Resumen
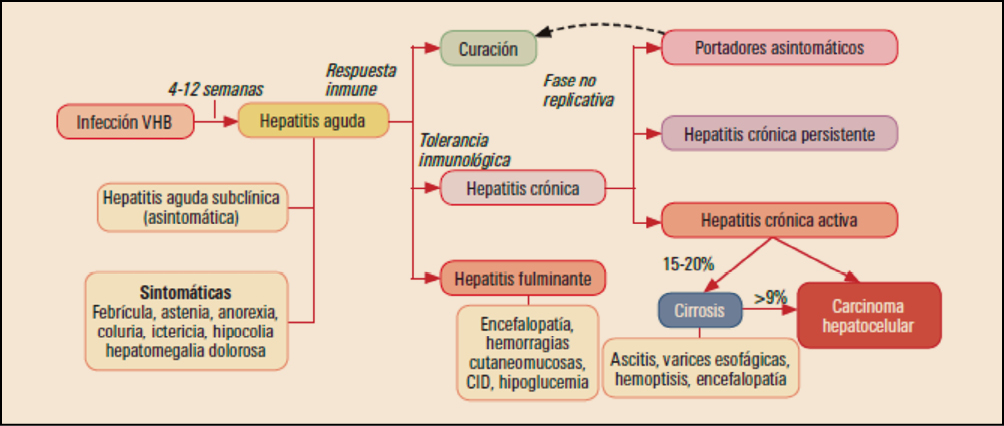
El benralizumab es un anticuerpo monoclonalque se une con elevada afinidad y especificidad a la subunidad α de los receptores para IL-5 (IL-5Rα) expresados específicamente en la superficie de eosinófilos y basófilos, induciendo la apoptosis de dichas células mediante el refuerzo de la citotoxicidad celular dependiente de anticuerpos (mediada sobre todo por células NK). Al impedir el acoplamiento de IL-5 con su receptor inhibe los efectos normalmente mediados por esta citocina sobre el ciclo biológico de los eosinófilos y es capaz de reducir su número en sangre. El medicamento ha sido autorizado para el tratamiento de mantenimiento adicional en pacientes adultos con asma grave eosinofílica no controlada a pesar de la administración de corticosteroides inhalados en dosis altas (CSI) y β-agonistas de acción prolongada (LABA).
Los datos derivados de tres ensayos clínicos pivotales de fase 3 muestran que la administración de benralizumab, en comparación con placebo, es capaz de reducir en torno al 28-51% la tasa de exacerbaciones clínicamente relevantes en pacientes con cuadros graves de asma eosinofílica mal controlados con altas dosis de corticosteroides inhalados o incluso con sistémicos, y permite una reducción del 75% de la dosis de los corticosteroides orales (vs. 25% con placebo). A ello cabe agregar ciertas mejoras en la función pulmonar y otras variables secundarias (como las puntuaciones de los cuestionarios de control asmático y calidad de vida asmática) a las que se concedió una relevancia clínica modesta. Desde el punto de vista de la seguridad, benralizumab presenta un perfil toxicológico relativamente benigno, con una incidencia de eventos adversos incluso inferior a la del placebo, fundamentalmente de carácter leve-moderado y transitorio, siendo los más frecuentes faringitis, cefalea, pirexia, reacciones de hipersensibilidad y reacciones en el lugar de la inyección.
A día de hoy no se dispone de comparaciones directas y, aunque las comparaciones indirectas solo pueden tener un carácter cualitativo, la evidencia disponible hace pensar que no hay diferencias sustanciales entre benralizumab y mepolizumab o reslizumab (anti-IL-5 que actúan sobre la misma vía de señalización). Tampoco hay datos concluyentes para establecer una ventaja sobre la adherencia al tratamiento con la pauta de administración aprobada para benralizumab.
En definitiva, benralizumab emerge como una nueva opción de tratamiento para pacientes con asma eosinofílica grave no controlada, como alternativa a mepolizumab o reslizumab, cuya indicación ha sido restringida a pacientes con mal control del asma a pesar del tratamiento con dosis altas de CSI, considerando que, de acuerdo a las guías actuales en los pacientes tratados con dosis medias de CSI se recomienda aumentar su dosis antes de iniciar el tratamiento con un anti-IL5 o con un antagonista de los receptores de IL5. No supone, pues, ninguna innovación disruptiva en el tratamiento de la patología.
ASPECTOS FISIOPATOLÓGICOS
El asma es un síndrome que comparte manifestaciones clínicas similares en apariencia pero de causas diferenciadas. Ello supone que sea complejo el alcanzar una definición precisa y exacta de la enfermedad, existiendo varias definiciones para la misma. De forma general, se define como una enfermedad inflamatoria crónica de las vías respiratorias, en cuya patogenia intervienen diversas células y mediadores de la inflamación, condicionada en parte por factores genéticos y que cursa con hiperrespuesta bronquial y una obstrucción variable al flujo aéreo, total o parcialmente reversible, ya sea por la acción medicamentosa o espontáneamente (Cuéllar, 2016).
El asma es un problema de salud de elevada prevalencia y, aunque las formas graves de la enfermedad solo suponen el 10% de todos los casos, en el resto hay importantes implicaciones en la esperanza y calidad de vida de las personas que la padecen, generando un importante consumo de recursos sanitarios y notables costes sociales. Según la Organización Mundial de la Salud (OMS), el asma es la séptima enfermedad más prevalente en el mundo, con cerca de 235 millones de afectados. La cifra de afectados en Europa es de alrededor de 29 millones. Además, supone la quinta causa de muerte en los países desarrollados.
