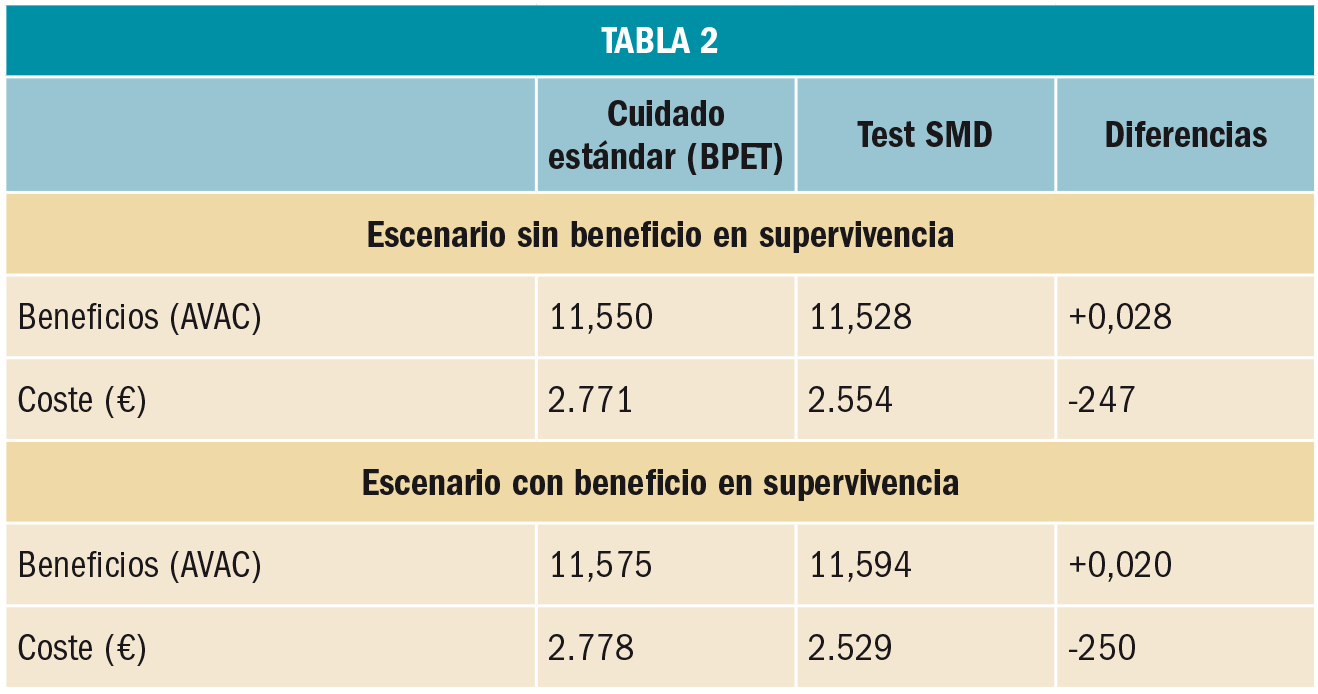
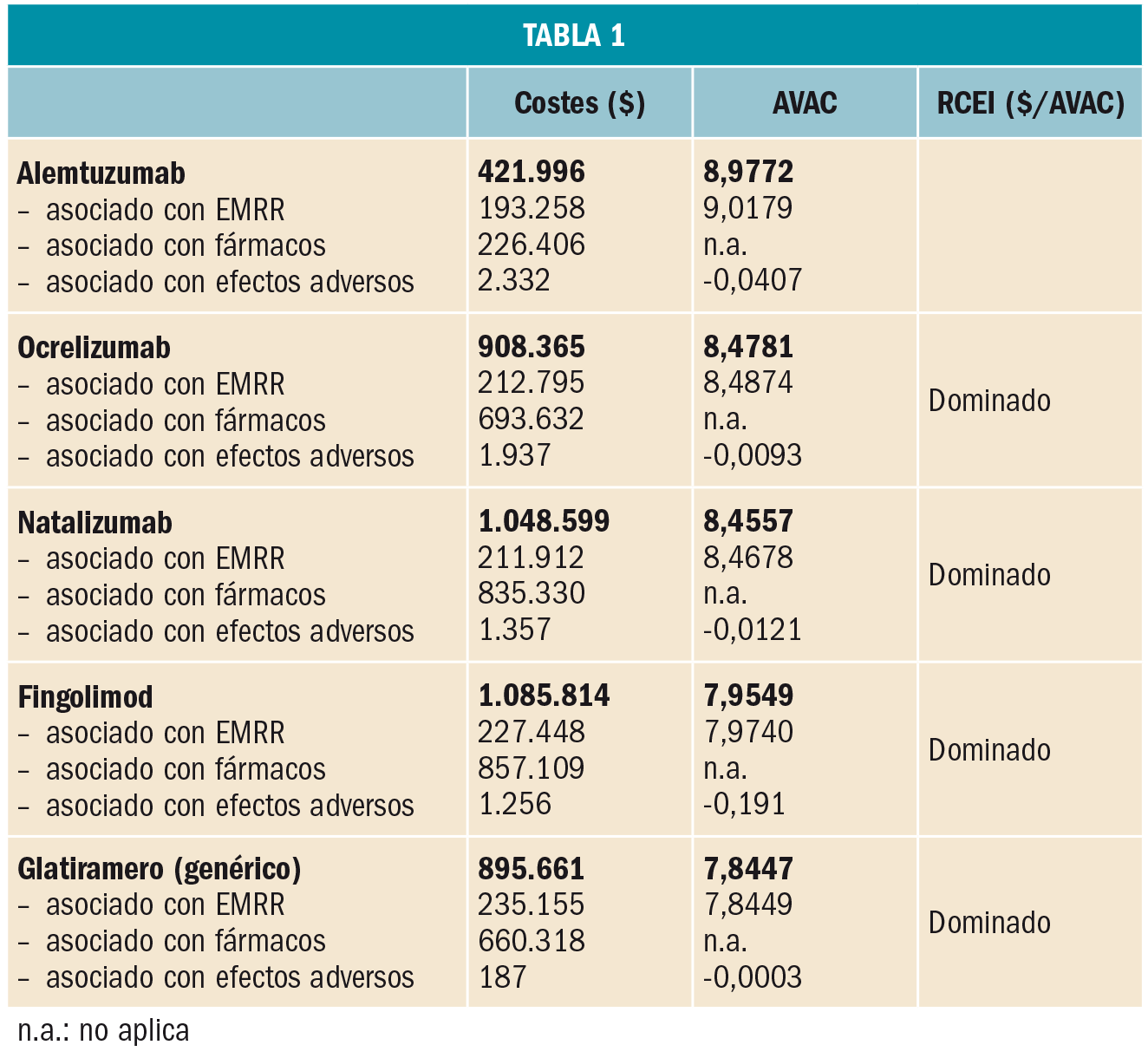
Un reciente ensayo clínico de fase IV ha demostrado la prometedora eficacia del cilostazol –un inhibidor de la fosfodiesterasa tipo 3 indicado en claudicación intermitente– como coadyuvante al donepezilo en el tratamiento de la enfermedad de Alzheimer. El cilostazol parece retrasar la disminución del metabolismo cerebral regional y mejorar o proteger la función cognitiva de los pacientes. Se requieren estudios más amplios y a largo plazo que permitan confirmar estos resultados.
El cilostazol es un antiagregante plaquetario que actúa inhibiendo las fosfodiesterasas intracelulares y, específicamente, la fosfodiesterasa tipo 3. En base a su efecto farmacológico, los medicamentos comerciales que lo contienen están indicados en el tratamiento sintomático de la claudicación intermitente.
Un reciente ensayo clínico de fase 4 ha evaluado, en pacientes con enfermedad de Alzheimer y lesiones en la sustancia blanca, su eficacia en combinación con donepezilo en comparación con donepezilo en monoterapia. Se trata de un estudio de 24 semanas de duración, aleatorizado, doblemente ciego, de grupos paralelos y controlado con placebo, que incluyó a un total de 36 pacientes que estaban en tratamiento con donepezilo y quienes fueron aleatorizados a recibir cilostazol (50 mg/12 h las dos primeras semanas y 100 mg/12 h hasta el final del estudio) o placebo (N=18 en cada grupo). Los participantes del ensayo se sometieron a exploraciones mediante PET-FDG (tomografía por emisión de positrones empleando como sonda la fluorodesoxiglucosa 18F) y a tres fases de pruebas clínicas y neuropsicológicas.
