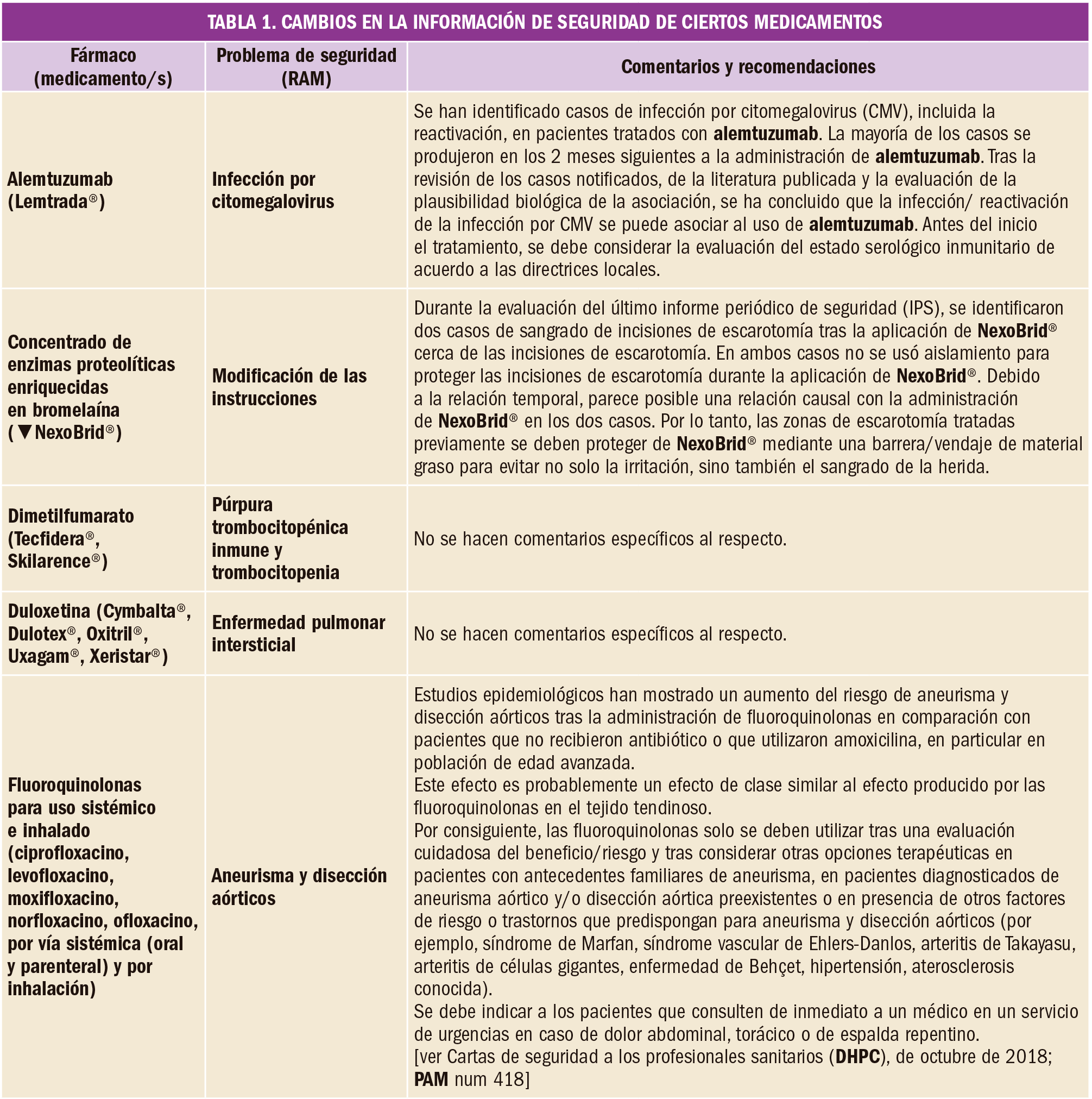
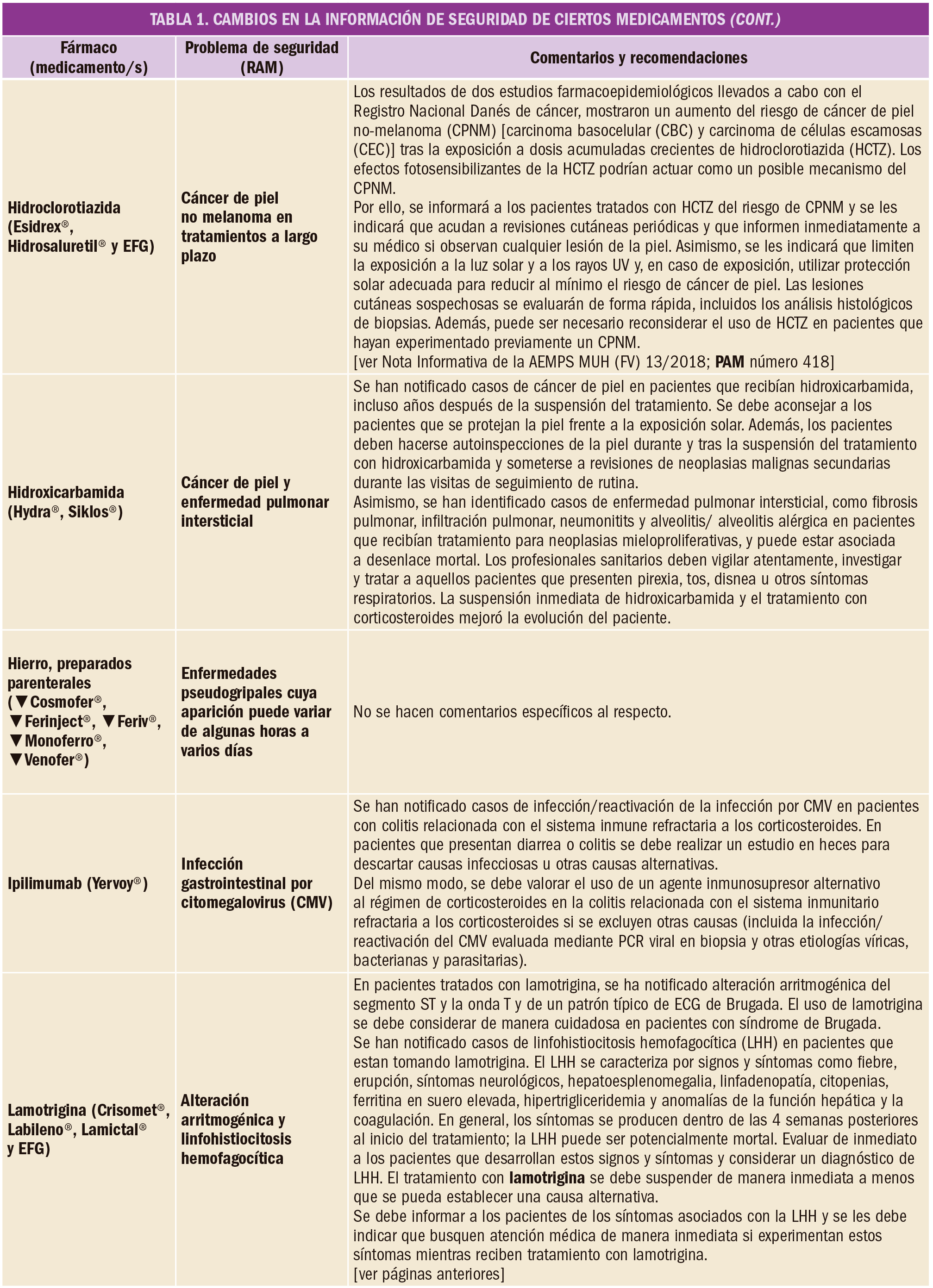
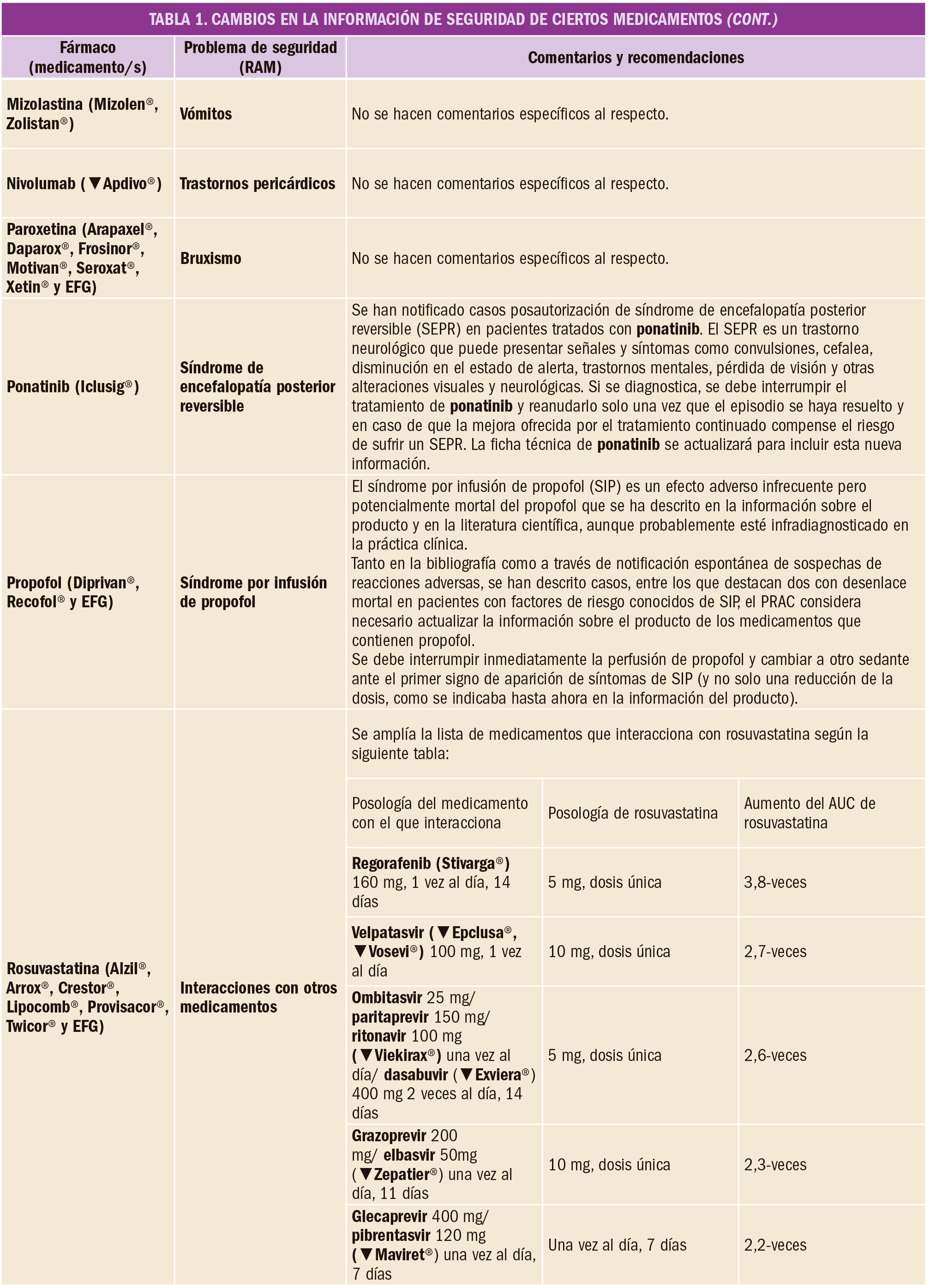
En un amplio estudio de cohortes, el tratamiento con alopurinol en dosis de al menos 300 mg/día se asoció con un menor riesgo de deterioro de la función renal. Por este motivo, se deberían considerar otras posibles causas cuando los pacientes con gota tratados con estas dosis de alopurinol experimentan disminución de la función renal.
Durante mucho tiempo se ha considerado que el empleo crónico de alopurinol, especialmente en altas dosis, en pacientes con gota podría agudizar el deterioro de