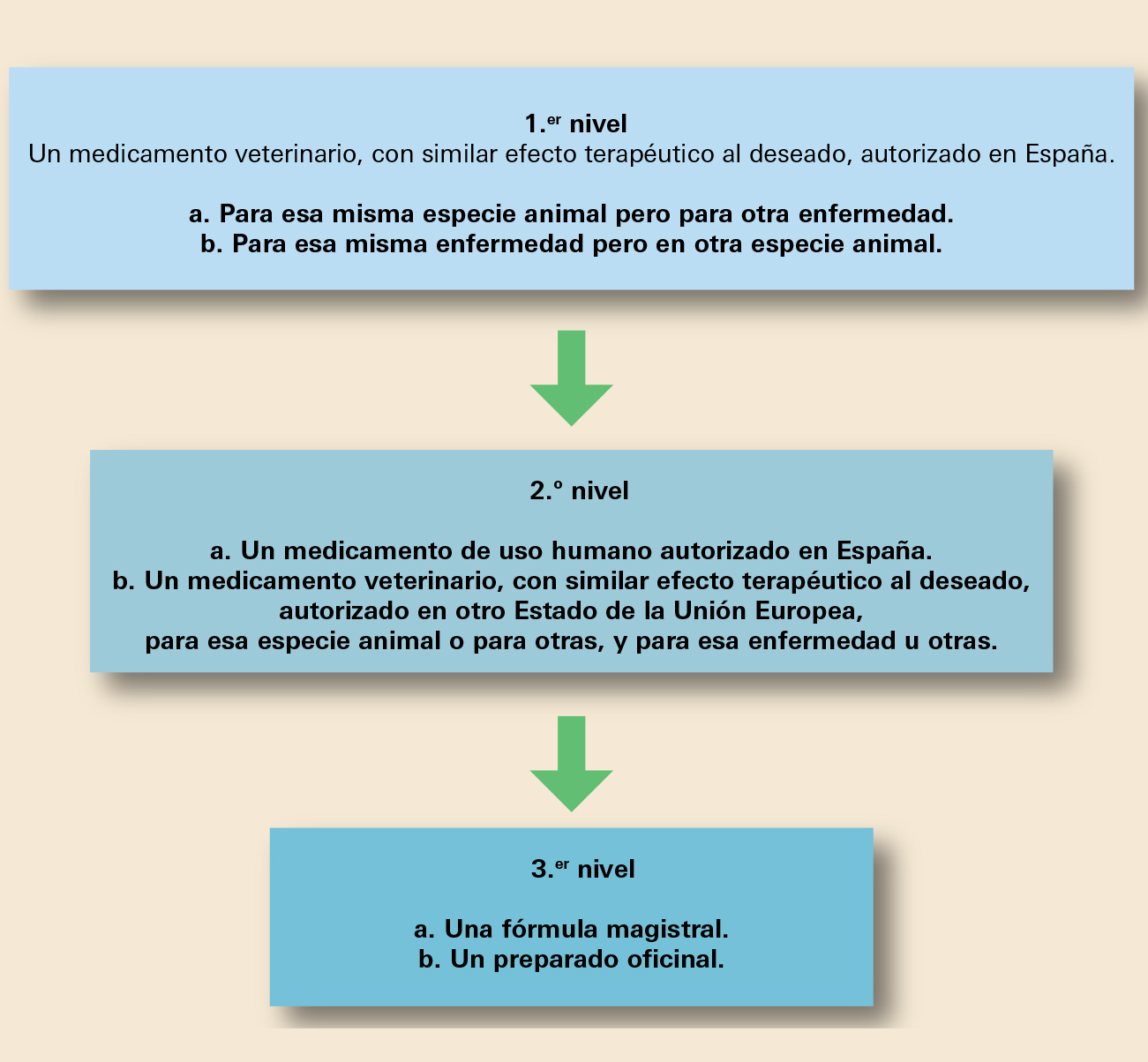
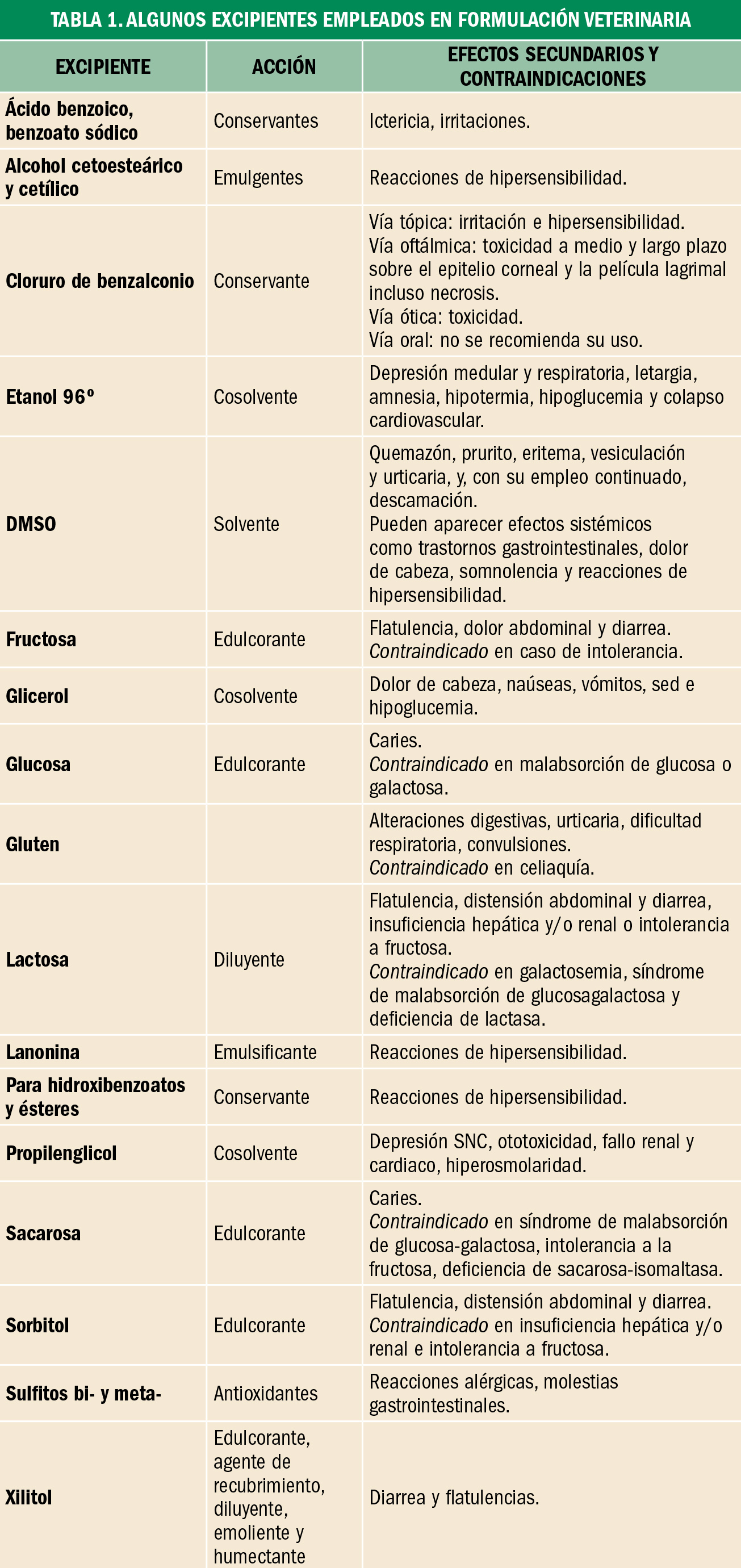
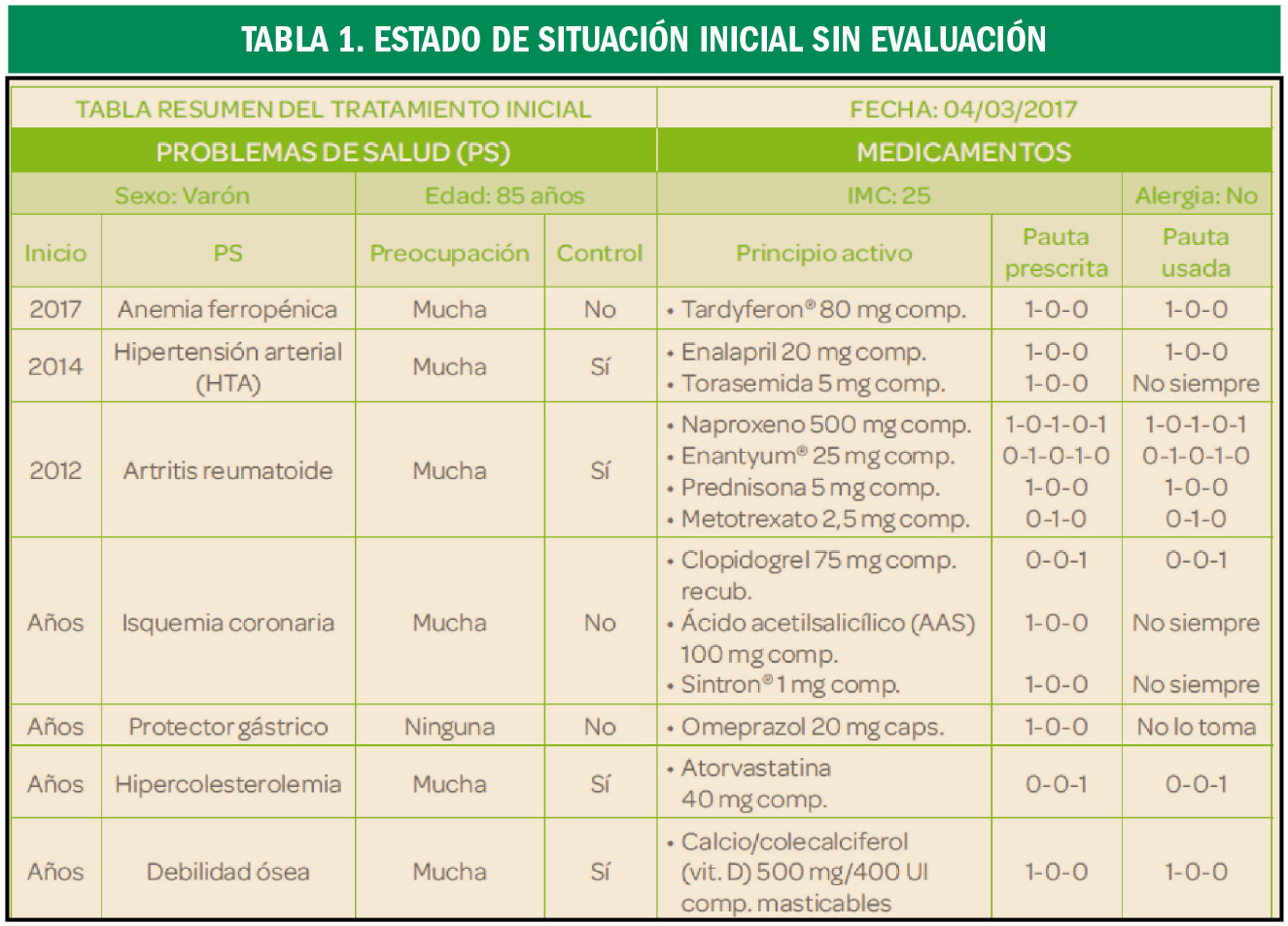
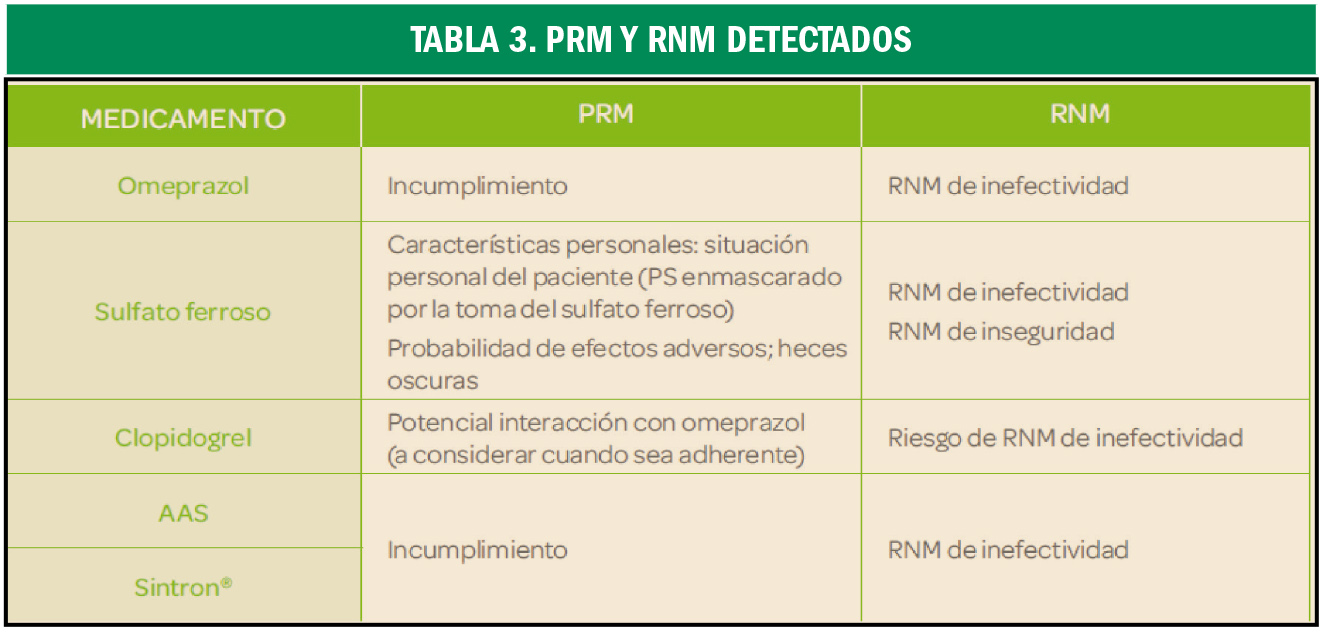
Hiperlipidemia
El exceso de selenio puede resultar peligroso
En un reciente estudio epidemiológico realizado en zonas rurales de China se correlacionaron los niveles elevados de selenio en suero con un aumento del riesgo de dislipidemia. Sin embargo, se requieren estudios más específicos que confirmen o desmientan estos primeros resultados.
El selenio es un antioxidante que previene los procesos oxidativos de las células originados por los radicales libres de oxígeno, retrasando el envejecimiento patológico. Es un componente de la glutation peroxidasa, enzima que actúa junto con la vitamina E en el sistema