Una extensa revisión bibliográfica confirma la evidencia favorable (de calidad moderada) sobre la eficacia de la adrenalina a dosis estándar, comparada con placebo, en el retorno de la circulación espontánea, la supervivencia al ingreso hospitalario y la supervivencia al alta. El empleo de altas dosis de adrenalina, de vasopresina o de la combinación de adrenalina más vasopresina parece no aportar un beneficio clínico adicional.
Tanto la adrenalina como la vasopresina han sido –y son– ampliamente empleadas para tratar a las personas con parada cardiorrespiratoria, aunque aún existen ciertas incertidumbres acerca de su seguridad, eficacia y de su dosis óptima. Con el objetivo de determinar si la administración de uno u otro fármaco, o incluso de ambos, aporta beneficio clínico expresado en términos de supervivencia de los pacientes, varios autores han llevado a cabo una extensa revisión sistemática en las principales bases de datos de ensayos clínicos (entre otras, Medline o la base de datos Cochrane).
La búsqueda de información se amplió, sin restricción de idiomas, a cualquier ensayo clínico aleatorizado que comparara los efectos de la dosis estándar de adrenalina frente a placebo, de la dosis estándar frente a dosis altas de adrenalina o de adrenalina frente a vasopresina, ante una situación de paro cardíaco por cualquier causa, tanto en adultos como en niños. Los últimos trabajos revisados datan de septiembre de 2018. Dos autores revisaron todos los estudios de forma independiente, y acordaron emplear los ratios de riesgo (RR) con un intervalo de confianza del 95% (IC95%) para comparar los resultados dicotómicos de los eventos clínicos, así como aportar los niveles de evidencia de acuerdo al sistema GRADE1 (Grading of Recommendations Assessment, Development, and Evaluation).
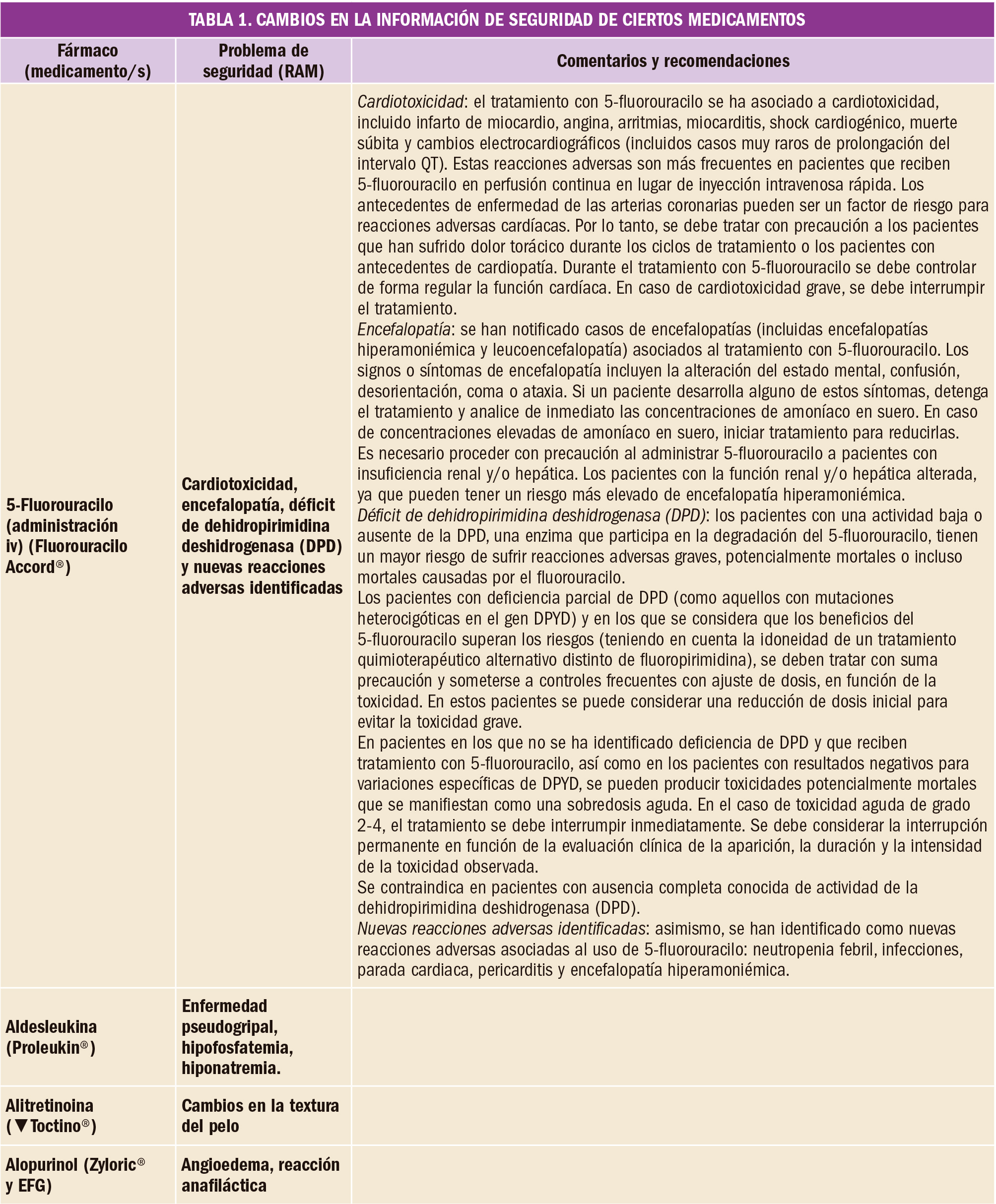
Se analizaron datos de un total de 26 ensayos clínicos (que englobaron a 21.704 participantes). Una evidencia de calidad moderada demuestra que la adrenalina incrementaba la supervivencia al alta hospitalaria en comparación con placebo (de 23 a 32 por cada 1.000 pacientes, IC95% 25 a 42; RR=1,44, IC95% 1,11 a 1,86), mientras que se hallaron incertidumbres respecto a la significación estadística de la supervivencia al alta hospitalaria