la función renal. Con el fin de aclarar esta cuestión, se ha llevado a cabo un estudio de cohorte prospectivo poblacional, basado en la puntuación de la propensión estratificada en el tiempo de individuos con gota recién diagnosticada que iniciaron un tratamiento con alopurinol (≥300 mg/día) en comparación con aquellos no tratados con este fármaco, utilizando la base de datos de registros de salud electrónicos del Reino Unido.
De los 4.760 pacientes que comenzaron el tratamiento con alopurinol (3.975 hombres, 785 mujeres) y el mismo número de aquellos que no comenzaron el tratamiento (3.971 hombres, 789 mujeres), 579 y 623, respectivamente, desarrollaron enfermedad renal crónica en estadio 3 o superior, con un tiempo medio de seguimiento de 5 y 4 años, siendo la edad promedio de 57 años y el índice de masa corporal (IMC) promedio de 30 para ambos grupos. El uso de alopurinol de al menos 300 mg/día se asoció con una reducción del 23% del riesgo de desarrollar enfermedad renal crónica en estadio 3 o superior en comparación con los no usuarios (HR=0,87; IC95% 0,77 a 0,97). Por el contrario, el empleo de dosis de alopurinol inferiores a 300 mg/día no se asoció con disminución de la función renal (HR=1,00; IC95% 0,91 a 1,09).
