiológicas de la molécula del factor VIII con la excepción de su unión a la proteína relacionada con el receptor de lipoproteínas de baja densidad (LDL), lo cual puede explicar el menor aclaramiento plasmático y el alargamiento de su vida media en 1,5 veces.
El damoctocog alfa (Jivi®, aún no comercializado en España) comparte con el anterior la técnica de la pegilación. Es también un factor VIII recombinante – producido en células BHK – a cuya molécula se le ha eliminado el dominio B y se le ha adicionado un codón de cisteína para permitir la conjugación sitio-específica de una única molécula de PEG de 60 KDa. Su vida media es de aproximadamente 19 h en comparación con las 13 h del fármaco original (octocog alfa: Advate®), es decir, 1,5 veces más prolongada. Turoctocog alfa pegol (Esperoct®) es el último representante de este grupo de factores VIII recombinantes pegilados, que ha sido muy recientemente autorizado por la FDA americana, pero que no dispone aún de autorización de comercialización en Europa. Producido en células CHO, incorpora un PEG de 40 KDa conjugado en la región enlazadora del dominio B, que está truncado por la glicopegilación sitio-específica.
En los estudios clínicos todos estos fármacos han demostrado una eficacia excelente cuando se emplean para el tratamiento o la profilaxis de hemorragias en pacientes con hemofilia A. No obstante, puesto que solo aportan una prolongación moderada de la vida media del factor VIII, una menor dosificación podría incrementar ligeramente el riesgo de hemorragias transitorias o subclínicas en ciertos pacientes susceptibles, por lo que se recomienda la frecuencia común de dosificación, especialmente en niños. Si bien la experiencia clínica con estos productos es limitada y se requiere una mayor evidencia, los datos disponibles no sugieren un mayor riesgo de desarrollo de inhibidores (Giangrande, 2017).
Con respecto a su seguridad, los productos pegilados han suscitado preocupación relativa a los efectos secundarios a largo plazo, debido a la posible acumulación de PEG. Por ejemplo, en estudios in vivo en animales, se ha descrito la vacuolización de ciertos tipos de células – células renales o macrófagos – y la acumulación de PEG en el plexo coroideo tras un tratamiento repetido con altas dosis. La dificultad de extrapolar a humanos y el diferente perfil de seguridad que puede tener la variedad de fármacos impiden confirmar el significado de tales hallazgos hasta que se aporte una mayor evidencia a partir de estudios observacionales a largo plazo.
Por otro lado, las citadas estrategias de pegilación y fusión proteica han permitido prolongar en mayor medida la vida media del factor IX recombinante: hasta en 3-6 veces respecto al factor IX recombinante original (nonacog alfa), frente a las 1,5-1,6 veces de los fármacos recombinantes del factor VIII. Probablemente es el acoplamiento entre factor VIII y el factor de von Willebrand en sangre lo que dificulta la prolongación de la semivida y acción del factor VIII al mismo nivel que para el factor IX.
Albutreprenonacog alfa (Idelvion®) es una proteína de fusión recombinante que ha sido diseñada por fusión genética del ADNc de la albúmina humana (que tiene una vida media de unos 20 días) y el ADNc del factor IX de la coagulación. Entre ambas regiones proteicas presenta una región bisagra escindible derivada del péptido de activación endógeno, de manera que el factor IX se activa cuando la albúmina se separa. Tiene una vida media en adultos de 95,3 h, entre 4 y 5 veces superior a la semivida plasmática del factor IX recombinante convencional (nonacog alfa: Benefix®), que es < 21 h. Por su parte, eftrenonacog alfa (Alprolix®) es otra proteína de fusión recombinante autorizada para el tratamiento y profilaxis de hemorragias en pacientes con hemofilia B. Ha sido desarrollada mediante la unión del factor IX a la porción Fc de una inmunoglobulina IgG1 y su vida media alcanza las 82,2 h.
Como ejemplo de fármacos pegilados para la hemofilia B, nonacog beta pegol (Refixia®) es un factor IX humano recombinante que presenta un PEG de 40 KDa unido a N-glicanos específicos en el péptido de activación. Tras dicha activación, tanto ese péptico como el PEG liberan la molécula activa de factor IX. Su vida media plasmática tras dosis repetidas en adultos ronda las 115 h, siendo más reducida en población pediátrica (70-89 h).
La mayor semivida plasmática de estos fármacos sí que permite una dosificación menos frecuente en la profilaxis de la hemofilia B y han permitido un cambio de paradigma, facilitando la adherencia terapéutica. Así, en estudios con adultos, se ha podido recuperar en sangre casi el 20% del fármaco una semana después de una única dosis de 50 UI/kg, y en algunos adultos se puede lograr un nivel aceptable más allá de 2 semanas tras una dosis de 80 UI/kg. Su empleo con una pauta de administración semanal en profilaxis desde etapas tempranas de la vida de un niño puede permitir evitar la necesidad de vías venosas centrales que implica la administración frecuente de concentrados convencionales. Eficaces tanto en el tratamiento como en la prevención de hemorragias, tampoco hay evidencias de que estos modernos fármacos anti-hemofilia B provoquen un aumento o disminución del riesgo de desarrollo de inhibidores, aunque una mayor experiencia clínica permitirá definir en detalle su perfil de beneficio-riesgo.
En los últimos años han emergido nuevos enfoques terapéuticos y profilácticos para la hemofilia, y especialmente para la hemofilia adquirida con presencia de inhibidores. Los nuevos fármacos, basados en mecanismos diferentes al reemplazo de los factores de coagulación, aportan como principales ventajas vías de administración diferentes a la intravenosa (vía subcutánea básicamente), efectos más prolongados (que permitirán una menor frecuencia de administraciones), y una potencial eficacia clínica independiente de la presencia de inhibidores. Sin embargo, no han sido aún comercializados en España o están en desarrollo clínico.
Emicizumab (Hemlibra®, autorizado en 2018) es un anticuerpo monoclonal biespecífico que mimetiza la función de la molécula del factor VIII activado y es capaz de unirse a el factor IXa y al factor Xa, uniéndolos y promoviendo la continuación natural de la cascada de la coagulación. Solo es eficaz, por tanto, en los pacientes con hemofilia A. La falta de similitud estructural de emicizumab con el factor VIII evita que su efecto sea reducido por los anticuerpos inhibidores. Administrado por vía subcutánea, tiene una vida media de 4-5 semanas.