de defensa antioxidante de la célula. Estimula el sistema inmunológico, reforzando la resistencia a las infecciones. Está presente también en otras enzimas como la formiato deshidrogenasa, la xantina deshidrogenasa, la glicina reductasa, etc. En el plasma circula en niveles fisiológicos de 0,03 a 0,59 mg/mL.
Su carencia produce dos enfermedades reconocidas, la enfermedad de Keshan, cardiomiopatía de la edad infantil, y la enfermedad de Kashin y Beck, que se caracteriza por trastornos graves del desarrollo óseo, con deformación de las articulaciones y debilidad muscular. Ambas se han observado en regiones de China muy escasas en selenio. También ha sido asociada con dolor y debilidad muscular, así como infertilidad masculina, disminución de la actividad inmunológica y caída del cabello. La exposición crónica a elevadas dosis de selenio ha sido asociada al desarrollo de cirrosis y de tumores hepáticos. Hay casos de intoxicación aguda que conducen a deficiencias de metionina y cisteína, debido a un intercambio de azufre por selenio. El exceso de este mineral puede provocar ennegrecimiento de las unas, inflamación cutánea y alteraciones nerviosas. Se estima que la dosis máxima segura es de 600 mg/día.
Como componente clave de la glutatión peroxidasa, se ha considerado que el selenio desempeña un papel importante en el metabolismo de los lípidos. Sin embargo, las asociaciones de concentraciones de selenio en suero con concentraciones de lípidos y dislipidemia son todavía controvertidas.
Un grupo de investigadores ha analizado los niveles séricos de selenio, las concentraciones de lípidos y otros índices relacionados en personas de 8.198 zonas rurales de China, encontrando unos niveles séricos medios de 0,120 μg/L, correlacionándose los superiores de forma estadísticamente significativa con niveles elevados de colesterol total, HDL-c, triglicéridos (TG) y LDL-c. En comparación con el quintil más bajo de selenio sérico, los participantes en los quintiles 3, 4 y 5 tuvieron un mayor riesgo de hipercolesterolemia (colesterol total y LDL-c). En los análisis estratificados por características antropológicas, las asociaciones selenio-dislipidemia fueron significativamente más robustas en las mujeres posmenopáusicas (odds ratio, OR=2,72; IC95% 1,97 a 4,17) y diabéticos (OR=9,40; IC95% 3,02 a 29,26).

{kind=link}