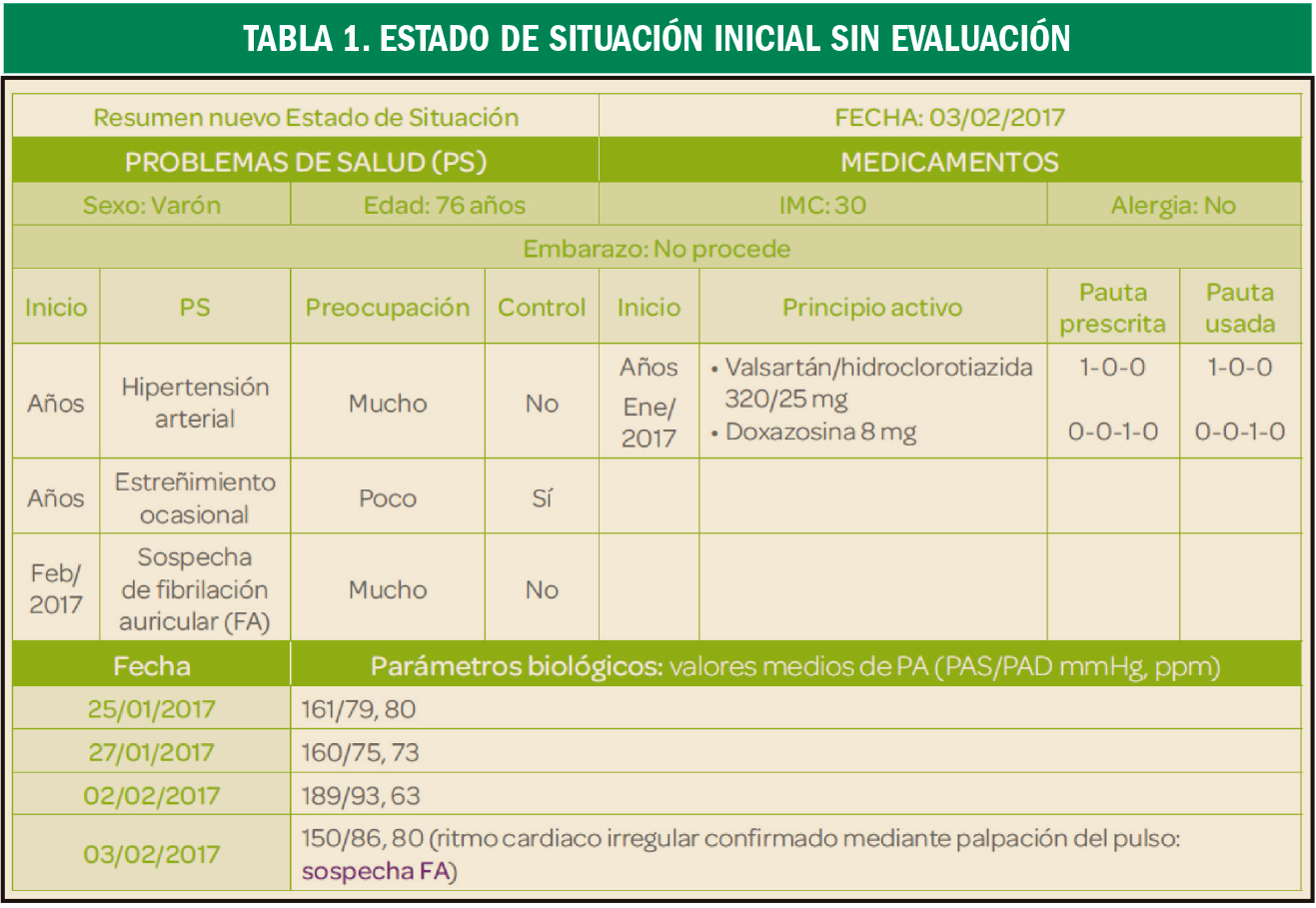
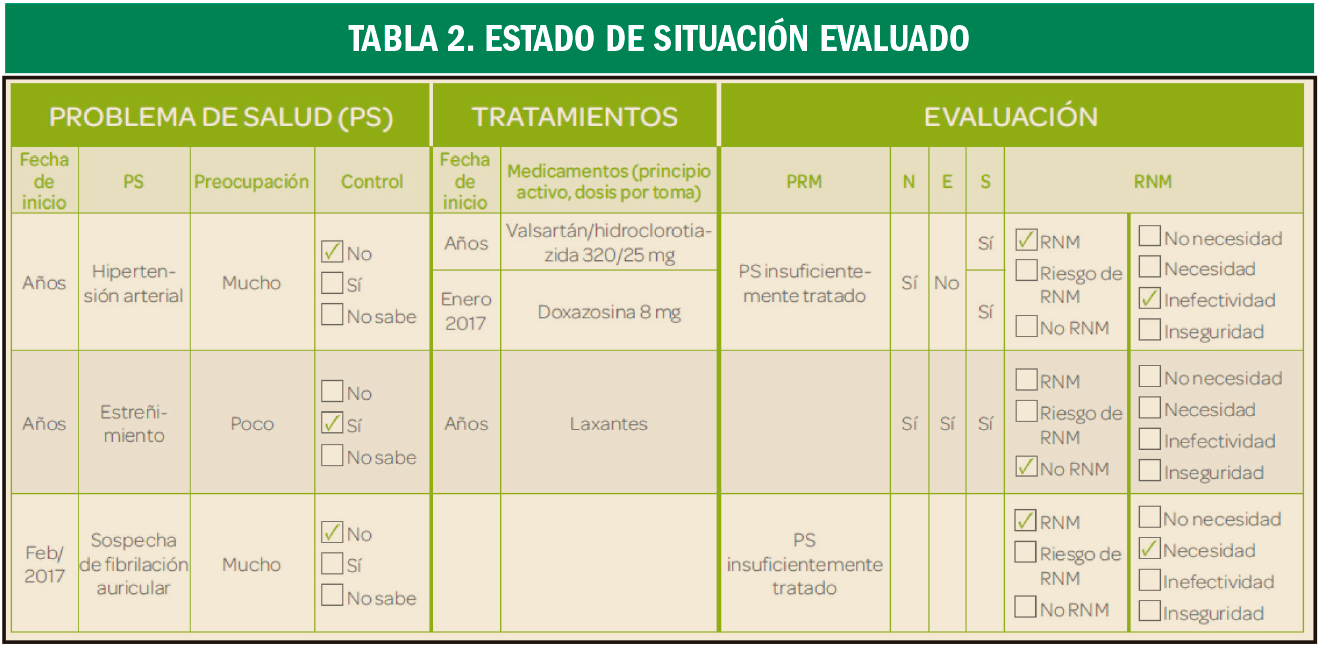
