Resumen
Guselkumab es un anticuerpo monoclonal que se une con elevada afinidad y especificidad a la subunidad p19 de la proteína interleucina 23 (IL-23). Al inhibir la acción proinflamatoria de esta citocina, impide la diferenciación y supervivencia de las células Th17, que serían las encargadas de liberar, por ejemplo, las IL-17A e IL-17-F, implicadas en la patogénesis de la psoriasis; el bloqueo de la acción biológica de IL-23 se traduce en la inhibición de la inflamación y de los síntomas clínicos de la psoriasis. El medicamento ha sido oficialmente autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamiento sistémico.
Los datos de eficacia y seguridad clínica derivan de tres amplios estudios pivotales de fase 3, aleatorizados, multicéntricos, doblemente ciegos y controlados con medicación activa (y, en dos de ellos, también con placebo) en pacientes con psoriasis en placas de moderada a grave candidatos a fototerapia o tratamiento sistémico. Con la pauta de administración autorizada (100 mg a tiempo 0 y a las 4 semanas, seguida de una dosis cada 8 semanas, vía subcutánea), guselkumab aportó una notable mejoría clínica frente a placebo a la semana 16 y a adalimumab (anti-TNFα ampliamente usado en psoriasis) a las semanas 16, 24 y 48, según los resultados obtenidos para las variables primarias de eficacia (IGA 0/1 y PASI 90). Además, guselkumab también demostró una superioridad estadísticamente significativa frente a ustekinumab (inhibidor de IL12 e IL23) en pacientes no respondedores a este fármaco que cambiaron su tratamiento (tras 16 semanas) frente a los que se mantuvieron en tratamiento con ustekinumab (media de 1,5 vs. 0,7 visitas en que los pacientes presentaban puntuación IGA 0/1 y mejoría de grado ≥2; p<0,001). Las mejorías observadas en variables secundarias –en términos de enfermedad regional (cuero cabelludo, manos y pies, y uñas), calidad de vida y síntomas referidos por los pacientes, entre otros– confirman el beneficio clínico aportado por guselkumab en el tratamiento de la psoriasis en placas moderada-grave.
En relación a su perfil toxicológico, el fármaco es relativamente bien tolerado. Los eventos adversos más comunes fueron nasofaringitis, infecciones del tracto respiratorio superior, dolor de cabeza, artralgia e hipertensión, la mayoría leves o moderados en gravedad. De forma similar a otros biológicos empleados en psoriasis, la tasa de efectos adversos graves fue muy baja, e incluso menor que la obtenida para adalimumab.
En definitiva, guselkumab es el primer fármaco que actúa única y selectivamente sobre IL-23, y emerge como una opción terapéutica alternativa –con eficacia clínica aparentemente más potente– a otros fármacos biológicos empleados en el tratamiento sistémico de la psoriasis moderada-grave, como los inhibidores de TNFα o ustekinumab. Es previsible que se posicione como fármaco de segunda línea, tras una respuesta inadecuada o contraindicación a tratamientos sistémicos convencionales o PUVA, aunque a la vista de los datos disponibles podría ser la primera opción entre los biológicos.
ASPECTOS FISIOPATOLÓGICOS
La psoriasis es una enfermedad inflamatoria de la piel de carácter crónico, aunque fluctuante, que puede afectar a la piel, uñas, articulaciones (artritis psoriásica) y, menos frecuentemente, a las mucosas. La lesión característica es una placa de color rojo oscuro, con escamas no adherentes de un peculiar tono blanco-nacaradas y con borde bien delimitado. Se manifiesta habitualmente de forma bilateral, siendo las localizaciones más frecuentes las superficies de extensión articular (codos y rodillas), la zona sacra y el cuero cabelludo. La afectación de las mucosas es muy rara, aunque se han citado casos localizados en los labios y en el pene. Las uñas están afectadas en un 20-50% de los casos, especialmente las de las manos; es aún más frecuente si hay afectación articular y en la forma eritrodérmica de psoriasis. Las lesiones más características son los hoyuelos o pits (depresiones puntiformes), manchas amarillentas debajo de la placa ungueal (mancha de aceite), fragilidad (onicolisis) e hipertrofia subungueal (Cuéllar, 2017).
La psoriasis es la más común de las enfermedades cutáneas crónicas humanas, con una incidencia del 2-4% en la población mundial. La prevalencia en niños varía desde el 0% en Taiwán al 2,1% en Italia, mientras que en los adultos oscila entre el 0,9% de Estados Unidos y el 8,5% de Noruega, con una incidencia entre las 78,9/100.000 persona-año en Estados Unidos y las 230/100.000 persona-año en Italia. Así, los datos sugieren que la aparición de la psoriasis varía según la edad y la región geográfica, siendo en general más frecuente en los países más distantes desde el ecuador. En España, la prevalencia de la patología se estima en torno al 1,4%.
Puede comenzar a cualquier edad, pero es rara en menores de 9 años. Presenta dos picos de máxima incidencia: la segunda década (de origen generalmente familiar) y los 55-60 años. Evoluciona con remisiones y recaídas espontáneas, y puede persistir toda la vida o durar solo unos meses. Si bien raramente llega a poner en peligro la vida del paciente, puede ser muy discapacitante, limitando considerablemente la calidad de vida.
Entre las principales características histológicas de la psoriasis pueden citarse la hiperplasia epidérmica, definida como una diferenciación anormal y maduración incompleta de los queratinocitos, es decir, un engrosamiento de la epidermis y una capa granular reducida o ausente. Todo ello es debido a la hiperproliferación y diferenciación de queratinocitos epidérmicos de evolución acelerada, cuyo ciclo vital es mucho más rápido y corto de lo normal: 7-10 días en lugar de 50-75 días. También se puede apreciar una infiltración de células del sistema inmune (linfocitos T) y de células dendríticas CD11+ en la dermis, así como de células CD8+ y neutrófilos en la epidermis.
Además de estas presencias celulares anómalas, también se puede observar un aumento en el proceso de formación de nuevos vasos sanguíneos (angiogénesis) y la inflamación de la piel. Aunque durante mucho tiempo se había pensado que la psoriasis es causada simplemente por la hiperproliferación de queratinocitos, actualmente se admite que el sistema inmune es un factor decisivo en el desarrollo de la enfermedad. En definitiva, hoy se considera que la psoriasis es una enfermedad inflamatoria crónica de origen autoinmune en la cual células dendríticas, linfocitos T, macrófagos y neutrófilos inducen hiperproliferaciones locales de los queratinocitos que, en última instancia, son los responsables de las lesiones de la piel.