En ensayos clínicos, una pauta profiláctica de emicizumab ha demostrado capacidad de reducir la tasa de eventos hemorrágicos de 23 a 3 (p<0,001) en pacientes con hemofilia A e inhibidores, siendo más activo que los agentes de baipás. En estudios con pacientes sin inhibidores, administrado a dosis de 1,5-3 mg/kg/semana redujo en un 96-97% los sangrados en comparación con un tratamiento a demanda; sin embargo, en estos pacientes se describieron hasta cuatro casos de eventos trombóticos graves y se requieren futuros estudios para elucidar la seguridad a largo plazo del fármaco. Por tanto, parece que emicizumab supone una innovación disruptiva en la profilaxis a largo plazo de la hemofilia A adquirida (que, en un futuro cercano, puede extenderse a la hemofilia A congénita sin inhibidores) al permitir una administración más sencilla y menos frecuente que los agentes de baipás.
Otros fármacos buscan mejorar la hemostasia plasmática regulando el equilibrio coagulación/anticoagulación mediante la reducción del efecto de los anticoagulantes naturales. A través de ese mecanismo serán eficaces, a priori, tanto en pacientes con hemofilia A como con hemofilia B, aunque cabe mencionar que también se plantea la incertidumbre del riesgo de alterar el delicado equilibrio de los sistemas procoagulante y anticoagulante –especialmente en situaciones como infecciones graves (que por sí mismas pueden crear un estado pro-coagulante)–, y de la posible necesidad de contrarrestar su riesgo de trombosis.
En este sentido, se han desarrollado anticuerpos monoclonales recombinantes contra el inhibidor de la vía del factor tisular (TFPI). Un ejemplo de ellos es concizumab, que es un anticuerpo humano de administración subcutánea capaz de impedir la unión del factor Xa al TFPI, lo que resulta en una generación amplificada del factor Xa y de trombina in vitro. Actualmente está siendo evaluado en un ensayo clínico de fase 2 de seguridad y eficacia en pacientes con hemofilia A y B con inhibidores. Otros fármacos con un mecanismo de acción similar están en etapas más precoces del desarrollo clínico.
Por su parte, fitusirán (ALN-AT3) es un agente de ARN de cadena pequeña diseñado para interferir con la producción de antitrombina, inclinando la balanza del lado de la anticoagulación con una producción aumentada de trombina. En estudios de fase 1/2, una administración mensual de fitusirán por vía subcutánea ha demostrado reducir, de forma dosis-dependiente, los niveles máximos de antitrombina en un 70-89% en comparación con el estado basal; también se ha evidenciado una mayor producción de trombina en pacientes de hemofilia A y B sin inhibidores. Actualmente está siendo evaluado en dos ensayos de fase 3 (Nogami, 2019).
A pesar de todo lo comentado hasta aquí, tanto el tratamiento actual como el resto de fármacos en investigación representan más bien un enfoque profiláctico que una cura. La única estrategia curativa10 –total o parcial– contemplada hasta la fecha sería la terapia génica, pues la condición de enfermedad monogénica convierte a la hemofilia en una teórica candidata ideal. De hecho, la terapia génica ha experimentado un significativo avance en los últimos años, con resultados prometedores sobre todo en hemofilia B.
La terapia génica en la hemofilia A ha mostrado como principal limitación la generación de vectores virales eficientes en la inserción de genes, pues el gran tamaño del ADN del gen F8 excede con mucho la capacidad de empaquetamiento normal de los vectores adenoasociados. El desarrollo de ADNc optimizado del gen F8 ha puesto de manifiesto que secuencias génicas más cortas pueden aumentar la eficacia de la terapia génica. Otros grupos también han intentado superar esa limitación con la producción de vectores virales adenoasociados duales o mediante el empleo de vectores lentivirales para la transferencia génica.
Así, por ejemplo, nueve hombres con hemofilia A severa recibieron la administración de un vector de virus adenoasociado que portaba un gen codificante para el factor VIII humano sin dominio B. Tal estrategia resultó en una normalización sostenida del nivel de actividad del factor VIII a valores normales (>50%) durante un período superior a 1 año en 6 participantes, algunos de los cuales también mostraron niveles especialmente elevados en algunas ocasiones; se redujo el número de hemorragias y los participantes necesitaron una dosificación de factor VIII significativamente menor (Rangarajan, 2017).
En hemofilia B, Nathwani y colaboradores publicaron los primeros resultados positivos de terapia génica en 2011. Utilizando un virus adenoasociado de serotipo 8, demostraron un aumento consistente del 5-7% en la actividad del factor IX a lo largo de un periodo de seguimiento de 1-4,5 años, sin describir efectos adversos graves a excepción del aumento de enzimas hepáticas (resuelto con prednisolona).
De forma similar, la administración de un vector viral adenoasociado monocatenario, consistente en una cápside diseñada por bioingeniería, un promotor específico del hígado y factor IX, dio como resultado una producción sostenida de factor IX en todos los pacientes tratados, con una actividad media del factor estabilizada en torno al 34% (±19%). En un seguimiento de 28 a 78 semanas, la tasa de sangrado anual se redujo significativamente de una media de 11,1 a 0,4 eventos (p=0,02), describiéndose ausencia de hemorragias en el 90% de los pacientes. También se redujo el uso de concentrados de factor (de una media de 2.908 a 49,3 UI/Kg), hasta el punto de que el 80% de los pacientes no lo utilizaron (George, 2017).
Deben aclararse aún incertidumbres relativas a la duración del efecto de las terapias génicas, la inmunogenicidad de los vectores virales (en administraciones repetidas), el riesgo de mutagénesis y de sobre-producción del factor, y la implicación de la existencia de anticuerpos inhibidores. No obstante, diversos ensayos clínicos con técnicas de terapia génica están a día de hoy en marcha, mayoritariamente en fases 1-2, empleando distintos tipos de vectores virales y secuencias génicas, por lo que podría esperarse que en un futuro a corto-medio plazo aparezca una solución curativa de la hemofilia. Además, se están desarrollando técnicas que dirigen los vectores específicamente al hígado, así como otras terapias celulares (Croteau, 2018).
La práctica totalidad de los medicamentos autorizados en España con la indicación de tratamiento y prevención de las hemorragias en pacientes hemofílicos –de administración intravenosa– son de uso hospitalario (a excepción de algunos medicamentos con ácido tranexámico y desmopresina). En base a ello puede comprenderse que el farmacéutico hospitalario jugará un papel destacado en la asistencia sanitaria a pacientes hemofílicos.
