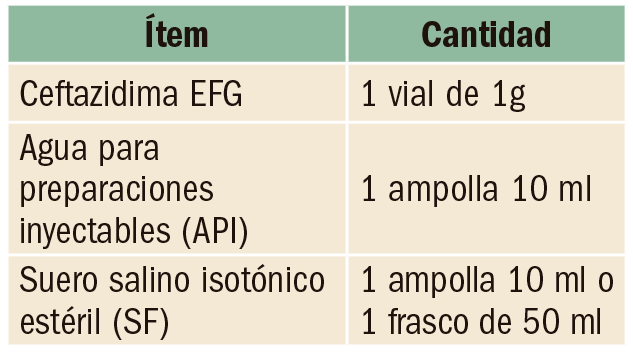
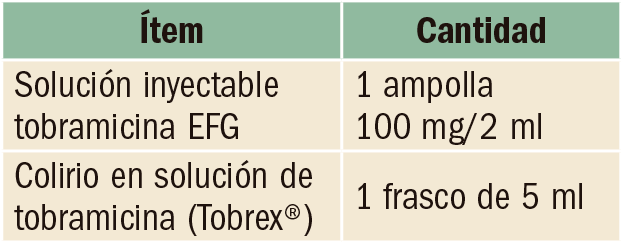
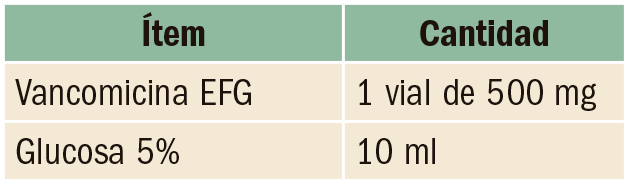
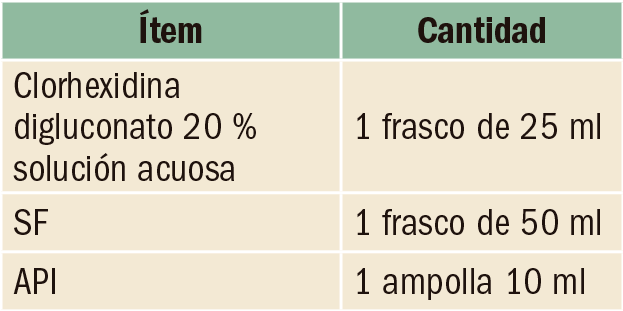
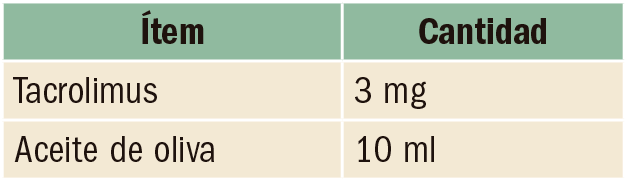
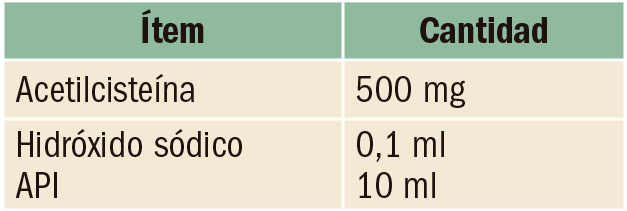
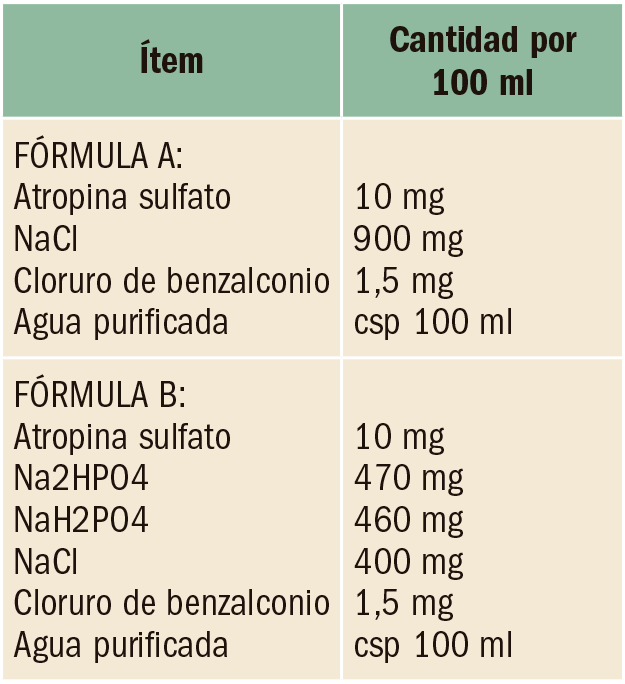
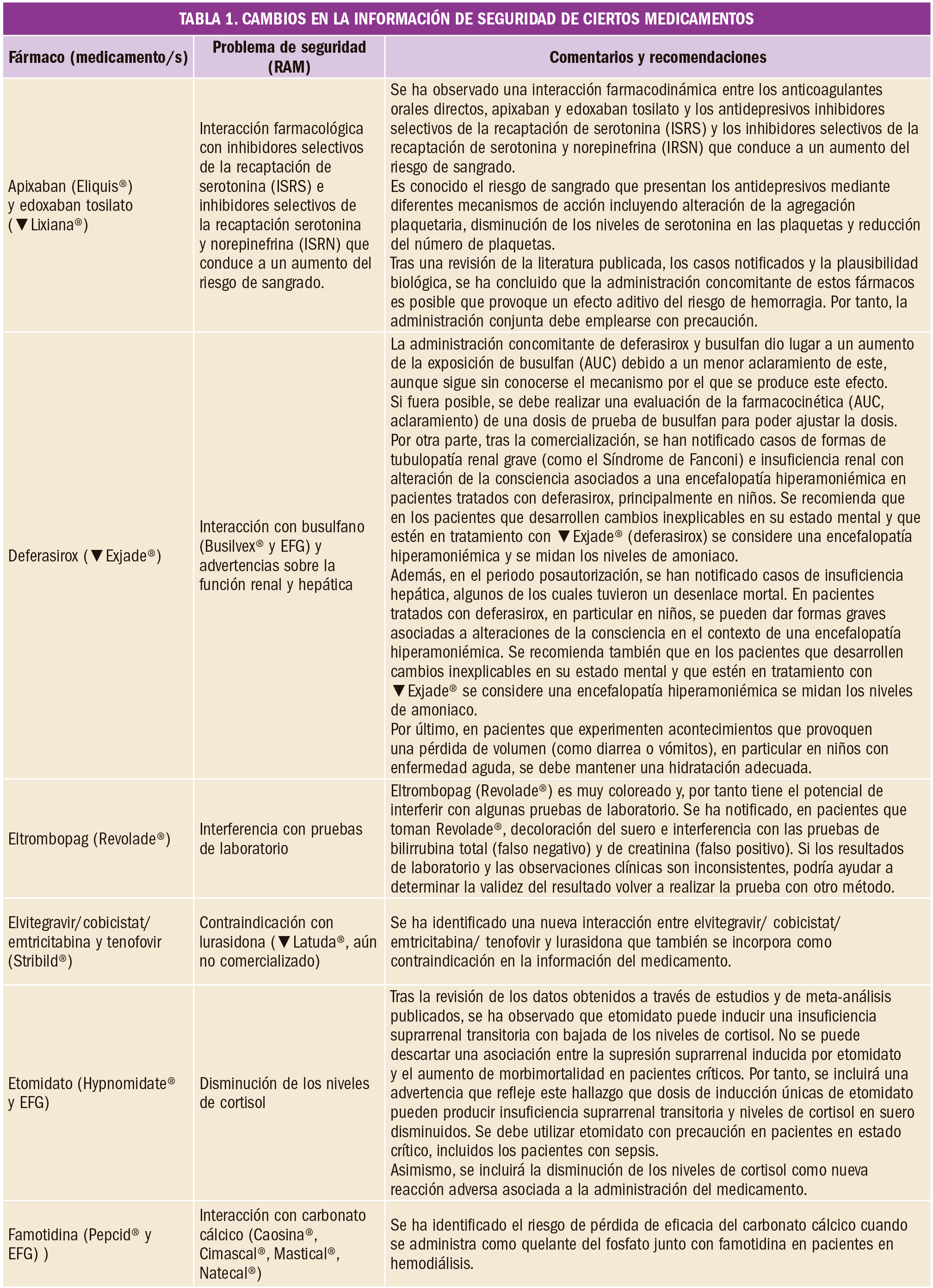
Anticoagulación
Anticoagulantes de acción directa vs. cumarínicos en la práctica clínica cotidiana
En un amplio estudios de cohortes de pacientes tratados con fármacos anticoagulantes en la práctica clínica cotidiana, se ha ha observado que el apixaban es el fármaco más seguro, con menores riesgos de hemorragia mayor, intracraneal y gastrointestinal en comparación con la warfarina. Sin embargo, el rivaroxabán y la dosis baja de apixabán se asociaron con un mayor riesgo de mortalidad por cualquier causa en comparación con la warfarina.
Con el fin de establecer el riesgo real de uso de los anticoagulantes orales directos en relació con warfarina, se llevó a cabo un estudio prospectivo de cohortes abiertas en Gran Bretaña, en el que participaron 132.231 personas que venían recibiendo warfarina, 7.744 dabigatran, 37.863 rivaroxabán y 18.223 apixaban durante al menos los últimos 12 meses anteriores al ingreso al estudio; 103.270 eran pacientes con fibrilación auricular y 92.791 sin fibrilación auricular. El estudio cubrió un periodo comprendido entre 2011 y 2016 y como variable principal se valoró la incidencia de hemorragia grave que condujese a la hospitalización del paciente o su muerte.