Las células presentadoras de antígenos (antigen presenting cells, APC) son un elemento clave del sistema inmunitario, implicado en la captación, procesamiento y presentación de moléculas de carácter antigénico sobre la membrana. Estas células permiten dar a conocer dichos antígenos al sistema inmunitario, especialmente por los linfocitos T, iniciando la cadena de respuestas antigénicas específicas. En concreto, la presentación del antígeno y la formación de la sinapsis inmunológica en las APC provoca la secreción de diversas citocinas e induce la diferenciación de los linfocitos T en células efectoras específicas (linfocitos T facilitadores o helper, Th), particularmente las Th1, Th2 y Th17, cada una de ellas secretando sus propias citocinas.
Se ha demostrado la participación directa de varias citocinas en el incremento de la proliferación de los queratinocitos en la psoriasis. Particularmente, el factor de necrosis tumoral alfa (tumor necrosis factor α, TNFα) activa el desarrollo de las lesiones mediante el aumento del número de moléculas que participan en la respuesta inflamatoria a las moléculas de adhesión. Los queratinocitos así activados producen citocinas y quimiocinas, que atraen a los linfocitos al sitio de la inflamación y que potencian su proliferación. De hecho, se suelen encontrar subpoblaciones de linfocitos Th1 y Th17 en las lesiones psoriásicas de la piel, además de queratinocitos, células dendríticas y células de Langerhans y, como consecuencia de todo ello, un aumento de la concentración de TNFα en las zonas de la piel afectadas. Es más, se ha observado que una disminución del TNFα, tanto en suero como en las lesiones, se relaciona con una mejora clínica, lo que sugiere un importante rol en la enfermedad.
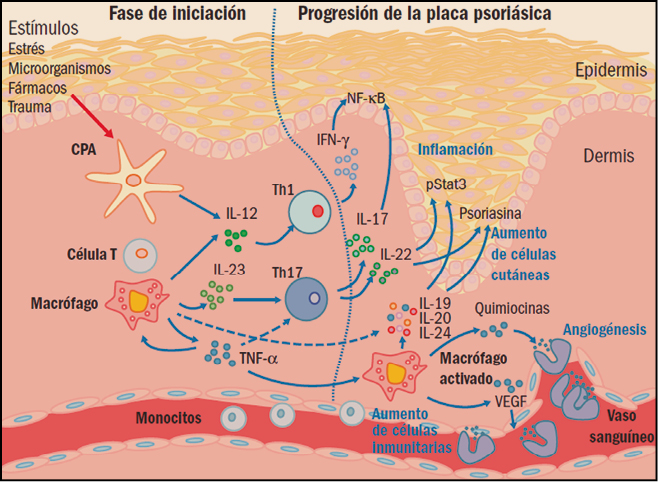
Figura 1. Iniciación y progresión de la lesión psoriásica. CPA: célula presentadora de antígenos; IFN-γ: interferón gamma; IL: interleucinas; NF-κB: factor nuclear κB; Th: linfocitos T helper (facilitadores); TNFα: factor de necrosis tumoral α; VEGF: factor de crecimiento endotelial vascular.
Asimismo, se ha observado que las interleucinas (IL) 12e IL-23 pueden tener también un importante papel patológico en la psoriasis. La IL-12 –producida por las células presentadoras de antígenos– es capaz de inducir la producción de nuevas poblaciones de linfocitos T e incrementar las respuestas de los linfocitos Th1, sobre todo en la producción de interferón (IFN). Estas células también parecen estimular la inmunidad mediada por células y la síntesis de anticuerpos fijadores del complemento. Por su parte, la IL-23 facilita la adquisición de memoria inmunológica por los linfocitos T, en especial de las subpoblaciones de linfocitos Th17, y parece tener un papel crítico en la patogénesis de la psoriasis. En este sentido, datos procedentes de modelos inflamatorios de la piel sugieren que los linfocitos Th17, que producen IL-17 e IL-22, podrían ser los inductores principales de la hiperplasia epidérmica, modificando la diferenciación epidérmica en la psoriasis.
Por otro lado, se ha observado que las anomalías en la regulación de IL-12 e IL-23 no solo se asocian a psoriasis, sino también a otras patologías de índole autoinmune, como la enfermedad de Crohn, la artritis reumatoide y la colitis ulcerosa, entre otras. De hecho, entre el 5% y el 7% de todos los pacientes con psoriasis y hasta un 40% de aquellos con la forma más grave (>10% de superficie corporal afectada) desarrollan artritis psoriásica, usualmente entre 5 y 10 años tras el inicio de la enfermedad cutánea. La afectación articular es más frecuente en los pacientes de 40-50 años, siendo la forma más común (50-70%) la oligoarticular asimétrica seronegativa, que afecta a las pequeñas articulaciones de algunos dedos de las manos.
También parece que las interacciones entre el antígeno asociado a la función leucocitaria de tipo 1 (LFA-1) y las moléculas de adhesión intercelular facilitan la patogenia de la psoriasis. En concreto, favorecen la migración de los linfocitos T desde la circulación sistémica a los tejidos de la dermis y epidermis, y su consiguiente reactivación. Todo ello conduce a una infiltración de las células T activadas en el tejido y a una proliferación anormal de los queratinocitos. Por su parte, la alta producción de factores de crecimiento endoteliales vasculares (vascular endothelial growth factors, VEGF) en los queratinocitos psoriásicos promueve la angiogénesis, lo que provoca un aumento de la vascularización y la inflamación local. Los neutrófilos se encuentran en grandes cantidades en las lesiones psoriásicas; de hecho, se ha demostrado que algunas citocinas, tales como la IL-8, causan la acumulación de neutrófilos en la piel.
Sin embargo, a pesar de todos los mecanismos bioquímicos mencionados, el origen específico de la enfermedad sigue siendo desconocido, toda vez que se ignora qué es lo que provoca la respuesta inmunológica y desencadena la hiperqueratosis y el resto de manifestaciones patológicas de la psoriasis. En la aparición de la psoriasis influyen significativamentealgunos factores genéticos, como lo demuestra la marcada agregación familiar, así como la concordancia en gemelos y la asociación a determinados antígenos principales de histocompatibilidad (HLA): es más frecuente en presencia del alelo Cw6 y, en pacientes con HLA B27, el inicio de la psoriasis es más precoz y la evolución más grave.