Los resultados del estudio muestran que los pacientes tratados con cilostazol no mostraban una disminución del metabolismo regional de la glucosa (medida indicativa de la viabilidad neuronal). Sin embargo, el metabolismo de la glucosa disminuyó significativamente en los lóbulos parietales y frontales de los pacientes del grupo placebo. Además, las medidas repetidas ANOVA, que mide las diferencias en los niveles de captación de la glucosa, evidenciaron que el metabolismo de la misma se preservaba de forma significativa en el giro frontal inferior izquierdo de los pacientes tratados con cilostazol en comparación con el grupo placebo (p<0,005).
Por otro lado, la media de las variaciones –desde el estado basal– en las escalas empleadas para evaluar la capacidad cognitiva y funcional de los pacientes (el Mini-Examen del Estado Mental; la subescala cognitiva del Test de Evaluación de la Enfermedad de Alzheimer; el Estudio Cooperativo de la Enfermedad de Alzheimer: Inventario de actividades de la vida diaria; y la Escala de Clasificación de Demencia Clínica por Suma de Recuadros) no difirió entre ambos grupos. No obstante, en el grupo de pacientes tratados con cilostazol, el aumento en el metabolismo de la glucosa se correlacionó con una mejora en la puntuación obtenida en la subescala cognitiva del Test de Evaluación de la Enfermedad de Alzheimer.
En resumen, los autores proponen que el la adición del cilostazol al tratamiento con donepezilo puede retrasar la reducción del metabolismo cerebral regional en los pacientes de enfermedad de Alzheimer con lesiones en la sustancia blanca, en mayor medida que la monoterapia con donepezilo. Los resultados publicados apuntan a la eficacia del cilostazol para mejorar o proteger la función cognitiva en la enfermedad de Alzheimer a través del aumento del metabolismo de la glucosa. Sin embargo, las limitaciones en el número de pacientes y el periodo de seguimiento en este estudio implican que la eficacia clínica real del cilostazol en la función cognitiva y su capacidad modificadora de la progresión de la enfermedad de Alzheimer debe evaluarse en ensayos más amplios y a largo plazo para poder extraer conclusiones definitivas.