Con la integración de estos profesionales en los equipos multidisciplinares y de gestión a nivel de hospitales, su papel asistencial ha ido ganando cada vez más peso. De manera similar a otras patologías que se abordan desde centros hospitalarios, el farmacéutico participará de los protocolos de actuación interdisciplinar en la toma de decisiones de selección de tratamientos y profilaxis anti-hemofílica, así como de las pautas posológicas individualizadas, aportando información relevante al equipo clínico sobre los medicamentos y colaborando en el análisis y resolución de problemas relacionados con su uso por parte de pacientes concretos. El tratamiento profiláctico a largo plazo con medicamentos de reemplazo de factores de coagulación implica unos costes económicos elevados, por lo que el farmacéutico hospitalario valorará, en el ámbito del equipo multidisciplinar, la eficiencia de los nuevos tratamientos.
No obstante, su labor va más allá, y se pueden identificar varias vías asistenciales en el abordaje del paciente hemofílico. Una creciente evidencia respalda el efecto que, sobre la calidad de vida del paciente hemofílico, ejerce una adecuada atención farmacéutica realizada por el farmacéutico especialista desde los centros de tratamiento de la hemofilia. A modo de ejemplo, el programa BE EMPOWERED –un programa de educación sanitaria multimodal desarrollado en Estados Unidos para pacientes con hemofilia B (o cuidadores, en el caso de pacientes pediátricos)– demostró que la intervención farmacéutica orientada a potenciar el conocimiento sobre la medicación y el autocuidado reducía significativamente el número de hemorragias y hemartrosis en adultos e incrementaba la utilización de la técnica RICE11 en niños, sugiriendo que podría asociarse a una optimización de los costes sanitarios (Blankenship, 2014).
La atención farmacéutica dirigida al paciente hemofílico será, por tanto, desarrollada por el farmacéutico hospitalario (en colaboración con el equipo multidisciplinar) durante la estancia del paciente en el centro por un posible episodio hemorrágico o para recibir su dosis periódica del tratamiento de sustitución de factor. Sin embargo, será continuada por el farmacéutico comunitario durante el periodo ambulatorio del paciente, ya que estos pacientes pueden estar polimedicados y acudir a la farmacia comunitaria para la dispensación de fármacos para otros problemas de salud o simplemente a solicitar consejo sanitario.
En ese sentido, cabe recordar que las farmacias comunitarias son los establecimientos sanitarios más accesibles y cercanos al ciudadano, por número, por distribución geográfica y por horarios de apertura: en España hay más de 22.000 oficinas de farmacia en las que trabajan más 50.000 farmacéuticos. Esto hace que el farmacéutico comunitario sea uno de los primeros profesionales sanitarios con los que interactúa un paciente cuando abandona el centro de tratamiento de hemofilia. Ofrecen, pues, una oportunidad excelente para que desde la farmacia comunitaria se puedan proporcionar diferentes servicios que den respuesta a las necesidades de las personas en relación a los medicamentos y otros aspectos relacionados con la salud.
En su actividad diaria, el farmacéutico trabaja para, entre otros objetivos, garantizar el acceso de los pacientes a medicamentos seguros, eficaces y de calidad, optimizar los resultados de dichos medicamentos, proporcionar información para que los usuarios conozcan para qué es el medicamento dispensado y cómo utilizarlo correctamente; además, puede identificar posibles problemas derivados del uso de la medicación, así como signos tempranos o factores de riesgo para determinadas enfermedades, y promover actividades relacionadas con la prevención de enfermedades y la promoción de la salud. Todo ello contando con la colaboración fundamental de otros profesionales de la salud.
Precisamente, la comunicación fluida y bidireccional entre el farmacéutico comunitario y el médico de atención primaria resulta imprescindible para poder identificar pacientes hemofílicos que estén bajo sospecha de padecer una hemorragia o de sufrir un problema relacionado con la medicación, y poder impulsar la ruta asistencial que conduzca a la evaluación por el especialista y a las decisiones terapéuticas pertinentes. A grandes rasgos, se pueden destacar varias líneas de actuación del farmacéutico comunitario en su labor asistencial al paciente trasplantado, todas las cuales son también compartidas, en su ámbito, por el farmacéutico hospitalario:
La incidencia mundial estimada por la Federación Mundial de Hemofilia para las coagulopatías hereditarias se corresponde con una tasa de 1 caso por cada 1.000 habitantes, lo que queda muy lejos del 0,065 por mil en que se traduciría los aproximadamente 3.000 casos de hemofilia confirmados en España. Esto es sugerente de que en nuestro país puede existir un grupo de pacientes no diagnosticados y, consecuentemente, no tratados ni asesorados genéticamente, con los evidentes riesgos y problemas que ello comporta. Por consiguiente, es muy importante realizar una labor de detección de este tipo de patologías y, desde luego, la oficina de farmacia es un centro sanitario que puede colaborar eficazmente en este cometido.
La idea de que las hemofilias tienen un carácter hereditario puede hacer bajar la guardia a muchas personas. Aunque, en efecto, la mayor parte de los casos se pueden explicar por los antecedentes familiares, conviene no olvidar dos aspectos muy relevantes:
Para poder contribuir al diagnóstico temprano de hemofilia, hay que tener presente que la expresión clínica más característica de todas las formas de hemofilia es la hemorragia, manifestada comúnmente en múltiples localizaciones (Tabla 4): muscular, sistema nervioso central, partes blandas y, muy especialmente, en las articulaciones. La hemartrosis o hemorragia articular es la forma de sangrado más frecuente (65-90%), hasta el punto de que constituye el sello distintivo de las hemofilias. Por ello, la aparición de forma reiterada de hemartrosis en tobillos, rodillas y/o codos es sugerente de la patología y hace especialmente aconsejable remitir el paciente –firmemente, pero sin alarmismos– a su médico para un estudio clínico, bioquímico y genético, si procede. En ocasiones, la artropatía hemofílica pasa desapercibida o simplemente confundida con otras formas de artritis (con las que puede coexistir), artrosis, anquilosis o incluso osteoporosis. Esté diagnosticado o no, en personas con artropatía hemofílica, cualquier intento no controlado de rehabilitación de la articulación podría provocar la ruptura de los vasos subsinoviales y de nuevos episodios de hemorragia articular.