Entre los factores externos desencadenantes pueden citarse traumatismos externos, determinadas infecciones (la forma “en gotas” aparece poco después de una faringitis estreptocócica), el uso de determinados fármacos (litio, betabloqueantes, antipalúdicos, antiinflamatorios no esteroideos (AINE), supresión del tratamiento con esteroides, etc.), bebidas alcohólicas, factores psicógenos (especialmente el estrés, que puede actuar como desencadenante o agravante), el clima (el clima cálido y la luz solar son beneficiosos mientras que el frío empeora las lesiones), factores metabólicos (hipocalcemia, alcoholismo, diálisis, etc.) y factores endocrinos (mayor incidencia en la pubertad y la menopausia, mejora en el embarazo).
FORMAS CLÍNICAS
- Psoriasis vulgar o en placas: es la forma clínica más frecuente (hasta el 90% de los casos), y su nombre hace referencia a las formaciones escamosas y de color rojizo presentes en las zonas de extensión (codos y rodillas, principalmente), así como en el cuero cabelludo. Se trata de placasbien delimitadas con una distribución simétrica en la mayoría de los casos, aunque pueden confluir y formar figuras policíclicas. El porcentaje del cuerpo afectado por las placas psoriásicas puede variar desde una forma leve (<2%) hasta las formas más graves (>10%), pasando por la forma moderada (2-10%). La enfermedad, considerada como crónica aunque de curso variable (con recaídas y remisiones de duración diversa), suele manifestarse por vez primera en dos grupos de edad: 16-22 y 57-60 años.
- La denominada psoriasis en gotas suele cursar con numerosas lesiones puntiformes (menores de 1 cm), sobre todo en el tronco. Es más común en niños y adolescentes, siendo típica su erupción aguda 10-14 días tras una infección estreptocócica, habitualmente de garganta, y que desaparece espontáneamente en 2-3 meses.
- Por su parte, la psoriasis invertida afecta a grandes pliegues (axilar, submamario, interglúteo) y presenta placas rojas lisas y brillantes, de color vivo, sin descamación y ocasionalmente con fisuras.
- La psoriasis pustulosa es una forma aguda y poco frecuente. Puede ser generalizada (tipo von Zumbusch), como la forma de comienzo de una psoriasis, o aparecer en el curso de una psoriasis crónica. Cursa con una brusca fiebre elevada, malestar general, eritema con pústulas en pocas horas, piel de color rojo escarlata seca y no descamativa. Sin tratamiento puede ser mortal. La forma localizada palmoplantar cursa con brotes repetidos de pústulas estériles sobre una base eritematosa en las palmas y las plantas, simétricas, y que suelen secarse, dejando escamas y costras marrones.
- Finalmente, la psoriasis eritrodérmica consiste en una forma generalizada y grave. Se instaura generalmente sobre cuadros de psoriasis crónica. Se presenta como una eritrodermia exfoliativa seca, que afecta todo el tegumento, incluyendo el pelo y sobre todo las uñas.
En general, la psoriasis se asocia con un aumento del riesgo de aterosclerosis y del riesgo de enfermedad cardiovascular, que se asocia con mayores tasas de morbilidad y mortalidad, especialmente en los pacientes de psoriasis más jóvenes y con formas más graves de la enfermedad, reduciendo su esperanza de vida. Los datos epidemiológicos también parecen apoyar una asociación de la psoriasis y de la artritis psoriásica con una mayor prevalencia de hipertensión. Además de las complicaciones vasculares, la psoriasis se ha relacionado con un incremento de la incidencia de algunas metabolopatías de alta incidencia, especialmente diabetes mellitus de tipo 2 y síndrome metabólico.
TRATAMIENTO
El tratamiento de la psoriasis es complejo, ya que no solo se lucha contra una enfermedad de etiología desconocida y con formas clínicas muy diversas, sino que está condicionada por diversos factores sociales. En principio, deben evitarse los factores desencadenantes y favorecedores conocidos: infecciones, golpes, tabaquismo y estrés. Se ha comprobado que el estrés del paciente tiende a agravar y a hacer más frecuentes las recaídas. Por el contrario, el sol tiene un efecto beneficioso, siendo capaz de producir una mejoría significativa de las lesiones; sin embargo, no existen evidencias sobre la posible eficacia de otros tratamientos no farmacológicos.
No existe un tratamiento curativo para la psoriasis, pero en la mayoría de los casos puede controlarse satisfactoriamente, aplicando diferentes tratamientos en función de la gravedad del caso. No obstante, la calificación de los resultados del tratamiento depende en buena medida de la aceptación de los pacientes, de sus criterios estéticos y de su propia personalidad. Por otro lado, las lesiones ungueales asociadas con la psoriasis son difíciles de tratar.
Los tratamientos tópicos son empleados en los casos más leves (afectación menor del 2% de la superficie corporal) y constituyen la forma más común de tratamiento de la psoriasis en placas, pero también es la menos eficaz en los casos graves. Carecen de utilidad en la artritis psoriásica o en la forma pustulosa o eritrodérmica.
Los agentes emolientes y queratolíticos son utilizados habitualmente como adyuvantes a otros tratamientos para hidratar, evitar la aparición de fisuras y eliminar las escamas. No deben aplicarse en pliegues. Entre los agentes queratolíticos, el ácido salicílico es el menos eficaz de todos los tratamientos disponibles, pero también el más barato y el mejor aceptado por los pacientes, por lo que constituye un paso indispensable en la terapéutica de la psoriasis en placas. La brea de hulla (coaltar) es algo más potente como queratolítico que el anterior, pero presenta como inconveniente el olor desagradable; sus efectos son lentos y de baja potencia, aunque produce remisiones generalmente prolongadas en los pacientes sensibles al tratamiento. El ditranol (antralina) es uno de los componentes activos de la brea de hulla; debido a su poder irritante para la piel y a su capacidad para manchar la ropa y teñir las uñas y la piel, muchos pacientes tienden a rechazar este tratamiento. Sin embargo, se trata de uno de los tratamientos tópicos más eficaces (más que los anteriores), cuyos efectos aparecen lentamente, aunque no tanto como los de la brea de hulla, y producen remisiones algo más cortas que ésta.