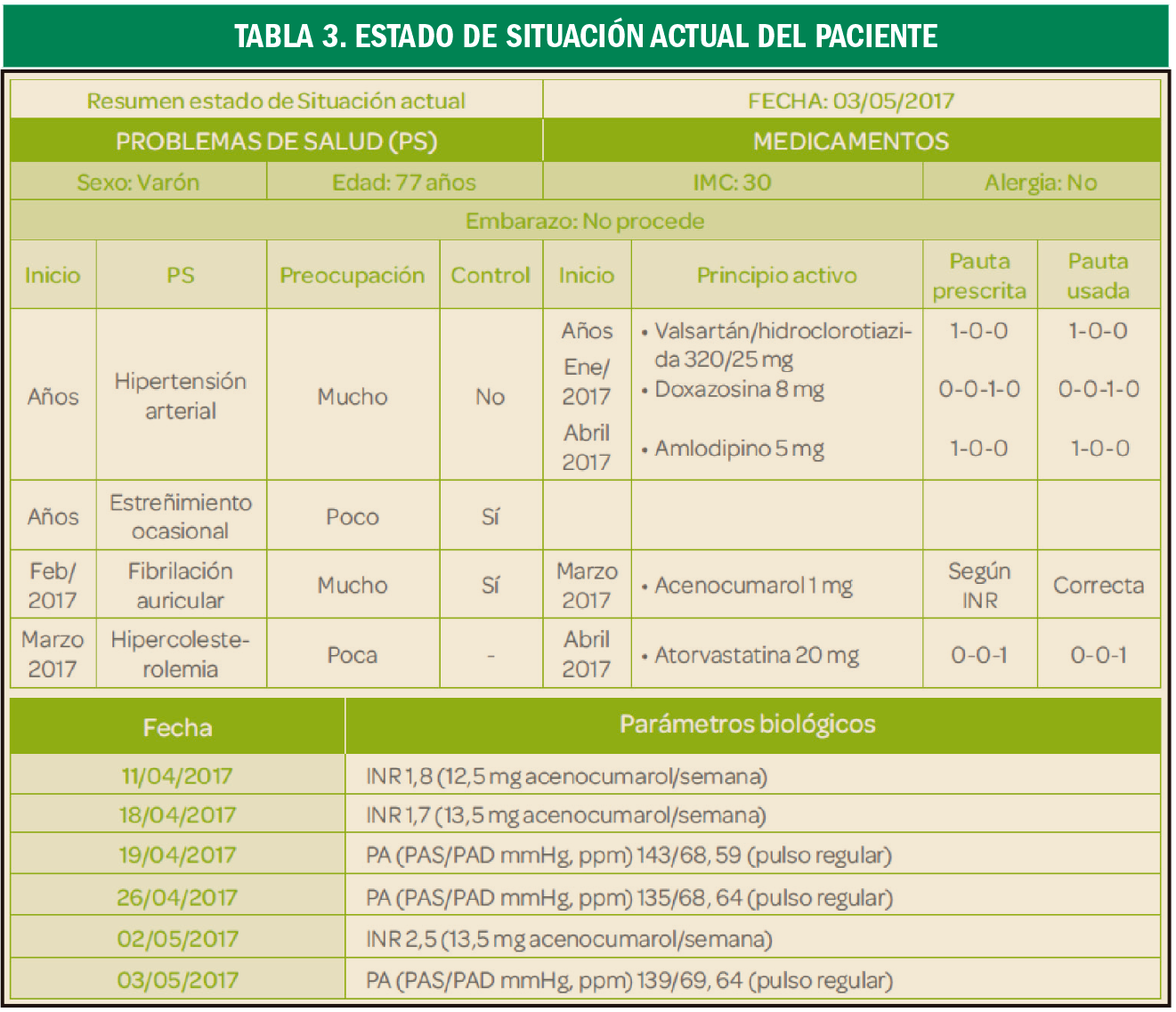