En el territorio español, la prevalencia de síntomas asmáticos en niños se ha mantenido constante en los niños de 13-14 años, mientras que ha sufrido un aumento significativo en el grupo de 6-7 años. En los adultos la prevalencia es inferior a la de los países anglosajones y centroeuropeos. Se estima que en España sufren asma aproximadamente al 4,9% de los adultos y a aproximadamente al 10% de los niños. El Estudio Europeo del Asma (GEEEA, 1996) constató en nuestro país unas tasas variables que van desde el 4,7% en Albacete hasta el 1% en Huelva, alcanzando en algunas zonas cifras cercanas al 14%. Y lo que es más preocupante: un 52% de las personas con asma no habían sido diagnosticadas y hasta un 26% de éstas, a pesar de padecer síntomas frecuentes, no seguía ningún tratamiento.
El asma en adultos se clasifica, según el grado de gravedad, en cuatro categorías: intermitente, persistente leve, persistente moderada y persistente grave. En los niños se definen dos patrones principales: asma episódica y asma persistente. El asma episódica puede ser ocasional o frecuente, dependiendo del número de crisis que presente. El asma persistente en el niño no puede considerarse leve, sino que al menos es moderada o grave. Existe, además, un fenotipo mixto EPOC-asma, caracterizado por una obstrucción no completamente reversible al flujo aéreo, acompañada de síntomas o signos de una reversibilidad aumentada de la obstrucción.
Los síntomas y signos clínicos característicos del asma son:
- Obstrucción bronquial reversible, con manifestación clínica como episodios recurrentes de disnea, sibilancias, opresión torácica y tos (particularmente por la noche o de madrugada). El término reversible indica que responde bien a la medicación broncodilatadora. Se considera aceptable una respuesta de FEV11> 15%.
- Inflamación: las alteraciones histológicas consisten básicamente en: a) infiltración de la mucosa por eosinófilos activados que segregan diversos productos citotóxicos (anión superóxido, proteína básica mayor, proteína catiónica, peroxidasa y neurotoxina), linfocitos T-helper (CD4+) y mastocitos; b) descamación de células epiteliales; y c) engrosamiento de la membrana basal por incremento de los depósitos de colágeno de los tipos I, III y V, junto con fibronectina.
- Hiperreactividad bronquial:la descamación y el daño del epitelio de las vías aéreas permite la exposición directa a irritantes de las terminaciones nerviosas subepiteliales, lo cual provoca reflejos axónicos locales y reflejos vagales que pueden producir broncoconstricción, hipersecreción de moco, tos y vasodilatación con extravasación, lo que ocasiona edema e infiltración de células inflamatorias. La hiperreactividad bronquial se ha atribuido a distintas causas (entre otras, a anormalidades de la inervación adrenérgica, a alteraciones en las catecolaminas circulantes, a defectos de los receptores β-adrenérgicos y/o al aumento del tono vagal).
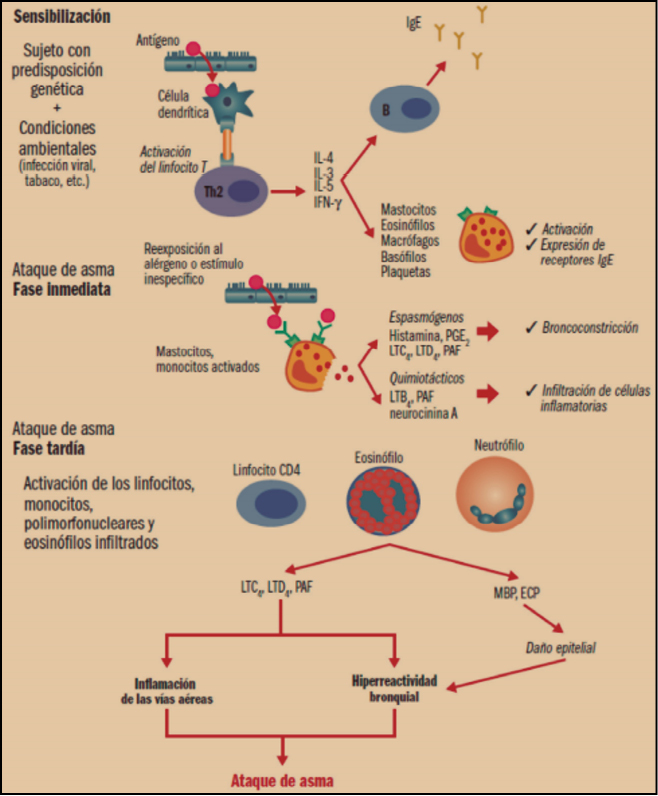
Figura 1. Eventos celulares en el asma. ECP: proteína catiónica del eosinófilo; IFN-γ: interferón γ; IgE: inmunoglobulina E; IL-3, 4, 5: interleucina 3, 4, 5; LTB4, LTC4, LTD4: leucotrienos B4, C4, D4; MBP: proteína básica mayor; PAF: factor activador de plaquetas; PGE2: prostaglandina E2; Th2: linfocitos T helper 2.