Los corticosteroides tópicos producen efectos rápidos y potentes, pero la duración de las remisiones es más bien corta. Se pueden considerar de primera elección en la psoriasis leve que no responde a otros tratamientos tópicos y en determinadas localizaciones como la cara, el cuero cabelludo, los pliegues, los genitales (localizaciones que no toleran otros tratamientos tópicos). Presentan el inconveniente de que, tras la suspensión del tratamiento, la enfermedad puede reactivarse. No es infrecuente la combinación de corticosteroides tópicos con agentes queratolíticos, de efectos menos potentes y rápidos, pero considerablemente más prolongados.
El calcipotriol y el tacalcitol son análogos hormonales de la vitamina D de aplicación tópica, similares al calcitriol. Su empleo en la psoriasis en placas se debe a la observación de que los análogos hormonales de la vitamina D son capaces de inhibir la proliferación y la diferenciación de los queratinocitos. Su eficacia es similar a la de los corticosteroides e incluso inducen periodos de remisión algo más largos que aquellos.
Cabe destacar que, en pacientes con psoriasis en el cuerpo y el cuero cabelludo, el tratamiento combinado con vitamina D y corticosteroides funciona mucho mejor que cualquiera de estos solos. Los análogos de la vitamina D producen por lo general mejores resultados que el alquitrán de hulla, pero los resultados en relación con el ditranol son dispares. Los corticosteroides son, como mínimo, igual de eficaces que los análogos de la vitamina D, pero se asocian con una menor incidencia de efectos adversos locales.
El empleo de lámparas de radiación ultravioleta (UV) constituye uno de los puntales en el tratamiento de la psoriasis. Sin embargo, la aplicación de radiación UV solo resulta útil en los casos de psoriasis en placas, resultando ineficaz en el resto de formas de psoriasis (artritis, etc.). Según la longitud de onda de la radiación se distinguen dos tipos básicos de radiación. La de longitud de onda más larga (UVA) tiene una menor capacidad de penetración en la piel; por este motivo, requiere la administración previa de sustancias que sensibilicen la piel (generalmente psoralenos), en lo que se conoce como terapia PUVA (psoralenos + UVA). Esta forma de tratamiento es conocida como fotoquimioterapia. Por su parte, la radiación de longitud de onda más corta (UVB) tiene una mayor capacidad de penetración y no requiere ninguna sustancia sensibilizante, aunque se suele emplear brea de hulla previamente; este método es conocido como fototerapia.
El método PUVA o fotoquimioterapia es el tratamiento más eficaz disponible para la psoriasis en placas. Su acción es lenta, pero produce periodos prolongados de remisión. Debido al riesgo de efectos adversos cutáneos se está empleando de forma mucho más restringida, para casos graves refractarios en pacientes de edad media (no en niños ni en jóvenes).
Por su parte, el tazaroteno es un retinoide que se utiliza por vía tópica y que presenta un eficacia similar a la de los corticosteroides tópicos en lo que se refiere a la elevación de las placas psoriásicas en los pacientes con psoriasis en placas, pero su efecto es algo menor en cuanto a la reducción del eritema. La combinación de tazaroteno y corticosteroides produce mejores resultados que el tazaroteno solo.
En el tratamiento sistémico se emplean agentes con efectos antiproliferativos sobre la epidermis, sobre todo fármacos inmunosupresores y derivados retinoides aromáticos. Son considerados como el segundo nivel de tratamiento, estando indicados en psoriasis extensas que no responden a otros tratamientos, formas eritrodérmicas y pustulosas y formas incapacitantes.
Los denominados fármacos antirreumáticos modificadores de la enfermedad (FAME o DMARD, disease modifiying antirheumatic drugs) son ampliamente utilizados como primera opción en el tratamiento de las formas activas moderadas o graves de la psoriasis, en particular en los pacientes con artritis psoriásica. Se trata de potentes inmunosupresores, entre los cuales el más utilizado es, sin duda, el metotrexato, considerado como el tratamiento de elección en las formas graves de psoriasis en placas, así como en la artritis psoriásica, la psoriasis pustulosa y la psoriasis eritrodérmica, por sus efectos antiproliferativo y antiinflamatorio. Por su parte, la ciclosporina –agente inmunosupresor que actúa inhibiendo especialmente la producción de anticuerpos dependientes de células T colaboradoras (aunque también inhibe la producción y liberación de linfocinas, sobre todo de IL-2)– tiene una eficacia clínica similar a la del metotrexato en la psoriasis en placas y en la psoriasis pustulosa, pero algo menor en la psoriasis eritrodérmica y en la artritis psoriásica.
En general, el metotrexato, la ciclosporina, los UVB y los PUVA son consideradas como las formas más eficaces de tratamiento de los casos graves o moderadamente graves de psoriasis, facilitando la desaparición prácticamente completa de las manifestaciones clínicas en gran parte de los pacientes. Una vez alcanzado este objetivo, el tratamiento puede ser reducido o incluso suspendido, al menos hasta que se produzca una recidiva (si es que llega a producirse).
Los retinoides son análogos estructurales de la vitamina A (ácido retinoico), pero de carácter aromático. Actúan sobre receptores específicos, reduciendo la producción de estímulos inflamatorios y la diferenciación y proliferación de los queratinocitos. Revierten los cambios típicos hiperqueratósicos de la psoriasis en placas, pero son potentes teratógenos, por lo que su uso debe ser estrictamente vigilado en mujeres. Actualmente solo está disponible la acitretina, que actúa sobre diversos procesos biológicos en la piel, incluyendo la proliferación y diferenciación celular, la función inmunológica, la inflamación y la producción de sebo. Los efectos de los retinoides son debidos a la activación de receptores específicos del ácido retinoico, conocidos como RAR (retinoic acid receptors). Tanto acitretina, como ciclosporina y metotrexato se asocian a toxicidad significativa sobre diversos órganos y tienen limitaciones en los tratamientos a largo plazo de formas crónicas de psoriasis.