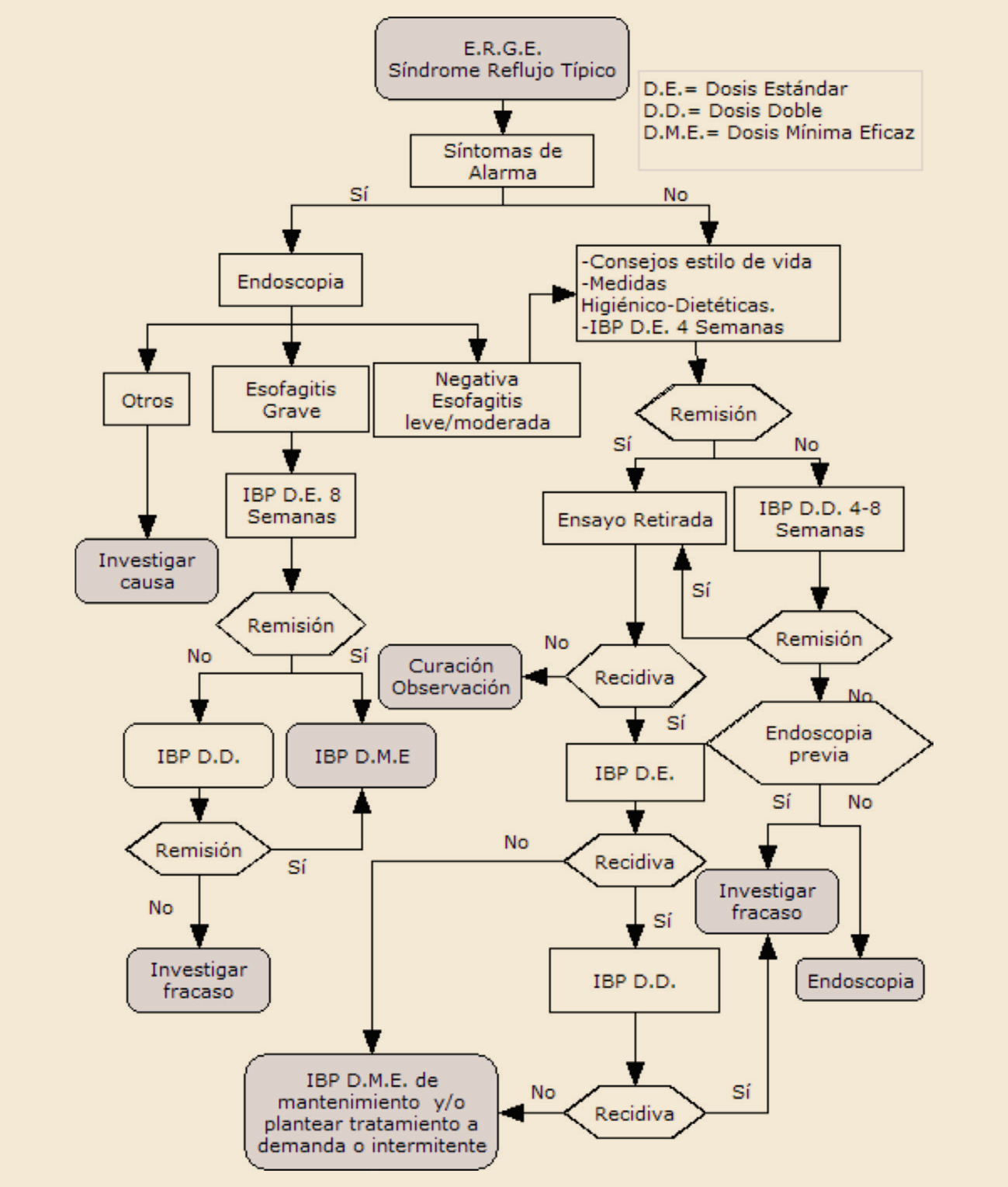
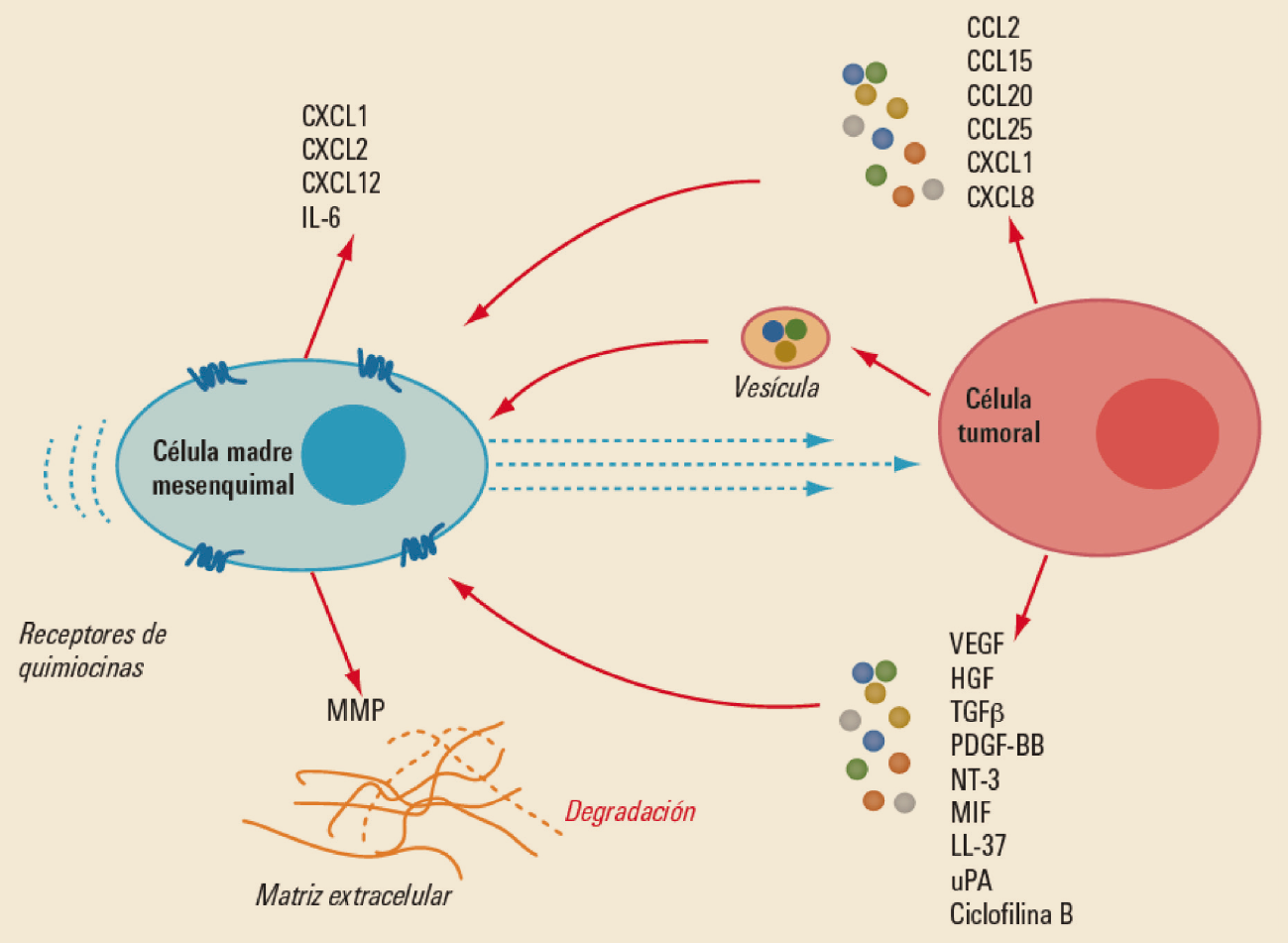