En un ataque de asma se produce un desequilibrio en la relación ventilación-perfusión con un aumento del gradiente alveolo-arterial de oxígeno, lo cual implica la disminución de la presión parcial de O2 en sangre. Además, este desacoplamiento ventilación-perfusión implica un aumento del espacio muerto fisiológico y, si la ventilación alveolar no aumenta lo suficiente, se produce un aumento de la presión parcial de dióxido de carbono (PCO2). En suma, la hipoxemia que acompaña el ataque de asma suele ser moderada y responde favorablemente al aumento de la concentración de oxígeno inspirado. La evolución de la PCO2 depende de la gravedad de la obstrucción al flujo aéreo y la aparición de fatiga muscular respiratoria.
La obstrucción bronquial que se produce en un ataque agudo de asma exige un aumento significativo del trabajo respiratorio como consecuencia del incremento en la resistencia pulmonar y en la carga elástica pulmonar, así como de una menor eficiencia en la contracción del diafragma debido a la pérdida parcial de su curvatura por la hiperinsuflación pulmonar.
En realidad, el asma no es una enfermedad única, sino más bien un síndrome con una amplia heterogeneidad de fenotipos ligados a diversos mecanismos celulares y biomoleculares. Ha pasado de ser considerada una reacción de hipersensibilidad de tipo I, en la que lo importante era el episodio de broncoespasmo desencadenado por la liberación de mediadores tras la desgranulación del mastocito al producirse la reacción alérgeno-inmunoglobulina E (IgE) específica, a ser interpretada en la actualidad como un proceso inflamatorio crónico de las vías aéreas puesto en marcha por una serie de factores desencadenantes, entre los que cabe destacar determinadas actividades profesionales, ejercicio físico, infecciones, fármacos (betabloqueantes y, especialmente, AINE, en particular el ácido acetilsalicílico), reflujo gastroesofágico y ciertos factores emocionales. Sin embargo, con mucho, el factor más frecuentemente desencadenante del asma es la alergia.
Los alérgenos más comúnmente involucrados en la patogénesis del asma son proteínas de los reinos animal y vegetal. En España, los procedentes de los ácaros (Dermatophagoides pteronyssinus y D. farinae) son los alérgenos más comunes. Otros alérgenos proceden de pólenes (gramíneas [Secale cereale, Triticum sativum, etc.], hierbas [Plantago lanceolata, Parietaria judaica], árboles [Corylus avellana, Olea sp.]), hongos [Alternaria sp., Aspergillus sp., Cladosporium sp., etc.] y animales [epitelios y fluidos biológicos de gato, perro, rata, etc.].
El proceso de asma alérgico comienza con la sensibilización. En individuos con predisposición genética (susceptibilidad a antígenos como el polen, el polvo doméstico, etc.) y en determinadas condiciones ambientales (infección viral, humo de tabaco, etc.), se produce la interacción del antígeno con las células dendríticas y la posterior activación de la población de linfocitos T, los cuales generan citocinas que promueven la diferenciación y activación de los eosinófilos (interleucina 5, IL-5), la expresión de receptores de la IgE en los mastocitos y esosinófilos (IL-4), la expresión por parte del epitelio de receptores que atraen a los eosinófilos (IL-4) y la producción y liberación de IgE por parte de linfocitos B.
La reexposición al alérgeno produce un episodio agudo o ataque de asma, el cual se presenta en dos fases –inmediata y tardía–, aunque en algunos sujetos sólo se produce una única fase. La fase inmediata se caracteriza por la aparición de un espasmo en el músculo liso bronquial. Se produce porque el antígeno interactúa con el mastocito que había expresado y fijado la IgE a sus receptores en la sensibilización, liberando principalmente histamina, leucotrienos C4y D4 (LTC4 y LTD4), responsables del broncoespasmo; también son liberados otros mediadores (prostaglandina D2 [PGD2], neurocinina A, LTB4) que provocan una migración de células inflamatorias –eosinófilos y monocitos– hacia esa zona anatómica. La fase tardía o respuesta diferida ocurre en un tiempo variable desde la exposición inicial al antígeno (6-8 horas), suele ser nocturna, y es claramente la progresión de una reacción inflamatoria iniciada en la primera fase que conlleva un acúmulo local de eosinófilos. Se piensa que los gránulos de los eosinófilos infiltrantes liberan mediadores citotóxicos que afectan al epitelio respiratorio ciliado.
Así pues, son numerosas las células inflamatorias implicadas en el asma:
- Eosinófilos. La prevalencia de la inflamación eosinofílica identifica un fenotipo de asma (asma eosinofílica), que representa el 40-60% de todos los casos de asma, cuyo nivel de gravedad se correlaciona con la cantidad de eosinófilos en sangre y esputo. En estos pacientes los eosinófilos activados, cuya apoptosis está inhibida, contienen enzimas inflamatorias responsables del daño epitelial y generan mediadores que amplifican la respuesta inflamatoria. Los eosinófilos disponen de receptores de tipo IgE que son capaces de internalizar los complejos antígeno-anticuerpo, liberando mediadores inflamatorios. Tienen gránulos que contienen diversos mediadores de las reacciones alérgicas, tales como histaminasa y arilsulfatasa. Asimismo, son capaces de segregar leucotrienos, que juegan un papel importante en la fisiopatología del asma, al provocar broncoconstricción e hipersecreción de moco. El ciclo biológico completo de los eosinófilos, desde su producción en la médula ósea hasta alcanzar el lugar donde se ha producido un proceso inflamatorio, de la producción a su reclutamiento, y de su activación a la apoptosis, está estrictamente modulado por la interleucina 5 (IL-5) y su receptor específico (IL-5R).