Teniendo en consideración la relevancia de la citocina TNFα en el desarrollo de la inflamación asociada a la psoriasis (previamente comentada), la terapia biológica anti-TNFα fue desarrollada precisamente para bloquear el TNFα e impedir o limitar su actividad y, en consecuencia, reducir las interacciones entre las células inmunes y los queratinocitos. La neutralización del TNFα impide su interacción con sus receptores (TNFR1) y, con ello, la subsiguiente cascada bioquímica que, entre otros efectos, desemboca en la activación del factor nuclear kappa B (NF-κB; factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas), un complejo proteico que se encuentra en la mayoría de los tipos de células animales y está implicado en la respuesta celular frente a estímulos como el estrés, las citocinas, la radiación UV y antígenos diversos. El NF-κB juega un papel clave en la regulación de la respuesta inmunitaria, dado que las cadenas ligeras kappa son componentes básicos de las inmunoglobulinas.
Asimismo, otra de las consecuencias del bloqueo del TNFα es el cambio en los niveles de las moléculas de adhesión responsables de la migración leucocitaria (ELAM-1, VCAM-1 e ICAM-1). Igualmente, se puede apreciar una disminución de los niveles de metaloproteinasas matriciales (MMP-1 y MMP-3), responsables de la remodelación tisular.
Los fármacos anti-TNFα actualmente comercializados en España que están indicados expresamente en la psoriasis son adalimumab, etanercept, infliximab y certolizumab pegol. Otro agente anti-TNF disponible en nuestro país es el golimumab, que está indicado en la artritis psoriásica, la artritis reumatoide, la colitis ulcerosa y la espondilitis anquilosante.
Por otra parte, tanto el secukinumab como el ixekizumab son anticuerpos monoclonales humanos que se unen y neutralizan a la interleucina 17A1 (IL-17A), una citocina proinflamatoria considerada como uno de los principales inductores de la hiperplasia y diferenciación epidérmica observada en la psoriasis, a través de la formación de factor nuclear κB (NFκB). Por su parte, el brodalumab se une al receptor de la IL-17A (IL-17AR), bloqueando la actividad de la IL-17A, además de la de IL-17F y del heterodímero IL-17A/F. Estos tres medicamentos han sido autorizados para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
El ustekinumab es un anticuerpo monoclonal frente a IL-12 e IL-232, autorizado para el tratamiento de la psoriasis en placas moderada o grave en adultos que no responden, tienen contraindicada o no toleran otras terapias sistémicas, incluyendo metotrexato, ciclosporina y PUVA. La IL-12 y la IL-23 contribuyen a la activación de los linfocitos natural killers (NK) y a la activación y diferenciación de los linfocitos CD4+ y su regulación parece estar alterada en pacientes con psoriasis y otras patologías de etiología autoinmne. De ahí que la formación del complejo de ustekinumab con dichas interleucinas impida la activación del receptor celular de éstas (IL-12Rβ1), tanto solo como formando parte de receptores complejos duales (IL-12Rβ1/β2 e IL-12Rβ1/23R), y en consecuencia, interrumpa la señalización bioquímica mediada por estos receptores, que participa en la secreción de citocinas inflamatorias implicadas en la psoriasis por parte de determinadas poblaciones de linfocitos.
Por último, el apremilast es un inhibidor selectivo de la fosfodiesterasa de tipo 4 (PDE4) que está indicado en el tratamiento de la psoriasis en placas crónica de moderada a grave en pacientes adultos que no han respondido, tienen contraindicado o no toleran otro tratamiento sistémico, incluyendo ciclosporina, metotrexato o psoraleno y luz ultravioleta A (PUVA). Al inhibir a la enzima PDE4, implicada en el metabolismo de AMPc, incrementa los niveles intracelulares de éste y facilita la reducción de la expresión de citocinas proinflamatorias, fundamentalmente TNFα e IL-12; asimismo, parece modular los niveles de otras citocinas, en este caso de carácter antiinflamatorio, como la IL-10.
Los datos publicados de varios ensayos clínicos –que han evaluado la adición de terapias tópicas a los fármacos biológicos con la intención de mantener las respuestas iniciales–, aunque limitados, sugieren que tal combinación es una estrategia eficaz y bien tolerada para controlar la psoriasis y mejorar la calidad de vida de los pacientes. De igual modo, una publicación reciente revisó la evidencia disponible sobre la combinación de fármacos biológicos y de fototerapia para la psoriasis moderada a grave y concluyó que 9 de cada 10 de los estudios publicados demostraban eficacia y seguridad favorables para la combinación de ambos tratamientos (EMA, 2017).
ACCIÓN Y MECANISMO
El guselkumab es un anticuerpo monoclonal que se une con elevada afinidad y especificidad a la subunidad p19 de la proteína interleucina 23 (IL-23), bloqueando las acciones biológicas mediadas por esta citocina proinflamatoria, lo que se traduce en la inhibición de la inflamación y de los síntomas clínicos de la psoriasis. El medicamento ha sido oficialmente autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamiento sistémico.
La IL-23 es una citocina reguladora heterodimérica principalmente producida y liberada por macrófagos, que afecta a la diferenciación, expansión y supervivencia de subgrupos de linfocitos T (por ejemplo, células Th17 y células Tc17) y subgrupos de células inmunitarias innatas, que representan fuentes de citocinas efectoras, como IL-17A, IL-17F e IL-22 (que, a su vez, inducen la enfermedad inflamatoria). Una creciente evidencia confirma que la vía de las IL-23/IL-17 contribuye significativamente a la fisiopatología de muchas enfermedades inmunomediadas, entre las que se incluyen, además de la psoriasis en placas, eritrodérmica y pustular, la espondilitis anquilosante o la enfermedad inflamatoria intestinal. La susceptibilidad a la psoriasis ha sido asociada, incluso, con polimorfismos genéticos en IL-23 y su receptor específico (IL-23R) (Puig, 2017).
Los estudios in vitro han demostrado que guselkumab inhibe la bioactividad de la IL-23 bloqueando su interacción con su receptor específico (IL-23R) de la superficie celular, lo que altera la señalización, la activación y las cascadas de citocinas mediadas por la IL-23, e impide la adquisición de memoria inmunológica por los linfocitos T: al neutralizar la IL-23 es capaz de inhibir la respuesta inmunitaria mediada por la IL-17 liberada por los linfocitos T helper 17. En seres humanos, se ha demostrado que el bloqueo selectivo de la IL-23 normaliza la producción y concentraciones séricas de ésta y otras citocinas relacionadas (IL-17A, IL-17F e IL-22), que se encuentran elevadas en la piel de los pacientes con psoriasis en placas.