para dosis altas de adrenalina en comparación con dosis estándar y para dosis estándar de adrenalina versus vasopresina más adrenalina, debido a la muy baja calidad de la evidencia disponible.
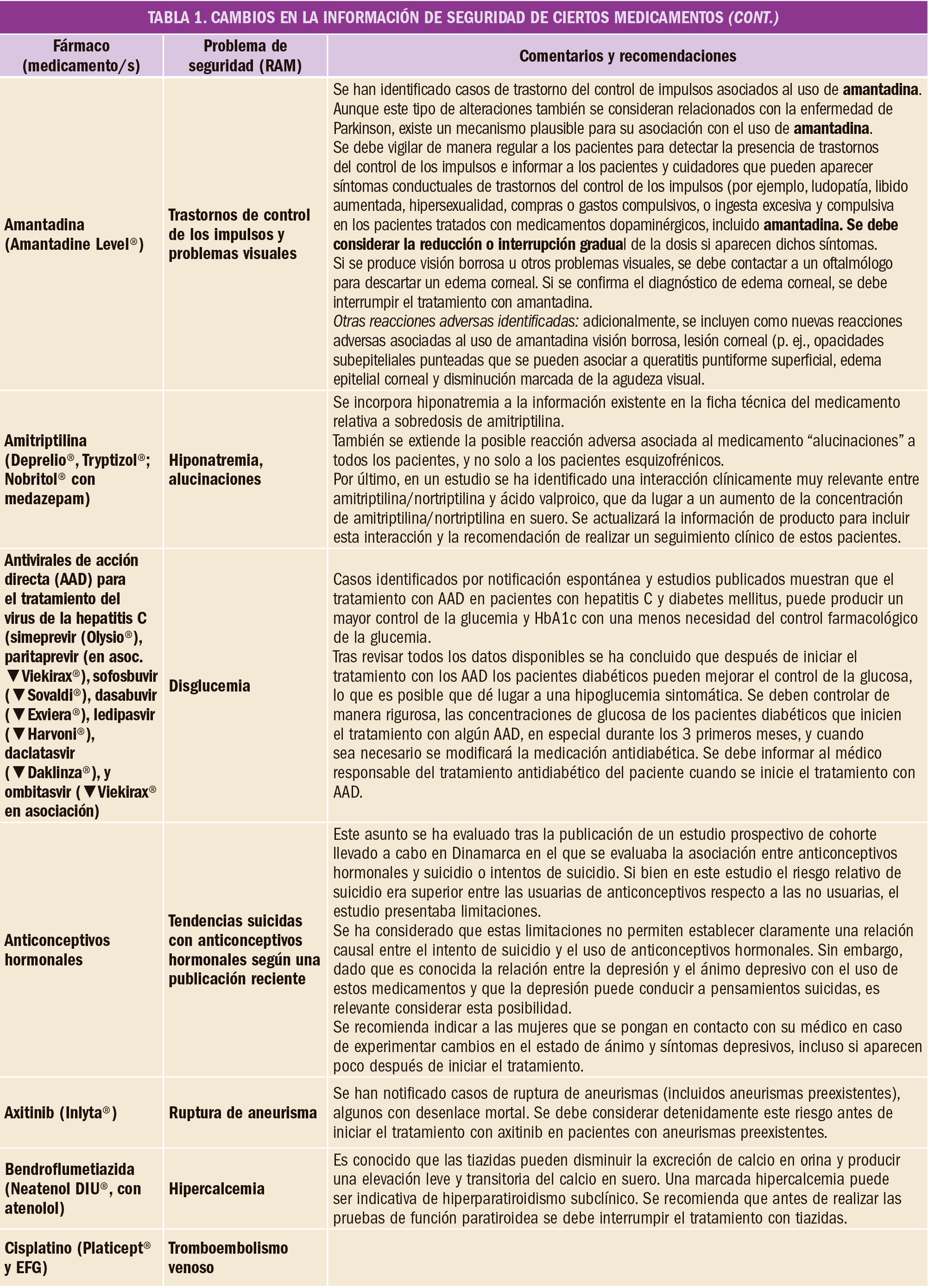
De forma similar, una evidencia de calidad moderada permitió a los autores concluir que la adrenalina incrementa la supervivencia al ingreso hospitalario (de 83 a 209 por cada 1.000 pacientes, IC95% 139 a 213; RR=2,51, IC95% 1,67 a 3,76) y aumenta la tasa de retorno de la circulación espontánea (de 115 a 329 por cada 1.000 pacientes, IC95% 254 a 427; RR=2,86, IC95% 2,21 a 3,71) en comparación con placebo. Sin embargo, el bajo nivel de evidencia impidió confirmar estos mismos efectos para la comparación de dosis estándar de adrenalina con dosis altas de adrenalina, con vasopresina (aunque cierta evidencia apuntaba a que la vasopresina sí podría incrementar la supervivencia al ingreso hospitalario) o con adrenalina más vasopresina. Por otro lado, no se encontró ninguna prueba de que la adrenalina o la vasopresina a cualquier dosis mejoren el estado neurológico de los pacientes supervivientes.