El farmacéutico puede sospechar de posible hemofilia en casos de pacientes con antecedentes de: familiares con hemofilia, aparición de hematomas durante la infancia, hemorragias espontaneas sin razón aparente (en especial en las articulaciones, músculos y tejidos blandos) o hemorragia excesiva posterior a un traumatismo o una cirugías. La hemorragia podría identificarse en una exploración física preliminar. Así, las hemorragias de piel y mucosas (como gingivorragia o hemorragia digestiva) se manifestarán típicamente con petequias y equimosis, bien simétrica o difusa, y son indicativos de una alteración de la hemostasia primaria. Por otra parte, los hematomas subcutáneos o musculares –que se evidencian con grandes equimosis o hemartros– o una hemorragia que se produce horas o días posteriores a un traumatismo o cirugía deben hacer pensar en una alteración de la hemostasia secundaria.
Una vez diagnosticado el paciente, los objetivos del tratamiento son prevenir la hemorragia y, en el caso de que aparezca, tratarla adecuadamente (así como sus complicaciones y secuelas), restaurar y mantener la función articular e integrar a los pacientes en la vida social normal. Para ello, a los pacientes con formas leves o moderadas de coagulopatía se les suele prescribir desmopresina o antifibrinolíticos (como el ácido tranexámico), que son medicamentos con receta médica. Sin embargo, en las formas graves, es preciso utilizar un tratamiento sustitutivo (de reemplazo o de restauración) con el factor correspondiente o concentrados de composición diversa, según sea el caso, tanto de origen extractivo como recombinante. Estos últimos son medicamentos hospitalarios que requieren prescripción por médicos especialistas y dispensación farmacéutica en hospital.
Puesto que la hemofilia es una patología para la que no se dispone aún de cura, el tratamiento profiláctico de sustitución del factor se instaura en muchos casos de por vida, y requiere de 1 a 3 administraciones semanales. La eficacia del tratamiento solo se alcanza si el paciente es adherente (y esa adherencia persiste en el tiempo), y son diversos los artículos científicos referidos a ese punto, identificando varios tipos de barreras a la adherencia terapéutica, relativas al sistema de atención médica, variables socioeconómicas, edad del paciente, régimen posológico, etc.
Si bien se han descrito tasas de adherencia al tratamiento anti-hemofílico en pacientes adultos y niños pequeños de hasta el 93%, éstas se ven reducidas en otros grupos, especialmente en adolescentes y adultos jóvenes (hasta valores tan bajos de adherencia como del 26-30%). Las consecuencias de la falta de adherencia pueden ir desde un empeoramiento de la calidad de vida del paciente, una falta de control de la enfermedad y una mayor probabilidad de hemorragias y complicaciones, hasta la aparición de efectos secundarios o incluso de mortalidad. Además, puede traducirse en ineficiencia del gasto farmacéutico y sanitario (Thornburg, 2017).
Las estrategias de promoción de la adherencia por parte del profesional farmacéutico deben desarrollarse con el paciente y la familia, centradas en los objetivos de tratamiento individualizados. Se necesitan estrategias personalizadas para reforzar la importancia y fomentar la confianza de los pacientes en la administración de las infusiones regulares necesarias para la terapia profiláctica. Estas estrategias pueden incluir información verbal y escrita, y recursos interactivos. Se debe informar a los pacientes de la aparición de factores de coagulación de vida media más prolongada y otros nuevos fármacos que requieren una infusión menos frecuente, lo cual pueden contribuir a mejorar la adherencia.
Parece ser que una de las etapas críticas de pérdida de adherencia a la profilaxis de hemofilia es la adolescencia y la juventud, cuando los pacientes pasan de estar bajo el cuidado de sus padres y deben ser ellos mismos los encargados de su tratamiento. La contribución del farmacéutico en esa etapa crucial –preservando la normalidad y erradicando estigmas sociales– puede conseguir que el paciente se sienta partícipe y tenga una actitud proactiva hacia su tratamiento (Lee, 2018).
En línea con la promoción de la adherencia, el farmacéutico, a través de la prestación de diferentes servicios asistenciales puede y debe contribuir a maximizar los beneficios de la farmacoterapia en el paciente hemofílico, tanto de la profilaxis específica como de los fármacos que puedan recibir para el tratamiento de otras patologías, pues hay que recordar que, en la mayoría de casos, el farmacéutico comunitario conoce toda la medicación que utiliza un paciente ambulatorio, así como el uso de medicamentos sin receta, complementos alimenticios, etc. A través de un adecuado seguimiento farmacoterapéutico, puede identificar y resolver posibles problemas relacionados con los medicamentos, tales como interacciones, contraindicaciones, reacciones adversas, duplicidades, etc.
Conviene recordar que hay varios medicamentos indicados en el tratamiento de la hemofilia que contienen los principios activos desmopresina y ácido tranexámico que sí son de dispensación en farmacia comunitaria. Ante una dispensación de inicio o de continuación de éstos, será importante tener presente algunas consideraciones sobre su perfil de seguridad:
Otros conceptos a tener en consideración en la atención farmacéutica a pacientes hemofílicos para maximizar los beneficios de la profilaxis y el tratamiento son los que se resumen a continuación:

Por último, el farmacéutico comunitario puede ejercer una importante labor a la hora de transmitir una serie de recomendaciones higiénico-dietéticas y de estilo de vida que contribuya a la prevención de las hemorragias y a una mejor calidad de vida. Por su condición de enfermos crónicos, los pacientes hemofílicos deben tener una mayor educación sanitaria que la población general, que puede implicar, por ejemplo, la necesidad de leer los prospectos de cualquier medicamento que tomen por primera vez.
Conviene recordar a los pacientes o sus cuidadores la conveniencia de llevar a cabo revisionesperiódicas, al menos una vez al año (los niños cada medio año), con su especialista en el centro de referencia, a fin de realizar estudios hematológicos, musculo-esqueléticos y psicosociales completos que permitan ajustar los detalles del plan individual de tratamiento integral. Además, se debe incidir en la conveniencia de que todos los pacientes lleven consigo una identificación (a la que se acceda fácilmente) que indique el diagnóstico y otros detalles –como la gravedad del trastorno, el estado de los inhibidores, el tratamiento y pauta posológica recibidas, etc.–, y que permita a los profesionales sanitarios conocer el mejor modo de actuación en situaciones de emergencia.
Debido a las frecuentes infusiones sanguíneas que los pacientes con hemofilia van a recibir a lo largo de su vida, el riesgo de exposición al virus de la hepatitis B está incrementado. Por ello, suele recomendarse de forma decidida la vacunación contra este virus.