Además, un estudio en fase I demostró –mediante análisis de ARNm obtenidos de biopsias cutáneas de lesiones de pacientes con psoriasis en placas– que el tratamiento con guselkumab reducía la expresión de los genes de la vía IL-23/Th17 y los perfiles de expresión de los genes asociados a la psoriasis.
Aspectos moleculares
Guselkumab es un anticuerpo monoclonal completamente humano de tipo inmunoglobulina G1 lamda (IgG1λ) producido en células de ovario de hámster chino (CHO) por tecnología del ADN recombinante.
La molécula intacta presenta un peso molecular aproximado de 143,6 kD y una fórmula molecular C6402H9864N1676O1994S42. Está formada por 2 cadenas pesadas idénticas de 447 aminoácidos (aproximadamente 49 kDa cada una) y 2 cadenas ligeras idénticas de 217 aminoácidos (aproximadamente 23 kDa cada una), que se unen entre ellas por enlaces disulfuro covalentes e interacciones proteína-proteína no covalentes. Los glicanos unidos a la posición N-terminal son bicatenarios.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de guselkumab han sido adecuadamente contrastadas en las indicaciones autorizadas mediante tres estudios clínicos confirmatorios de fase 3: VOYAGE 1 (PSO3001), VOYAGE 2 (PSO3002) y NAVIGATE (PSO3003). Se trata de ensayos pivotales aleatorizados, multicéntricos, multinacionales, doblemente ciegos y controlados con medicación activa en pacientes con psoriasis en placas de moderada a grave que eran candidatos a fototerapia o tratamiento sistémico. Los tres ensayos enrolaron a un total de 2.700 adultos. Las principales características basales de las poblaciones de estudio estuvieron bastante balanceadas en los tres ensayos clínicos (Tabla 1).
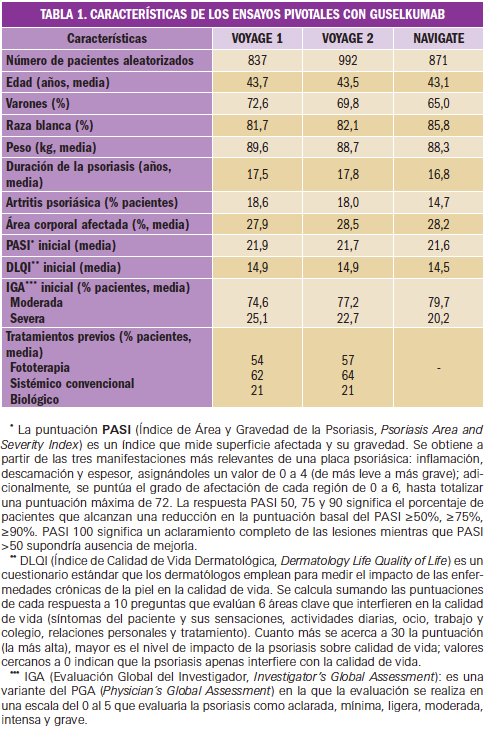
Por un lado, en los estudios VOYAGE 1 y VOYAGE 2 se evaluó la eficacia y la seguridad de guselkumab comparado con placebo y adalimumab en un total de 1.829 pacientes adultos. Los 825 pacientes asignados al grupo de guselkumab recibieron 100 mg del fármaco en las semanas 0 y 4, y luego cada 8 semanas hasta la semana 48 (VOYAGE 1) y la semana 20 (VOYAGE 2), mientras que 582 pacientes recibieron 80 mg del comparador activo adalimumab en la semana 0 y 40 mg en la semana 1, seguido de 40 mg cada 2 semanas hasta la semana 48 y la semana 23, respectivamente. En VOYAGE 2, además, los pacientes del grupo adalimumab que no alcanzaron PASI 90, comenzaron ser tratados con guselukmab a las semanas 28 y 32, y luego cada 8 semanas. Entre ambos ensayos, 422 pacientes fueron aleatorizados en el grupo de placebo, que implicaba, tras varias dosis de placebo, la administración de 100 mg de guselkumab en las semanas 16 y 20 y luego cada 8 semanas.
En los dos ensayos se realizó un seguimiento máximo a los pacientes de 48 semanas desde la primera administración, si bien la eficacia y seguridad a largo plazo de guselkumab se está actualmente evaluando en las extensiones de 4 años, con lo que ambos estudios tendrán una duración total de 5 años.
Según los primeros resultados publicados para VOYAGE 1 (Blauvelt, 2018) y VOYAGE 2 (Reich, 2017), guselkumab demostró aportar una mejoría significativa de las medidas de la actividad de la enfermedad –evaluando enfermedad cutánea global, enfermedad regional (cuero cabelludo, manos, pies y uñas), calidad de vida y resultados comunicados por los pacientes– en comparación con placebo y con adalimumab en la semana 16 y en comparación con adalimumab en las semanas 24 y 48. Como variables co-primarias de eficacia se emplearon el PASI75, PASI90 y PASI100, la tasa de respondedoressPGA3y la de PSI4(Tabla 2).
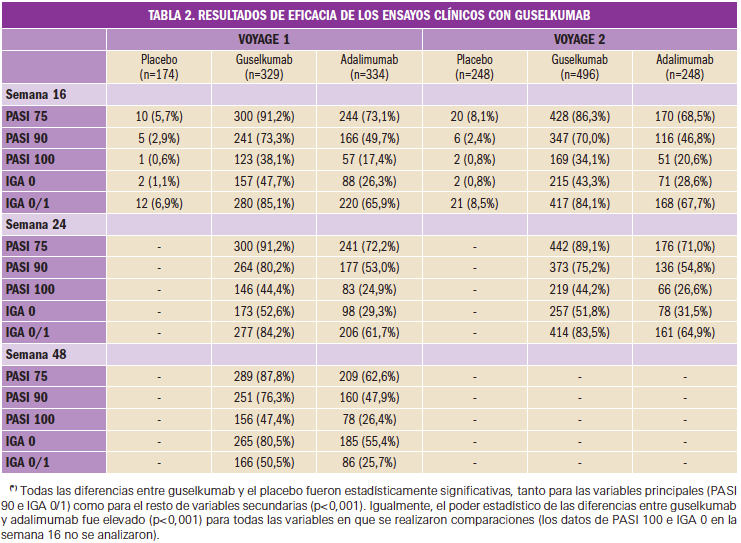
Cabe destacar que guselkumab demostró eficacia de manera rápida, con una mejoría porcentual significativamente mayor del PASI en comparación con placebo ya en la semana 2 (p<0,001). El porcentaje de pacientes que consiguieron una respuesta PASI 90 fue numéricamente mayor con guselkumab que con adalimumab a partir de la semana 8, hallándose la diferencia máxima en la semana 20 (VOYAGE 1 y 2) y se mantuvo hasta la semana 48 (VOYAGE 1).