- Células dendríticas. Existen dos poblaciones bien diferenciadas, la mieloide (tipo 1), originada en la médula ósea y la plasmocitoide (tipo 2), originada en tejido linfoide. Los asmáticos presentan un exceso de plasmocitoides. El alérgeno es capaz de activarlas de forma directa, ya que pueden desplegar en su superficie receptores de alta afinidad para la IgE. Dada la cercanía que existe entre los cuerpos de las células dendríticas, las células epiteliales y las terminales nerviosas no mielinizadas, es posible que estas células desempeñen un rol en los fenómenos de hiperreactividad bronquial e inflamación neurogénica.
- Células epiteliales. El epitelio de las vías aéreas no ejerce una simple función de barrera, sino que es capaz de sintetizar diversas sustancias con acción biológica como el ácido araquidónico, óxido nítrico, endotelinas, citocinas y factores de crecimiento. Así, es capaz de modular la función del músculo liso.
- Linfocitos T. Están elevados en la vía aérea, con un desequilibrio en la relación de células CD4+ en comparación con las CD8+.
- Mastocitos. Están aumentados, tanto en el epitelio como infiltrando el músculo liso de la pared. Su activación da lugar a la liberación de mediadores con efecto broncoconstrictor y proinflamatorio. Producen citocinas que mantienen y promueven la inflamación
- Neutrófilos. Están elevados en la vía aérea de algunos pacientes con asma grave, durante las exacerbaciones, en caso de tabaquismo y en casos de asma relacionada con el trabajo.
- Macrófagos. Pueden ser activados por alérgenos a través de receptores de baja afinidad para la IgE y liberar sus mediadores, que amplifican la respuesta inflamatoria.
- Fibroblastos. Por los estímulos de citocinas como la IL-4 y la IL-13, se convierten en células inflamatorias activadas, participando tanto en la inflamación como en la remodelación. El número de fibroblastos y miofibroblastos activados en casos de asma se halla aumentado, fenómeno que adquiere aún mayor intensidad después de una provocación con alérgenos.
El objetivo principal del tratamiento del asma es lograr y mantener el control de la enfermedad lo antes posible, además de prevenir las exacerbaciones y la obstrucción crónica al flujo aéreo y reducir al máximo su mortalidad.
Las exacerbaciones (ataques o crisis) de asma son episodios agudos o subagudos caracterizados por un aumento progresivo de uno o más de los síntomas típicos (disnea, tos, sibilancias y opresión torácica) acompañados de una disminución del flujo espiratorio (FEV1). Según la rapidez de instauración de las crisis, existen dos tipos: las de instauración lenta2 (normalmente en días o semanas) y las de instauración rápida3 (en menos de 3 horas), que deben identificarse por tener causas, patogenia y pronóstico diferentes. La intensidad de las exacerbaciones es variable, cursando en ocasiones con síntomas leves e indetectables por el paciente y en otras con episodios muy graves que ponen en peligro su vida.
El objetivo inmediato del tratamiento de una crisis es preservar la vida del paciente revirtiendo la obstrucción al flujo aéreo y la hipoxemia si está presente, de la forma más rápida posible, y posteriormente instaurar o revisar el plan terapéutico para prevenir nuevas crisis. La pauta de tratamiento en la exacerbación leve debe incluir la administración de broncodilatadores agonistas β2-adrenérgicos de acción corta (SABA), glucocorticoides orales y oxígeno (si es necesario).
Los SABA inhalados son los fármacos broncodilatadores más eficaces y rápidos en el tratamiento de la exacerbación asmática. Si en las primeras 2 horas del tratamiento se constata una evolución favorable (desaparición de síntomas, FEV1 superior al 80% del teórico o del mejor valor personal del paciente) y ésta se mantiene durante 3-4 horas, no son necesarios más tratamientos. Sin embargo, en pacientes no controlados adecuadamente con combinaciones inhaladas de corticosteroides y SABA administradas a demanda, puede recurrirse a combinaciones similares con agonistas β2 de acción prolongada (LABA), como salmeterol, formoterol, vilanterol, etc.
En el tratamiento de mantenimiento a largo plazo del asma se distinguen varios escalones de complejidad creciente, que deben ajustarse a la respuesta y el nivel de control que manifieste el paciente, siendo necesaria una evaluación periódica del paciente para determinar si se cumplen los objetivos terapéuticos. En todos los escalones de tratamiento, los corticosteroides inhalados (CSI) son de elección para el control a largo plazo del asma, a dosis crecientes según la gravedad del cuadro asmático, y empleándose junto con un LABA en los casos más graves. Además, a estos puede asociarse tiotropio (anticolinérgico) y/o un antagonista de los receptores de leucotrienos (LTRA) y/o administrar corticosteroides orales (CSO), si la gravedad de la patología así lo recomienda (GEMA, 2018). En todos los escalones, no obstante, se pueden utilizar agonistas β2 adrenérgicos de acción corta administradas a demanda.