En pacientes con fibrilación auricular, en comparación con la warfarina, el apixaban se asoció con una disminución del 34% en el riesgo de hemorragia grave (cociente de riesgo ajustado, HR=0,66; EC95% 0,54 a 0,79) y del 60% en la hemorragia intracraneal (HR=0,40; IC95% 0,25 a 0,64); el dabigatrán se asoció con una disminución del riesgo de hemorragia intracraneal del 55% (HR=0,45; EC95% 0,26 a 0,77). Se observó un aumento del 19% en el riesgo de mortalidad por todas las causas en pacientes que tomaban rivaroxaban (HR=1,19; EC95% 1,09 a 1,29) y del 27% con las dosis más bajas de apixaban (HR=1,27; EC95% 1,12 a 0,79 (1,27, 1,12 a 1,45). En pacientes sin fibrilación auricular, en comparación con warfarina, apixaban se asoció con una reducción del 40% del riesgo de hemorragia grave (HR=0,60; EC95% 0,46 a 0,79), un 45% menos de hemorragia gastrointestinal (HR=0,55; EC95% 0,37 a 0,83 y de hemorragia digestiva alta (HR=0,55; EC95% 0,36 a 0,83); rivaroxaban se asoció con una disminución del 46% del riesgo de hemorragia intracraneal (HR=0,54; EC95% 0,35 a 0,82. Se observó un aumento del 51% del riesgo de mortalidad por todas las causas en pacientes que tomaban rivaroxabán (HR=1,51; EC95% 1,38 a 1,66) y un 34% en aquellos que usaban las dosis más bajas de apixaban (HR=1,34%; EC95% 1,13 a 1,58).