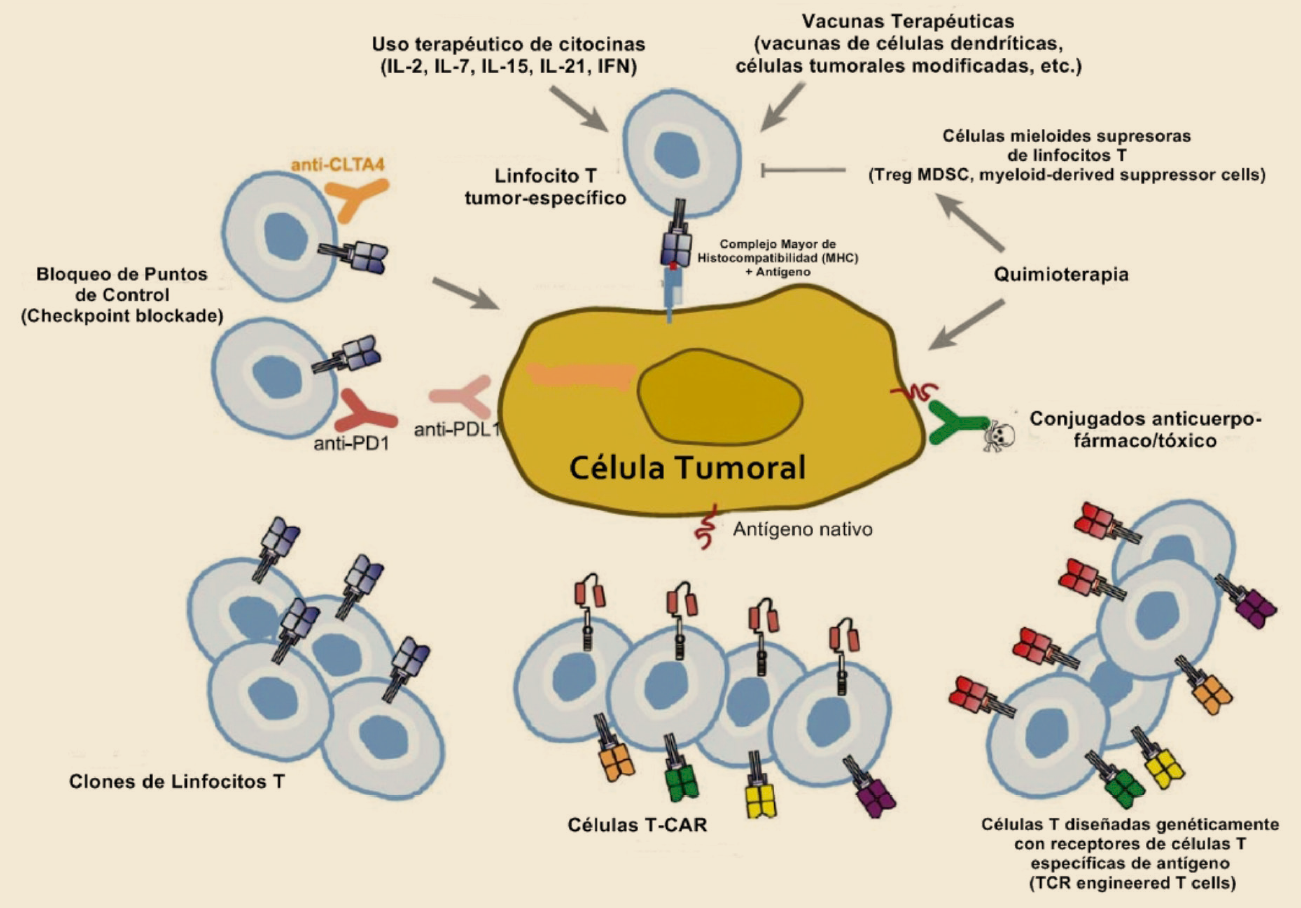
{kind=link}
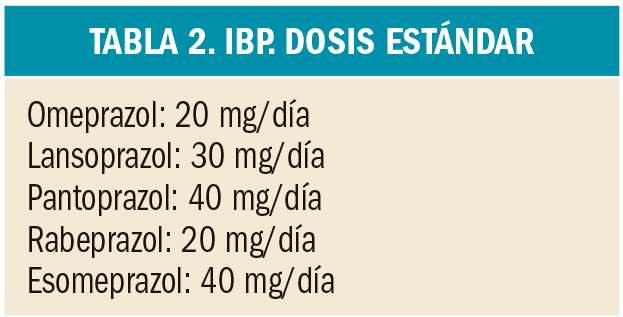{kind=link}