El uso de glucocorticoides sistémicos acelera la resolución de las exacerbaciones. Excepto en crisis muy leves, deben administrarse siempre, especialmente si: a) no se consigue una reversión de la obstrucción de las vías respiratorias con SABA; b) el paciente estaba tomando ya CSO; c) el paciente ha tratado ya su pérdida de control previa con otras opciones terapéuticas sin éxito; d) existen antecedentes de exacerbaciones previas que requirieron CSO. Es importante señalar que el uso prolongado de corticosteroides sistémicos suele asociarse con efectos adversos, en ocasiones graves.
Además de los fármacos anteriormente indicados, los anticuerpos monoclonales han abierto un importante capítulo en el tratamiento de mantenimiento del asma. Básicamente, los fármacos biotecnológicos ya autorizados contemplan las siguientes vías inmunofarmacológicas:
- El bloqueo de la producción o actividad de citocinas proinflamatorias implicadas, como la IL-5: mepolizumab y reslizumab. Están autorizados como tratamiento adicional en pacientes adultos con asma grave no controlada eosinofílica.
- Impedir la unión de la Ig E a sus receptores en los mastocitos y basófilos inhibiendo la reacción de hipersensibilidad inmediata y la subsiguiente cascada inflamatoria: omalizumab. Está autorizado como tratamiento adicional en asma persistente grave alérgica mediada de forma convincente por IgE.
Cabe recordar que el asma grave no controlada se define como la enfermedad asmática que persiste mal controlada4 a pesar de recibir tratamiento con una combinación de CSI a dosis elevadas junto con un LABA en el último año, o bien CSO durante al menos seis meses del mismo periodo. En nuestro medio, la prevalencia de pacientes con asma grave no controlada es aproximadamente del 3,9% del total de la población asmática, de entre los cuales el asma eosinofílica representa aproximadamente el 25% de los casos (y el 40-60% de todos los casos de asma). Esta última se caracteriza por la presencia de eosinófilos en biopsias bronquiales y esputo (mayores niveles de eosinófilos se asocian a mayor gravedad), a pesar de dosis altas de corticosteroides; suele ser de inicio tardío y se relaciona con pólipos nasales, rinosinusitis e infecciones del tracto respiratorio.
Finalmente, la inmunoterapia con alérgenos específicos es un tratamiento de mantenimiento indicado en el asma alérgica bien controlada, siempre que se haya demostrado una sensibilización mediada por la inmunoglobulina E frente a aeroalérgenos comunes, y se utilicen extractos bien estandarizados.
ACCIÓN Y MECANISMO
El benralizumab es un anticuerpo monoclonal humanizado antieosinófilos que se une con elevada afinidad y especificidad a la subunidad α de los receptores para IL-5 (IL-5Rα), los cuales se expresan específicamente en la superficie de eosinófilos y basófilos. Ha sido autorizado para el tratamiento de mantenimiento adicional en pacientes adultos con asma grave eosinofílica no controlada a pesar de la administración de corticosteroides inhalados en dosis altas y agonistas β de acción prolongada.
Mediante su unión a IL-5Rα, benralizumab inhibe la hetero-oligomerización de las subunidades alfa y beta del receptor y bloquea el desencadenamiento de la señal de transducción. De esta forma, se opone a que la citosina IL-5 ejerza sus efectos sobre eosinófilos y basófilos, inhibiendo el crecimiento, diferenciación, reclutamiento y activación de eosinófilos, pues todo el ciclo biológico de los mismos está estrictamente mediado por IL-5.
Por otro lado, la ausencia de fucosa en el dominio Fc del anticuerpo determina una gran afinidad por los receptores FcɣRIIIa, los principales receptores de Fc expresados en células efectoras inmunitarias, como las células NK y los macrófagos. Esto conlleva a la apoptosis de eosinófilos y basófilos –en cuya superficie está unido el benralizumab– mediante un refuerzo de la citotoxicidad celular dependiente de anticuerpos. Así, contrarresta la inhibición de la apoptosis de eosinófilos que tiene lugar en pacientes con asma eosinofílica, y es capaz de impedir que éstos liberen las enzimas pro-inflamatorias (responsables del daño epitelial) y los mediadores que amplifican la respuesta inflamatoria, todo lo cual revierte en la reducción de la inflamación.
El tratamiento con benralizumab produce una depleción casi completa (≥95%) de los eosinófilos en sangre en las 24 horas siguientes a la primera dosis, que se mantiene durante todo el periodo de tratamiento. Ese efecto se acompaña de una reducción de los basófilos en sangre (aunque en menor medida), así como de una disminución en suero de las proteínas granulares eosinofílicas, de la neurotoxina derivada de eosinófilos, de la proteína catiónica de los eosinófilos, lo que demuestra que no induce por sí mismo la activación o la necrosis de eosinófilos, sino que depende de la citotoxicidad ejercida por las células NK (Matera, 2017).
También se ha demostrado que benralizumab consigue reducir el número de eosinófilos de la mucosa de las vías respiratorias con respecto al momento basal, en más del doble que el placebo.
ASPECTOS MOLECULARES
El benralizumab es un anticuerpo monoclonal humanizado de tipo IgG1κ compuesto de dos cadenas pesadas idénticas, de aproximadamente 49.400 Da cada una, y dos cadenas ligeras también idénticas entre sí, de unos 23.500 Da cada una. Presenta una N-glicosilación en el dominio CH2 de la cadena pesada, en la asparagina en posición 301 (Asn301) formado por un complejo bicatenario de oligosacáridos.