Un tema común de consulta a los profesionales sanitarios por parte de estos pacientes suele ser el relacionado a la actividad física y el deporte. Algunas personas no practican ejercicio por creer que puede promover hemorragias pero, en cambio, se ha comprobado que el ejercicio y una buena condición física (que implica un peso saludable) con desarrollo neuromuscular normal pueden ayudar a prevenir las hemorragias, pues un músculo fuerte protegerá del daño en las articulaciones y de hemorragias espontáneas. El deporte es una actividad importante, especialmente en niños y jóvenes, porque también contribuye al bienestar social, la autoestima y al desarrollo de la concentración mental y la coordinación.
A este respecto, en línea con lo recomendado por la Federación Mundial de Hemofilia, desde la farmacia se puede fomentar la práctica de actividades que no impliquen contacto o riesgo de heridas, como el senderismo, la natación, el golf, el bádminton, el ciclismo, el remo, la navegación o el tenis de mesa, evitando deportes de alto impacto como el fútbol, el hockey, el rugby, el boxeo, el motociclismo o el esquí. Además, los pacientes hemofílicos pueden presentar una densidad ósea disminuida y, en esos casos, se deben promover los ejercicios con peso para contrarrestar el riesgo de fracturas, en tanto y en cuanto la salud de sus articulaciones se lo permita. Siempre deberán informar a los compañeros para organizar y planificar posibles situaciones de emergencia en caso de hemorragias.
No obstante, lo idóneo es que cada paciente se someta a una valoración física por un fisioterapeuta especializado en hemofilia, para la correcta indicación de unos ejercicios específicos que se adapten a su demanda clínica. De hecho, la fisioterapia desempeña un papel fundamental en el diseño de estrategias de prevención que minimicen las situaciones de riesgo ante posibles hemorragias que puedan derivar en problemas músculo-esqueléticos. Es recomendable que el paciente –sobre todo aquellos con artropatías– consulte a su fisioterapeuta de referencia antes de participar en cualquier actividad física a fin de analizar si es apropiada, el equipo de protección necesario, la profilaxis correspondiente y la condición física que se requiere antes del inicio. En general, se acepta que las articulaciones diana pueden protegerse con sujetadores o entablillados durante la actividad, en especial ante la ausencia de cobertura con el factor de coagulación. Después de una hemorragia, las actividades deben retomarse gradualmente para minimizar las probabilidades de una nueva hemorragia.
Bibliografía
El colesterol es un nutriente común en la dieta humana y los huevos son una fuente importante de colesterol en la misma. En la sociedad de hoy, la hipercolesterolemia (y su asociación con epidemias como la obesidad y el síndrome metabólico) representa un problema sanitario de primera magnitud. En los últimos tiempos, se ha generado un controvertido debate sobre si el consumo de colesterol o huevos en exceso en la dieta está relacionado con la enfermedad cardiovascular (ECV) y una mayor mortalidad por esta causa.
A fin de esclarecer esa relación, un reciente estudio realizado en EE.UU. ha analizado los datos –recopilados desde 1985 hasta 2016– de 29.615 participantes procedentes de 6 cohortes prospectivas (edad media de 51,6 años, 44,9% de hombres y 31,1% de raza negra). Los datos de las dietas individuales se armonizaron según un protocolo estandarizado, describiendo la ingesta de colesterol en mg/día y el consumo de huevos en número/día. Los investigadores analizaron la razón de riesgo (HR) y la diferencia de riesgo absoluta (DRA) –durante todo el periodo de seguimiento– de la incidencia de ECV (medida compuesta de enfermedad cardíaca coronaria letal y no letal, accidente cerebrovascular, insuficiencia cardíaca y otras muertes por ECV) y de la mortalidad por todas las causas; ajustaron los resultados por factores demográficos, socioeconómicos y de comportamiento.
Durante una mediana de seguimiento de 17,5 años, se describieron hasta 5.400 eventos de ECV y 6.132 muertes por todas las causas. La asociación entre el consumo de colesterol o huevo en la dieta con eventos de ECV y la mortalidad por todas las causas fueron monotónicas. Así, el consumo adicional de 300 mg/día de colesterol en la dieta se asoció –con un poder estadísticamente significativo– con un mayor riesgo de evento de ECV [(HR: 1,17; IC95% de 1,09 a 1,26) (DRA: 3,24%; IC95% de 1,39 a 5,08)] y de mortalidad por todas las causas [(HR: 1,18; IC95% de 1,10 a 1,26) (DRA: 4,43%; IC95% de 2,51 a 6,36)].
De igual modo, el consumo adicional de medio huevo por día se asoció –con un poder estadísticamente significativo– con un mayor riesgo de incidencia de ECV [(HR: 1,06; IC95% de 1,03 a 1,10) (DRA: 1,11%; IC95% de 0,32 a 1,89)] y de mortalidad por todas las causas [(HR: 1,08; IC95% de 1,04 a 1,11) (DRA: 1,93%; IC95% de 1,10 a 2,76)]. Sin embargo, la asociación entre el consumo de huevos y la incidencia de ECV o la mortalidad por todas las causas no era significativa cuando se ajustaba por el consumo de colesterol en la dieta.
En conclusión, los resultados de este estudio demuestran que, al menos entre los adultos estadounidenses, la mayor ingesta de colesterol y el mayor consumo de huevos en la dieta se asocian, de forma estadísticamente significativa, con un mayor riesgo de enfermedad cardiovascular y de mortalidad por todas las causas. Además, esa asociación es directamente dependiente de la cantidad (de colesterol o de huevos) ingerida. Tales hallazgos deben ser tenidos en cuenta en el desarrollo de futuras recomendaciones dietéticas, considerando que pueden ser extrapolables a otras cohortes poblacionales.
En 1998 se publicó en la revista Lancet un estudio que establecía una relación entre la vacuna contra el sarampión, las paperas y la rubeola (triple vírica) y la aparición de autismo en niños. Generó gran controversia entre la comunidad científica internacional y una gran polémica sanitaria en la sociedad. A pesar de que dicho estudio fue retractado y que estudios posteriores refutaron sus conclusiones, las tasas de vacunación contra el sarampión se han visto afectadas en la última década, lo cual se ha traducido en la aparición de diversos brotes de la enfermedad en nuestro entorno en los últimos años.