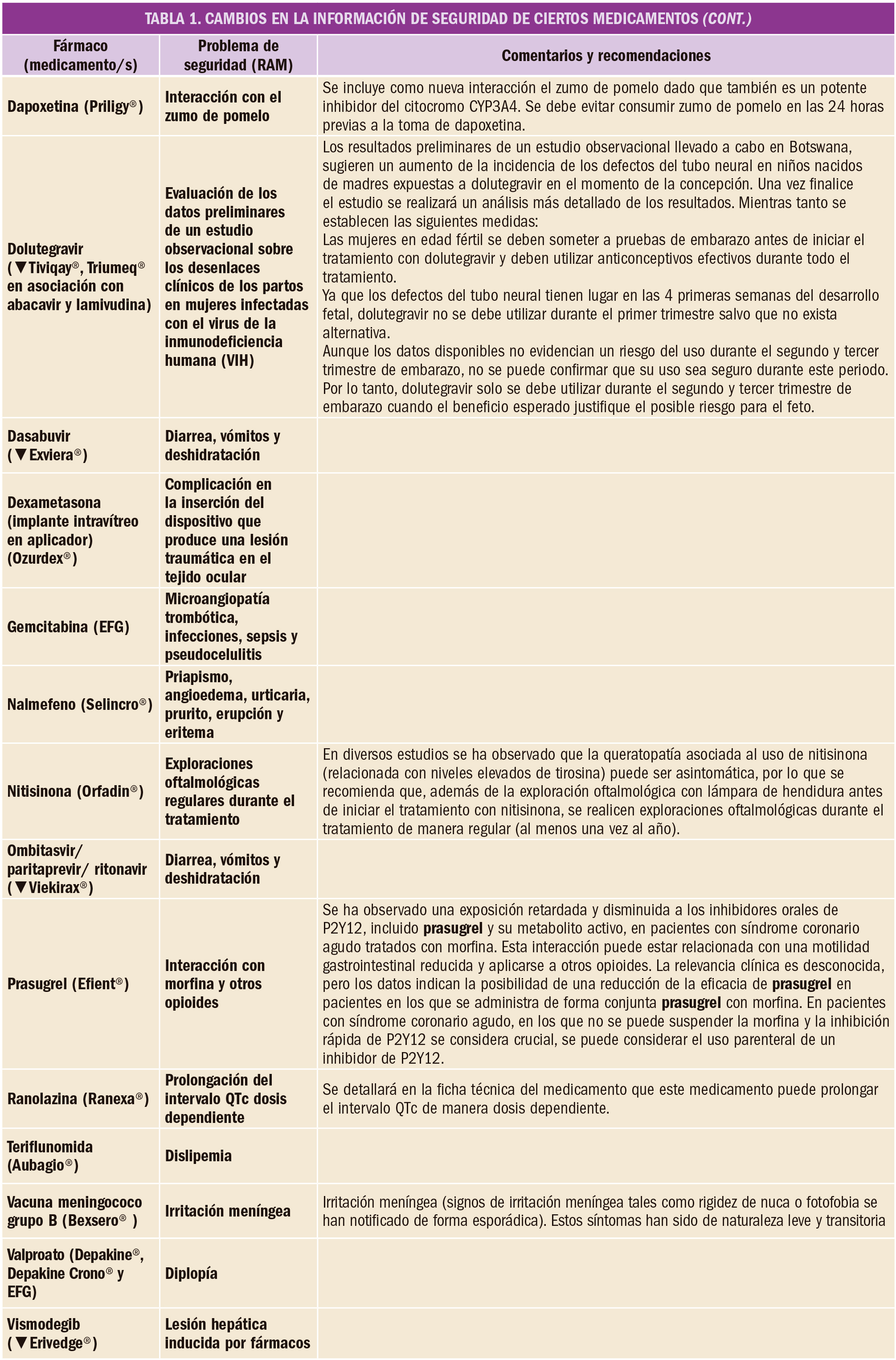
En definitiva, este vasto meta-análisis aporta evidencia de calidad moderada para los efectos positivos de la adrenalina a dosis estándar, en comparación con placebo, sobre el retorno de la circulación espontánea y la supervivencia (tanto al ingreso como al alta hospitalaria) de los pacientes que han sufrido un paro cardíaco, si bien no se confirma que la adrenalina mejore los resultados neurológicos. Tampoco se dispone de evidencia convincente para recomendar la utilización en clínica de dosis altas de adrenalina, de vasopresina sola, o de la combinación de adrenalina más vasopresina. Cabe destacar, por último, que muchos de los ensayos clínicos revisados se realizaron hace más de 20 años y que, teniendo en cuenta que el tratamiento ha cambiado en los últimos años, la evidencia aquí comentada puede no reflejar la práctica actual.