La línea celular de células de ovario de hámster chino (en que se produce el fármaco mediante tecnología de ADN recombinante) ha sido modificada adecuadamente por ingeniería genética para eliminar la competencia de la fucosilación y prevenir que ocurra en la molécula de benralizumab.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de benralizumab han sido adecuadamente contrastadas en las indicaciones autorizadas mediante tres ensayos clínicos pivotales de fase 3 (confirmatorios de eficacia y seguridad), multicéntricos, doblemente ciegos, de grupos paralelos, aleatorios y controlados con placebo, de 28 a 56 semanas de duración, en pacientes de 12 a 75 años.
Dos de ellos evaluaron el efecto sobre las exacerbaciones (estudio D3250C00017 –SIROCCO– y D3250C00018 –CALIMA–) y el tercero analizó la reducción en el uso de corticosteroides orales (estudio D3250C00020 –ZONDA–) de una pauta de benralizumab consistente en tres dosis de 30 mg administradas una cada cuatro semanas, seguido de una dosis de 30 mg cada 4 u 8 semanas. El tratamiento experimental fue administrado a los pacientes con asma grave no controlada siempre por vía subcutánea, como tratamiento adicional al tratamiento de base, constituido en todos los casos por corticosteroides más LABA inhalados.
Aunque con ciertas diferencias entre los tres ensayos, todos los pacientes aleatorizados debían de tener antecedentes de asma que requiriera dosis medias-altas de CSI+LABA durante al menos un periodo de 12 meses, así como tratamiento documentado con CSI+LABA durante los últimos 3-6 meses, con tratamientos adicionales para el control del asma durante, al menos, 30 días (CSO de forma continuada en los 6 últimos meses en el estudio ZONDA). Además, debían presentar asma documentada mediante reversibilidad de la obstrucción al flujo aéreo (FEV15 pos-broncodilatador ≥12% y 200 ml), obstrucción persistente al flujo aéreo (FEV1 pre-broncodilatador <80% del valor predicho) y al menos una exacerbación en el año previo.
Las características más relevantes de dichos ensayos clínicos se resumen en la Tabla 1: en la primera mitad con fondo azul, se muestran las relativas a la población de pacientes incluida en el estudio y, con fondo verde, se destacan los principales resultados de eficacia clínica obtenidos. Si bien en los estudios de exacerbaciones no se empleó como criterio de selección de pacientes el carácter eosinofílico de la patología, el análisis primario de los datos se realizó en la población de pacientes tratados con altas dosis de CSI y recuentos de ≥300 eosinófilos/μl de sangre.
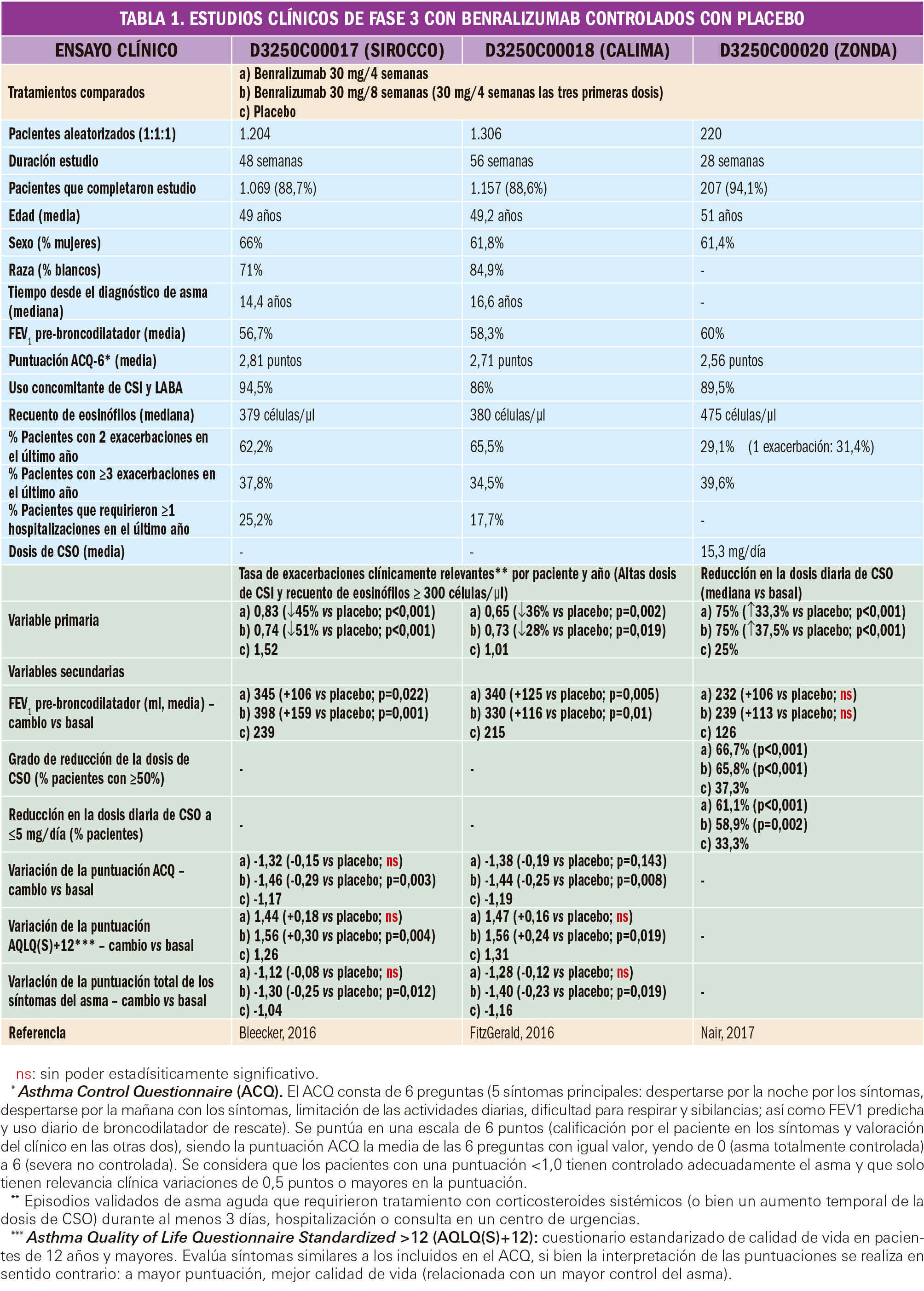
Además de estos datos, cabe destacar que, en los subanálisis post-hoc derivados de los ensayos SIROCCO y CALIMA, se observó una mayor reducción en la variable primaria en aquellos pacientes que habían presentado 3 o más exacerbaciones en el año anterior (reducción relativa del riesgo de hasta el 57% frente a placebo). No obstante, en la subpoblación de pacientes con recuento de linfocitos en sangre <300 células/μl, el poder estadístico obtenido para tasa anual de exacerbaciones es insuficiente para detectar diferencias entre los brazos de tratamiento. En línea con estos resultados, tampoco se evidenció un efecto remarcable de benralizumab sobre la tasa de exacerbaciones que requirieron visita a urgencias/hospitalización en el ensayo CALIMA, puesto que la baja tasa de eventos en el grupo placebo (0,10) no permite extraer conclusiones al respecto. Se observa, además, que una dosificación del fármaco más frecuente (una dosis cada cuatro semanas) no aporta un beneficio clínico adicional sobre el régimen de dosificación recomendado.