Con el objetivo de evaluar si la vacuna triple vírica incrementa el riesgo de autismo en niños, bien en algunos subgrupos de éstos o bien en diversos periodos de tiempos posvacunación, un estudio de cohortes restrospectivo –realizado por investigadores del Statens Serum Institut de Copenhague (Dinamarca)– ha evaluado los datos de un total de 657.461 niños nacidos en Dinamarca desde 1999 hasta 2010. El seguimiento de estos pacientes se realizó desde su primer año de vida y hasta el 31 de agosto de 2013.
Los autores utilizaron registros de información sobre el estado de vacunación (tanto de la triple vírica como de otras vacunas infantiles), los diagnósticos de autismo, el historial de autismo entre hermanos y los factores de riesgo de autismo en los niños de la cohorte. Partiendo del tiempo hasta el diagnóstico, se empleó el análisis de supervivencia, también conocido como regresión de Cox, para estimar los ratios de riesgo de autismo según el estado de vacunación con la triple vírica ajustado por diversos factores (edad, año de nacimiento, sexo, otras vacunas infantiles, riesgo de autismo, etc.).
Habida cuenta de que el 95% de los niños habían recibido la vacuna triple vírica, los resultados muestran que, durante un total de 5.025.754 persona-año de seguimiento, 6.517 niños fueron diagnosticados con autismo (tasa de incidencia de 129,7 casos por 100.000 persona-año). La comparación de niños vacunados frente a los niños no vacunados con la triple vírica arrojó un índice de riesgo de autismo muy ajustado (HR: 0,93; IC95% de 0,85 a 1,02). Si bien las diferencias no fueron estadísticamente significativas, los resultados apuntaban a que los niños que recibieron la vacuna tenían un 7% menos de probabilidades de desarrollar un autismo respecto a aquellos que no la habían recibido.
En la misma línea, no se observó un aumento del riesgo de autismo después de la vacunación con triple vírica en el análisis por subgrupos de niños, definidos éstos en base al historial de autismo de los hermanos, los factores de riesgo de autismo u otras vacunaciones infantiles, o durante períodos de tiempo específicos después de la vacunación. El estudio sí que reveló, no obstante, que los niños con hermanos diagnosticados de autismo tienen 7 veces más probabilidades de padecer la enfermedad, que los niños tienen 4 veces más probabilidades que las niñas, y que los niños que no recibieron ninguna vacuna de las recomendadas en la infancia tenían un riesgo un 17% mayor que los niños a quienes se les administraron las vacunas recomendadas.