De forma interesante, en VOYAGE 2, de los 112 pacientes que no respondieron a adalimumab en la semana 28 (no alcanzaron PASI 90) y que se pasaron a tratamiento con guselkumab, el 66% alcanzó respuesta significativa (PASI 90) tras 20 semanas con el nuevo fármaco. El ensayo VOYAGE 2 demostró, además, que en respondedores a guselkumab en la semana 28 a los que se les retiró el fármaco, se atenuaba considerablemente la mejoría clínica de la psoriasis a la semana 48 con la administración de placebo respecto a los pacientes que se mantuvieron en tratamiento con guselkumab (PASI 90 de 36,8% vs. 88,6%; p<0,001).
Los resultados obtenidos para las variables secundarias de eficacia no hicieron sino confirmar los hallazgos comentados para las variables primarias: el tratamiento con guselkumab demostró mejorías significativas a la semana 16 frente a placebo (p < 0,001), tanto en las pruebas de evaluaciones específicas de enfermedad regional (en cuero cabelludo, manos y/o pies, y en las uñas) como de la calidad de vida (medida con el índice DLQI) y de los 10 síntomas y signos de psoriasis comunicados por los pacientes. Guselkumab también aportó un beneficio clínico en las semanas 24 y 48 (p<0,05) superior a adalimumab en la psoriasis del cuero cabelludo y de manos y pies.
Por su parte, el estudio NAVIGATE evaluó la eficacia y seguridad de guselkumab en pacientes con respuesta inadecuada a tratamiento con ustekinumab tras la semana 16 (habiendo recibido en régimen abierto dos dosis de ustekinumab a las semanas 0 y 4), definida como valor de IGA≥2. A partir de ese momento, los pacientes se aleatorizaron para seguir tratamiento con 45 mg de ustekinumab cada 12 semanas (90 mg si >100 kg; n=133) o iniciar guselkumab (n=135), que sería administrado en dosis de 100 mg en las semanas 16 y 20 y posteriormente cada 8 semanas.
Como variable primaria de eficacia, en este caso se midió el número de visitas posteriores a la aleatorización (de entre las 4 visitas realizadas entre las semanas 12 y 24) en las que los pacientes obtuvieron una puntuación en IGA 0/1 y experimentaron una mejoría de grado ≥2. Los resultados publicados para el ensayo NAVIGATE (Langley, 2018) indican que, entre los pacientes con una respuesta inadecuada inicial a ustekinumab, se observó un beneficio clínico significativamente superior en los que cambiaron a guselkumab frente a los que siguieron el tratamiento con ustekinumab (media de 1,5 vs. 0,7 visitas; p<0,001). Entre las variables secundarias, destacan el notable incremento aportado por el tratamiento con guselkumab frente a ustekinumab en la proporción de pacientes que alcanzaron PASI 90 (48,1% vs. 22,6%; p<0,001) o IGA 0/1 con una mejoría de grado ≥2 (31,1% vs. 14,3%) en la semana 28 de seguimiento; tales diferencias eran significativas ya a las 4 semanas pos-aleatorización y fueron máximas a la semana 24.
En cuanto a la seguridad clínica de guselkumab, los datos derivan de un total de 1.748 pacientes con psoriasis en placas moderada-severa que fueron tratados con el fármaco durante los ensayos de fase 2 y 3 de su desarrollo clínico. En general, el perfil toxicológico de guselkumab fue comparable al de placebo hasta la semana 16, momento en que la proporción de pacientes con acontecimientos adversos en el brazo de guselkumab fue de 49,2%, en el de placebo de 46,7% y en el de adalimumab de 49,9%. Esa tendencia se mantuvo hasta el final del período de seguimiento (48 semanas en VOYAGE 1 y VOYAGE 2). La tasa de eventos adversos para el brazo de guselkumab fue comparable a la de adalimumab, e incluso inferior para los casos de eritema en el lugar de inyección, prurito, dolor o efectos adversos derivados de la psoriasis.
Los efectos adversos más comunes fueron: nasofaringitis (32,84 casos/100 pacientes-año), infecciones del tracto respiratorio superior (17,24/100 pacientes-año), dolor de cabeza (7,29/100 pacientes-año), artralgia (5,95/100 pacientes-año) e hipertensión (5,13/100 pacientes-año) (EMA, 2017). Se estimó que solo el 0,7% de las inyecciones de guselkumab (vs. 0,3% de las inyecciones de placebo) se asociaron con reacciones en el lugar de inyección, que fueron leves-moderadas. La frecuencia general de efectos adversos que llevaron a la discontinuación del tratamiento fue baja (≤0,2/100 pacientes-año) para el grupo de guselkumab y menor que para el grupo de adalimumab a las semanas 24 y 48.
Por último, en cuanto a su inmunogenicidad, menos del 6% de los pacientes tratados con guselkumab desarrollaron anticuerpos antifármaco durante un período de hasta 52 semanas de tratamiento, y los títulos de anticuerpos fueron bajos. Entre los que sí los desarrollaron, solo el 7% tenían anticuerpos neutralizantes, lo que equivale al 0,4% de todos los pacientes tratados con guselkumab. Los anticuerpos antifármaco no se asociaron con una menor eficacia ni con el desarrollo de reacciones en el lugar de inyección.
ASPECTOS INNOVADORES
El guselkumab es un anticuerpo monoclonal que se une con elevada afinidad y especificidad a la subunidad p19 de la proteína interleucina 23 (IL-23), bloqueando las acciones biológicas mediadas por esta citocina proinflamatoria, lo que se traduce en la inhibición de la inflamación y de los síntomas clínicos de la psoriasis. El medicamento ha sido oficialmente autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamiento sistémico.