Respecto al ensayo ZONDA, se podría también destacar que el tratamiento con benralizumab permitió la reducción de ≥90% en la dosis diaria de CSO en el 37% de los pacientes (vs. 12% en el grupo placebo), siendo significativa la diferencia en el número de pacientes que pudieron retirar completamente los CSO de su medicación antiasmática (52% con benralizumab vs. 19% en el grupo placebo).
Desde el punto de vista de la seguridad, se tienen datos de 2.514 pacientes que han recibido al menos una dosis de benralizumab en los ensayos de fase II y III durante el desarrollo clínico del medicamento; el grueso de los datos deriva, no obstante, de los dos ensayos pivotales más amplios (SIROCCO y CALIMA). El perfil toxicológico de benralizumab es relativamente benigno, con una incidencia de eventos adversos similar a la del placebo (73,6-73,8% con los dos regímenes experimentales de benralizumab y 78% con placebo), fundamentalmente de carácter leve o moderado, y transitorios.
Los eventos adversos más frecuentes fueron cefalea (8,6% con benralizumab en el régimen autorizado vs. 6,3% con placebo), faringitis (5,0% vs. 3,4%), reacciones de hipersensibilidad (3,2% en ambos brazos), pirexia (2,9% vs. 1,7%) y reacciones en el sitio de inyección (2,2% vs. 1,9%). La frecuencia de acontecimientos adversos graves (incluyendo los casos mortales) fue mayor en los brazos placebo que el los de tratamiento experimental, por lo que no se pueden relacionar directamente con el fármaco. Ninguno de los fallecimientos que se produjeron en los ensayos de fase III fue relacionado con el tratamiento por parte del investigador, salvo un caso en el estudio ZONDA debido a neumonía con insuficiencia respiratoria.
Por último, 107 pacientes (13%) de los tratados con la pauta posológica autorizada desarrollaron anticuerpos frente a benralizumab (la mayoría fueron neutralizantes y persistentes), lo cual se relacionó con un aumento en el aclaramiento del fármaco y en los niveles de eosinófilos. Con todo, no hay datos suficientes para relacionar la presencia de anticuerpos con variaciones de los perfiles de eficacia y seguridad.
ASPECTOS INNOVADORES
El benralizumab es un anticuerpo monoclonal humanizado antieosinófilos que se une con elevada afinidad y especificidad a la subunidad α de los receptores para IL-5 (IL-5Rα) expresados específicamente en la superficie de eosinófilos y basófilos, induciendo la apoptosis de dichas células mediante el refuerzo de la citotoxicidad celular dependiente de anticuerpos (mediada, principalmente, por células NK). Ha sido autorizado para el tratamiento de mantenimiento adicional en pacientes adultos con asma grave eosinofílica no controlada a pesar de la administración de corticosteroides inhalados en dosis altas y agonistas β de acción prolongada.
Los datos procedentes de dos ensayos clínicos confirmatorios, SIROCCO y CALIMA, muestran que benralizumab es capaz de reducir la tasa de exacerbaciones del asma en un 51% y un 28%, respectivamente, en comparación con placebo en la subpoblación de pacientes con niveles plasmáticos elevados de eosinófilos (≥ 300 células/μl) y en tratamiento con altas dosis de CSI. En ambos estudios, los pacientes tratados con benralizumab también mostraron mejorías notables de la función pulmonar.