En conclusión, el estudio apoya firmemente que la vacunación con la vacuna triple vírica no aumenta el riesgo de autismo, no desencadena el autismo en niños susceptibles y no se asocia con una mayor agrupación de casos de autismo tras de la vacunación; sino más bien lo contrario. Este se suma a otros estudios previos, aportando un significativo poder estadístico adicional al abordar las hipótesis de subgrupos susceptibles y agrupación de casos. Parece evidente que es una imprudencia por parte de los padres saltarse la vacuna triple vírica de sus hijos por miedo al desarrollo de autismo.
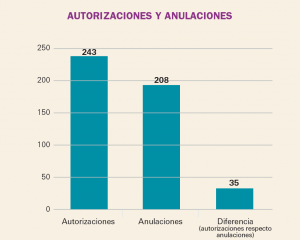
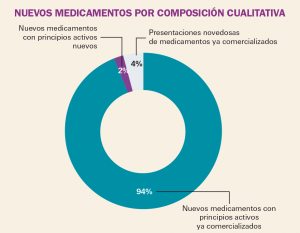
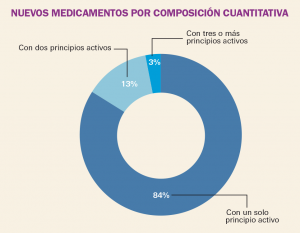
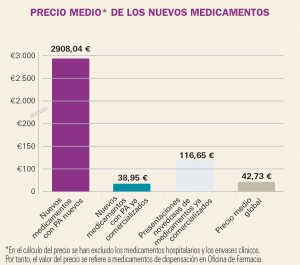
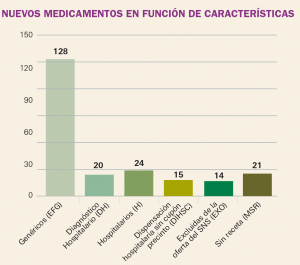
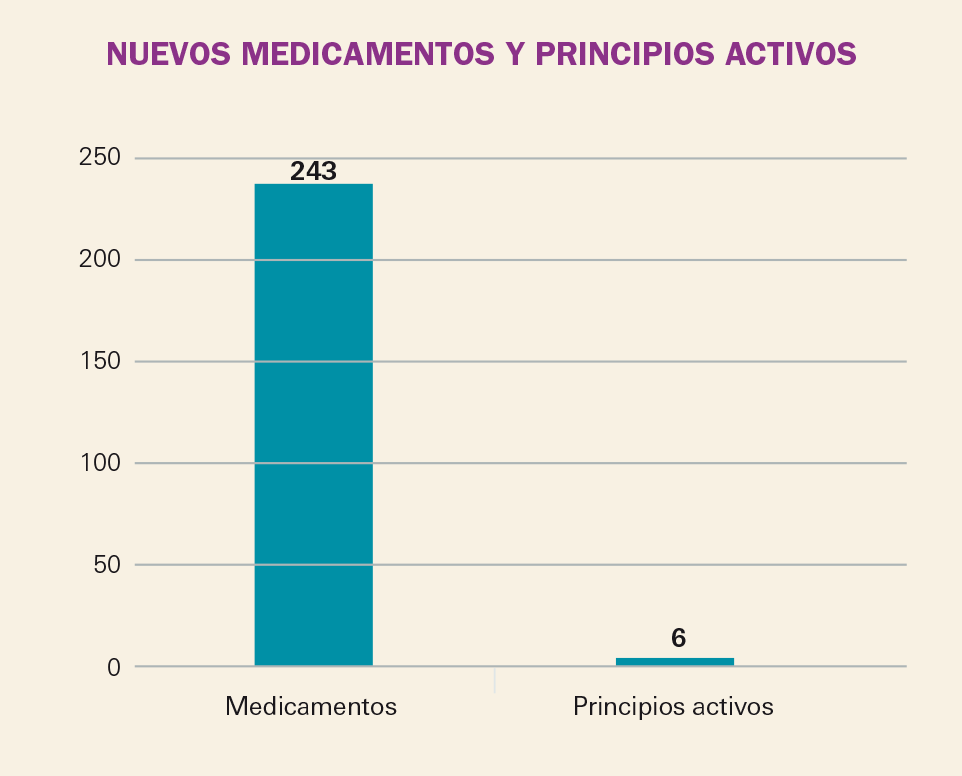{kind=link}
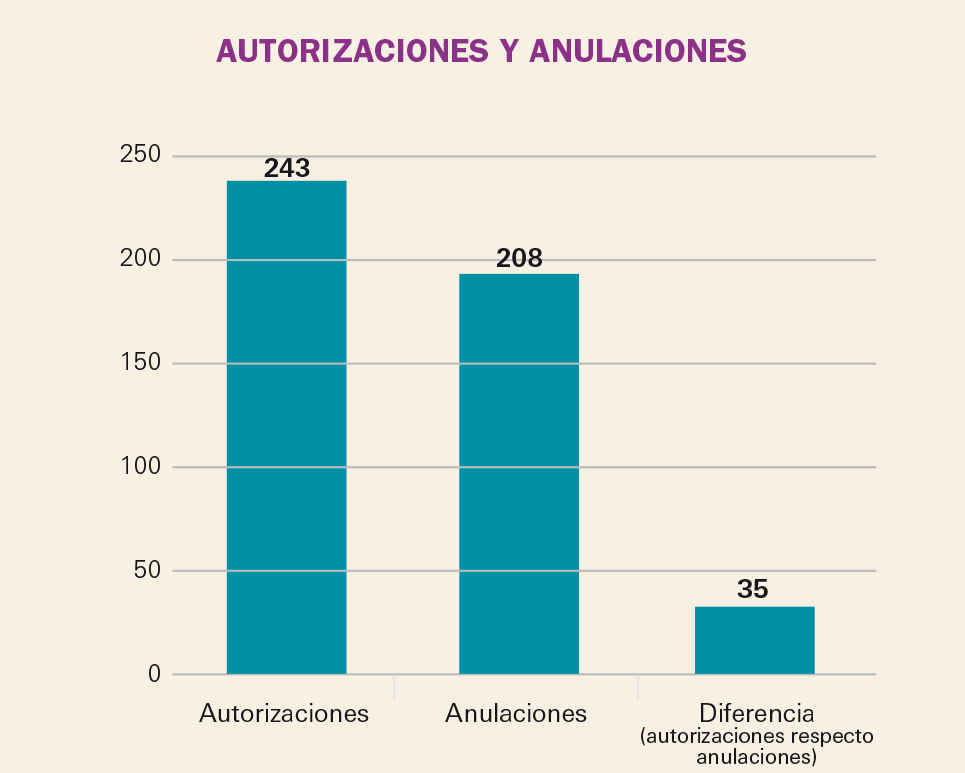{kind=link}
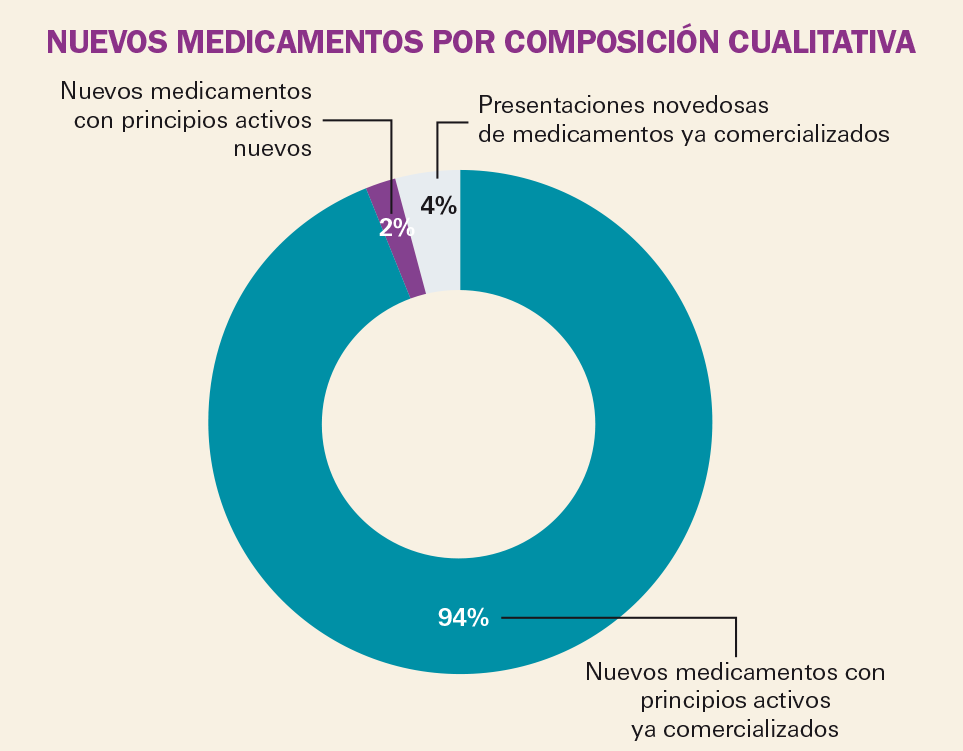{kind=link}
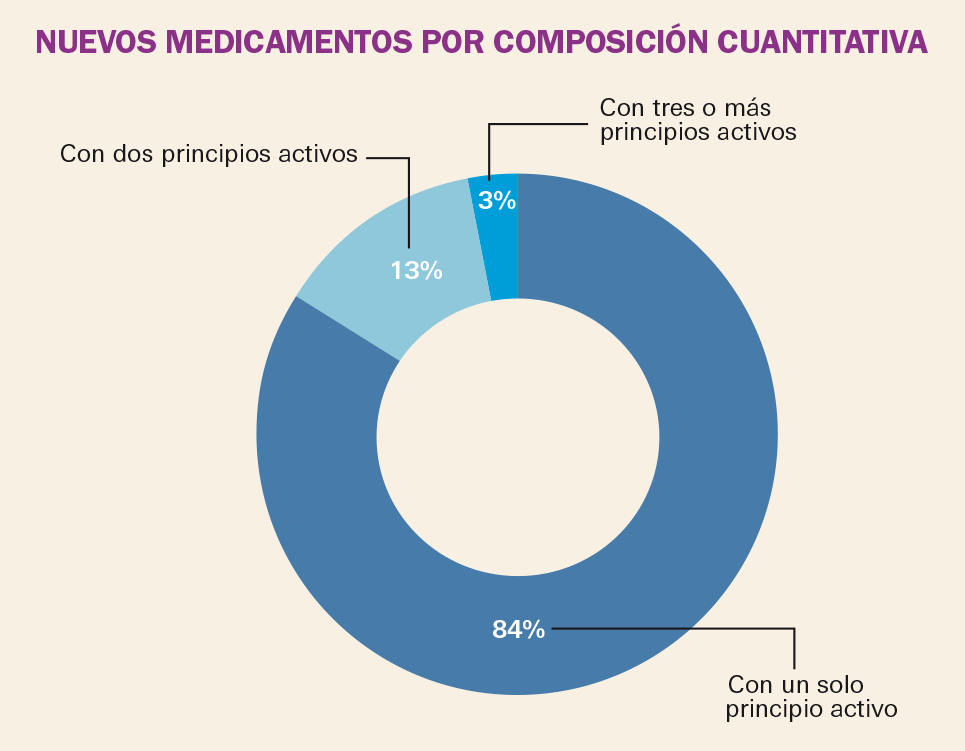{kind=link}
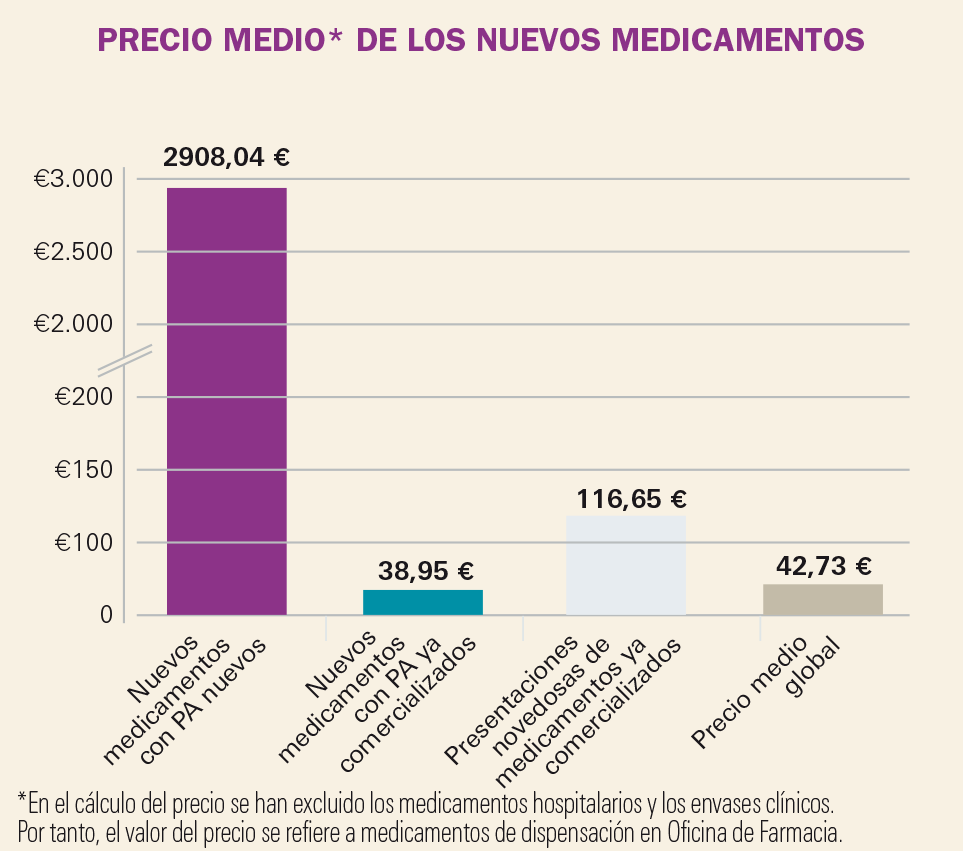{kind=link}
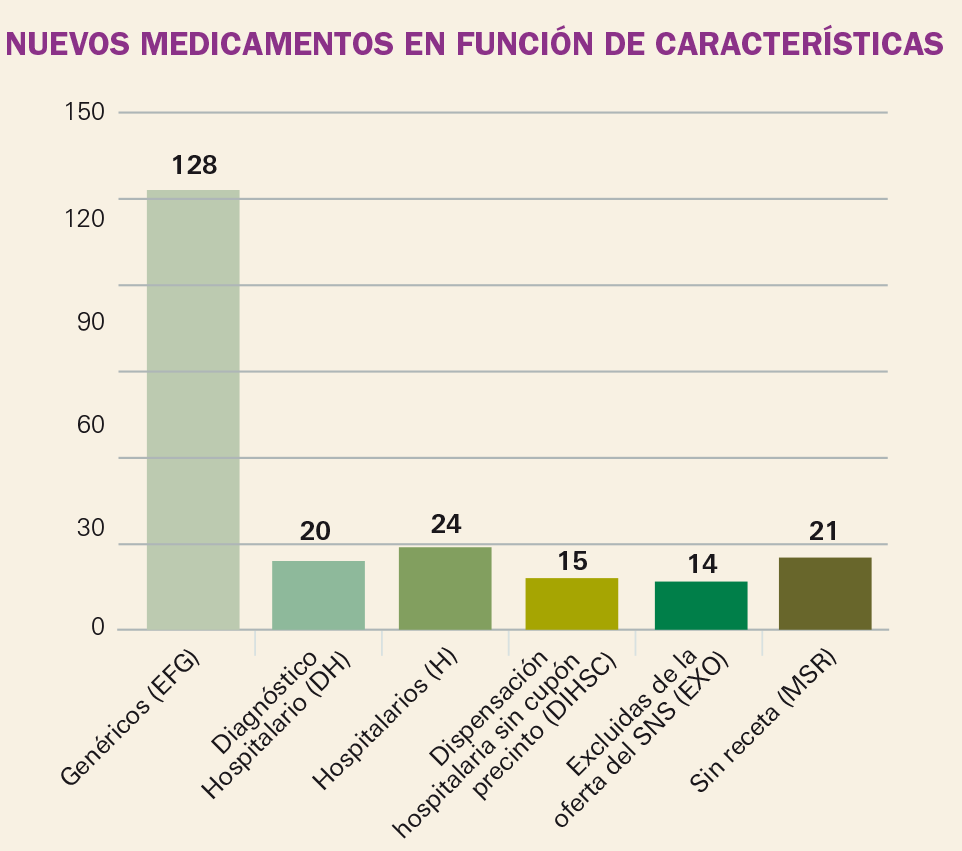{kind=link}