Los datos clínicos que han permitido verificar la eficacia y seguridad clínicas de guselkumab en su pauta posológica autorizada (administrado por vía subcutánea en dosis de 100 mg a tiempo 0 y a las 4 semanas, seguida de una dosis de mantenimiento cada 8 semanas) proceden de tres amplios estudios pivotales de fase 3 aleatorizados, multicéntricos, multinacionales, doblemente ciegos y enmascarados, controlados con comparador activo (adalimumab y ustekinumab) y, en dos de ellos, también con placebo hasta la semana 16.
En ese punto temporal, aproximadamente el 70-73% de los pacientes que recibieron guselkumab (588 de 825) en los estudios VOYAGE 1 y VOYAGE 2 experimentaron un beneficio clínico –expresado como una reducción de al menos el 90% en las puntuaciones PASI–, en comparación con el 48% de los que recibieron adalimumab (282 de 582) y menos del 3% que recibieron placebo (11 de 422). La mejoría en los signos y síntomas de la psoriasis se mantuvo durante más de 48 semanas con el tratamiento con guselkumab, siendo significativamente superior a la obtenida con adalimumab en los mismos periodos. Cabe destacar, por ejemplo, que en torno al 40% de los pacientes tratados con guselkumab (37,4% en la semana 16, 49,8% en la semana 32 y 47,4% en la semana 48) tuvieron un aclaramiento completo de la piel (PASI 100), frente al 26-28% de los pacientes tratados con adalimumab en la semana 16. Además, guselkumab se mostró eficaz en pacientes que habían fallado previamente a la terapia con adalimumab.
En el tercer ensayo, NAVIGATE, guselkumab también demostró una superioridad estadísticamente significativa frente a ustekinumab en los pacientes no respondedores a este fármaco que cambiaron su tratamiento (tras 16 semanas) frente a los que se mantuvieron en tratamiento con ustekinumab (media de 1,5 vs. 0,7 visitas en que los pacientes presentaban puntuación IGA 0/1 y mejoría de grado ≥2; p<0,001). En general, en los tres ensayos clínicos, las mejorías observadas en las variables primarias fueron confirmadas por las medidas de variables secundarias, en términos de enfermedad regional (cuero cabelludo, manos y pies, y uñas), calidad de vida y síntomas referidos por los pacientes, entre otros.
La eficacia clínica de guselkumab ha sido demostrada con independencia de la edad, sexo, raza, peso corporal, localización de las placas, intensidad basal del PASI, artritis psoriásica concomitante y tratamiento previo con un fármaco biológico. Guselkumab resultó eficaz en pacientes no tratados previamente con fármacos sistémicos convencionales, y en tratados y no tratados previamente con fármacos biológicos, como ustekinumab. Los resultados apuntan, además, a que tras la retirada de guselkumab, se mantiene una eficacia residual (decreciente) antipsoriásica hasta transcurridas unas 15 semanas.
Por otro lado, el perfil toxicológico de guselkumab es relativamente benigno y está en la línea de lo esperable a la vista de la seguridad clínica de otros medicamentos biológicos anti-interleucinas indicados en psoriasis. Se dispone de datos suficientes (incluyendo 728 pacientes tratados por período de un año con guselkumab) y los eventos adversos más comúnmente reportados han sido nasofaringitis, infecciones del tracto respiratorio superior, dolor de cabeza, artralgia e hipertensión, la mayoría leves o moderados en gravedad.
Cabe indicar que, de manera interesante, la tasa de efectos adversos graves fue baja para guselkumab (1,03/100 pacientes-año a la semana 48), siendo inferior que la de adalimumab (1,73/100 pacientes-año) y similar a los datos disponibles para ustekinumab (otro anti-IL23). Sin embargo, no se han establecido aún conclusiones sobre el efecto en la seguridad clínica de guselkumab de los anticuerpos neutralizantes que desarrollan un 6% de los pacientes. La ausencia de datos sobre su uso a largo plazo (> 2 años) impide evaluar el efecto de este tipo de terapias inmunológicas sobre la incidencia de reacciones de hipersensibilidad generalizadas o tumores malignos, pues el período de inducción de los mismos puede ser mayor; este riesgo potencial debe ser considerado en futuros estudios de más largo seguimiento, así como su seguridad en pacientes pediátricos, embarazadas, o con insuficiencias renal o hepática graves.
Desde el punto de vista mecanístico, guselkumab comparte diana biológica –IL-23– con ustekinumab (comercializado en España en 2010), pero aporta la relativa innovación de unirse a la subunidad p19 de la proteína, de forma que su especificidad frente a IL-23 es elevada, mientras que ustekinumab se une a la subunidad p40 de dicha citocina, compartida también por la IL-12. Esta especificidad de guselkumab frente a IL-23 podría explicar su mayor eficacia en comparación con ustekinumab en pacientes que no tienen una buena respuesta a este último.
La IL-23 es una citocina reguladora implicada en la diferenciación y funcionalidad de subgrupos de linfocitos T, que serán los encargados de producir citocinas efectoras como IL-17 (A y F) e IL-22, claves en la hiperplasia epidérmica. En el mercado español, se dispone de otros fármacos antipsoriásicos que actúan sobre esa misma vía farmacológica, tales como anti-IL 17A (secukinumab e ixekizumab) y anti-receptor de IL17 (como brodalumab, que también bloquea los efectos de IL-17F). Guselkumab puede, teóricamente, contribuir a una mayor eficacia clínica que los anteriores porque bloquea la diferenciación previa de las células T, impidiendo así que sinteticen y liberen IL-17, en lugar de bloquearla una vez liberada (lo que se puede presumir menos eficaz). Además, la inhibición de la producción y liberación de IL-22 contribuye a sus efectos inmunomoduladores. Los resultados clínicos parecen confirmar esa mayor eficacia clínica, pues ha demostrado superioridad clínica en comparación directa frente a un inhibidor de TNFα (adalimumab) y frente a otro anti-IL23 como es ustekinumab.
En definitiva, a pesar de la disponibilidad de diversos tratamientos frente a la psoriasis, el manejo clínico de formas crónicas moderadas-graves sigue siendo un reto. Guselkumab emerge como una alternativa –con una eficacia clínica aparentemente más potente– a otros fármacos biológicos empleados en el tratamiento sistémico de la psoriasis moderada-grave. A pesar de que su indicación oficial no lo recoge así, es previsible que se posicione como fármaco de segunda línea, tras una respuesta inadecuada o contraindicación a tratamientos sistémicos convencionales o PUVA, aunque a la vista de los datos disponibles podría ser la primera opción entre los biológicos.