No obstante, la reducción en términos absolutos de –0,78 y –0,29 exacerbaciones/paciente/año, respectivamente, son consideradas de relevancia modesta desde el punto de vista clínico (EMA, 2017). A ello cabe agregar que las mejoras significativas en las puntuaciones de los cuestionarios de control asmático y calidad de vida asmática se situaron por debajo de la diferencia mínima considerada clínicamente relevante, que se sitúa en 0,5 puntos.
Un tercer ensayo –ZONDA– demostró que el tratamiento con benralizumab permite una reducción significativa en la dosis de corticosteroides orales adicionales, permitiendo en un importante porcentaje de pacientes la retirada completa de los CSO (52% vs. 19% en el brazo de placebo). Confirmó, además, la consistencia de los resultados obtenidos en los otros dos ensayos sobre la función pulmonar, la puntuación de los síntomas de asma y la calidad de vida de los pacientes con asma grave eosinofílica.
Por otro lado, el perfil toxicológico aceptable de benralizumab es equiparable al que han mostrado los otros anticuerpos monoclonales que actúan sobre la misma vía de señalización, esto es, mepolizumab y reslizumab (introducidos en el mercado español en los últimos 3 años). Sin embargo, no se han establecido aún conclusiones sobre el efecto en la seguridad clínica del fármaco de los anticuerpos neutralizantes que desarrollan un 13% de los pacientes tratados con benralizumab. Además, los ensayos clínicos principales no incluyeron pacientes con enfermedades pulmonares graves, infecciones respiratorias del tracto respiratorio superior o inferior demandantes de tratamiento o con patologías cardíacas, lo cual abre una ventana de incertidumbre en su uso a largo plazo en la población real con asma eosinofílica.
Merece una reseña el hecho de que benralizumab inaugura una nueva diana farmacológica al actuar sobre el receptor de IL-5 (IL-5R) en lugar de sobre la propia molécula de IL-5 (como hacen mepolizumab y reslizumab). Sin embargo, sus efectos farmacológicos serán los mismos, en tanto que ambos mecanismos inhabilitan la vía de la IL-5, que regula el ciclo biológico completo de los eosinófilos, desde su producción a su activación y su apoptosis.
Los dos anticuerpos anti-IL5 se autorizaron, igual que benralizumab, como tratamiento adicional en pacientes con asma grave no controlada eosinofílica, en pautas de dosis cada 4 semanas. Hasta el momento, no se dispone de comparaciones directas entre la eficacia clínica de benralizumab y la de esos dos anti-IL-5 para la misma indicación, ni frente a omalizumab en aquellos pacientes que cumplen a la vez con el criterios de asma alérgica persistente mediada por Ig E (en torno al 30% de los pacientes con asma eosinofílica).
Las comparaciones indirectas se ven limitadas por la variabilidad en las características basales de las poblaciones incluidas en los ensayos clínicos. En todo caso, si se analizan los resultados de eficacia de mepolizumab y reslizumab sobre la tasa de exacerbaciones en pacientes con asma eosinofílica (Cuéllar, 2016; Cuéllar, 2017), se observan reducciones significativas con valores cercanos al 50% frente a placebo. Son datos muy similares al obtenido para benralizumab, por ejemplo, en el ensayo SIROCCO. Todo hace pensar, por tanto, que no hay diferencias sustanciales entre benralizumab, mepolizumab y reslizumab.
Benralizumab podría presentar una ventaja relativa frente a reslizumab respecto a la vía de administración, pues la vía subcutánea se suele preferir por su comodidad frente a la perfusión intravenosa. No obstante, tiene la desventaja frente a mepolizumab (que se puede administrar en niños desde los 6 años) de que solo está autorizado en pacientes adultos. Además, mepolizumab –cabeza de serie que inauguró la vía farmacológica de la IL-5– demostró una reducción significativa en la dosis adicional de CSO, por lo que este efecto de benralizumab tampoco implica un avance extraordinario. La pauta administración cada 8 semanas de benralizumab sí podría suponer una ventaja frente a los anti-IL-5 en cuanto a la adherencia al tratamiento, si bien no existen datos concluyentes y hay que recordar que sus tres primeras dosis se administran cada 4 semanas.
En definitiva, la evidencia apunta hacia un mayor beneficio del tratamiento con benralizumab en aquellos pacientes con asma grave que hayan presentado mayor número de exacerbaciones (3 o más) en el año y con mayor concentración de eosinófilos. De hecho, considerando factores de eficacia y eficiencia, el IPT (AEMPS, 2019) sugiere que para el tratamiento con inhibidores de la vía de IL-5 (incluyendo benralizumab) se debe priorizar a los pacientes con asma eosinofílica refractaria grave que presenten eosinofilia ≥500 células/μL en sangre.
Benralizumab emerge como una nueva opción de tratamiento para pacientes con asma eosinofílica grave no controlada, como alternativa a mepolizumab o reslizumab, pues también actúa mediante la inhibición de la vía de la IL-5. La indicación ha sido restringida a pacientes con mal control del asma a pesar del tratamiento con dosis altas de CSI, considerando que, de acuerdo a las guías actuales en los pacientes tratados con dosis medias de CSI se recomienda aumentar la dosis antes de iniciar el tratamiento con un anti-IL-5 o con un antagonista de los receptores de IL-5. No supone, pues, ninguna innovación disruptiva en el tratamiento de la patología.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}