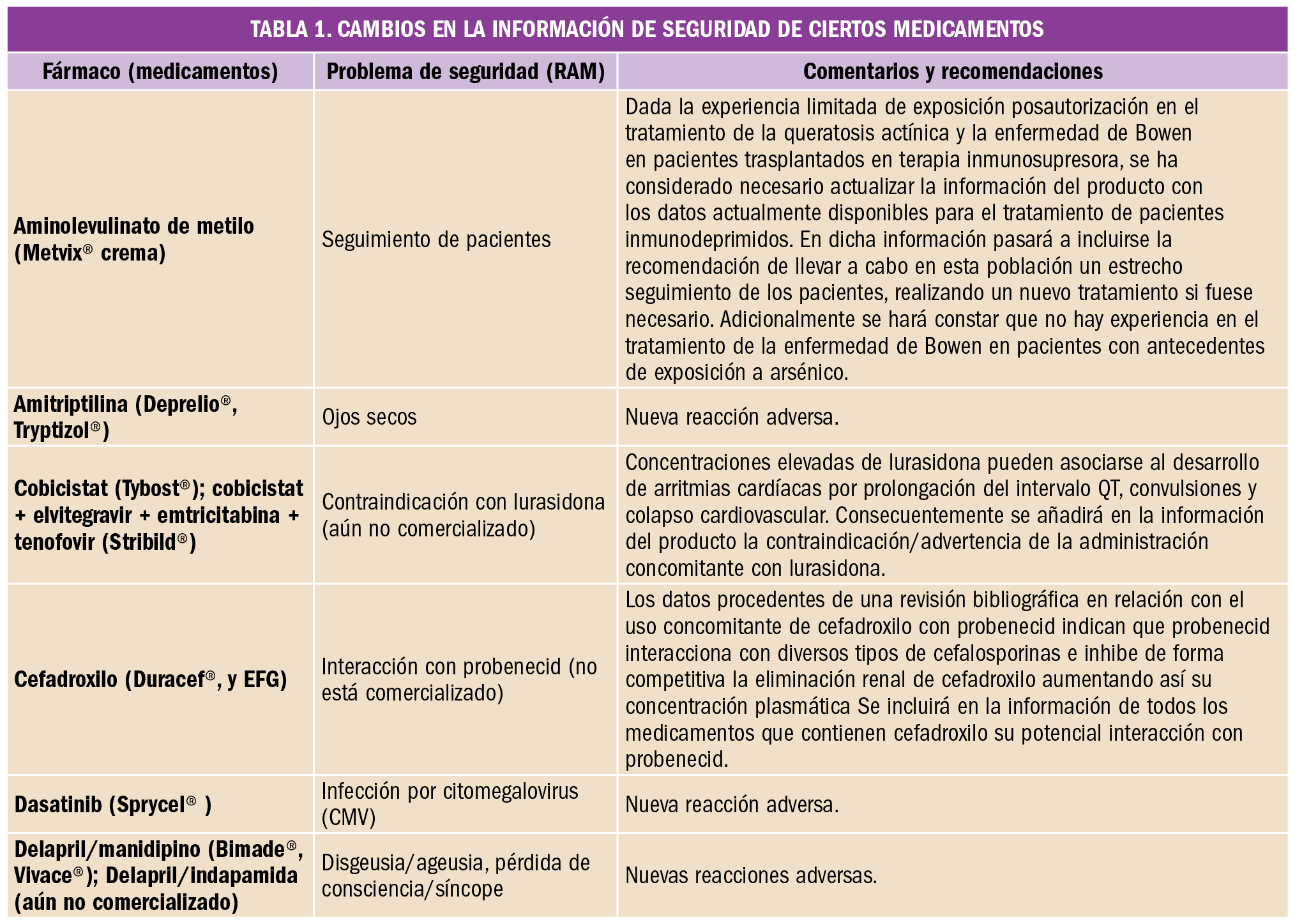
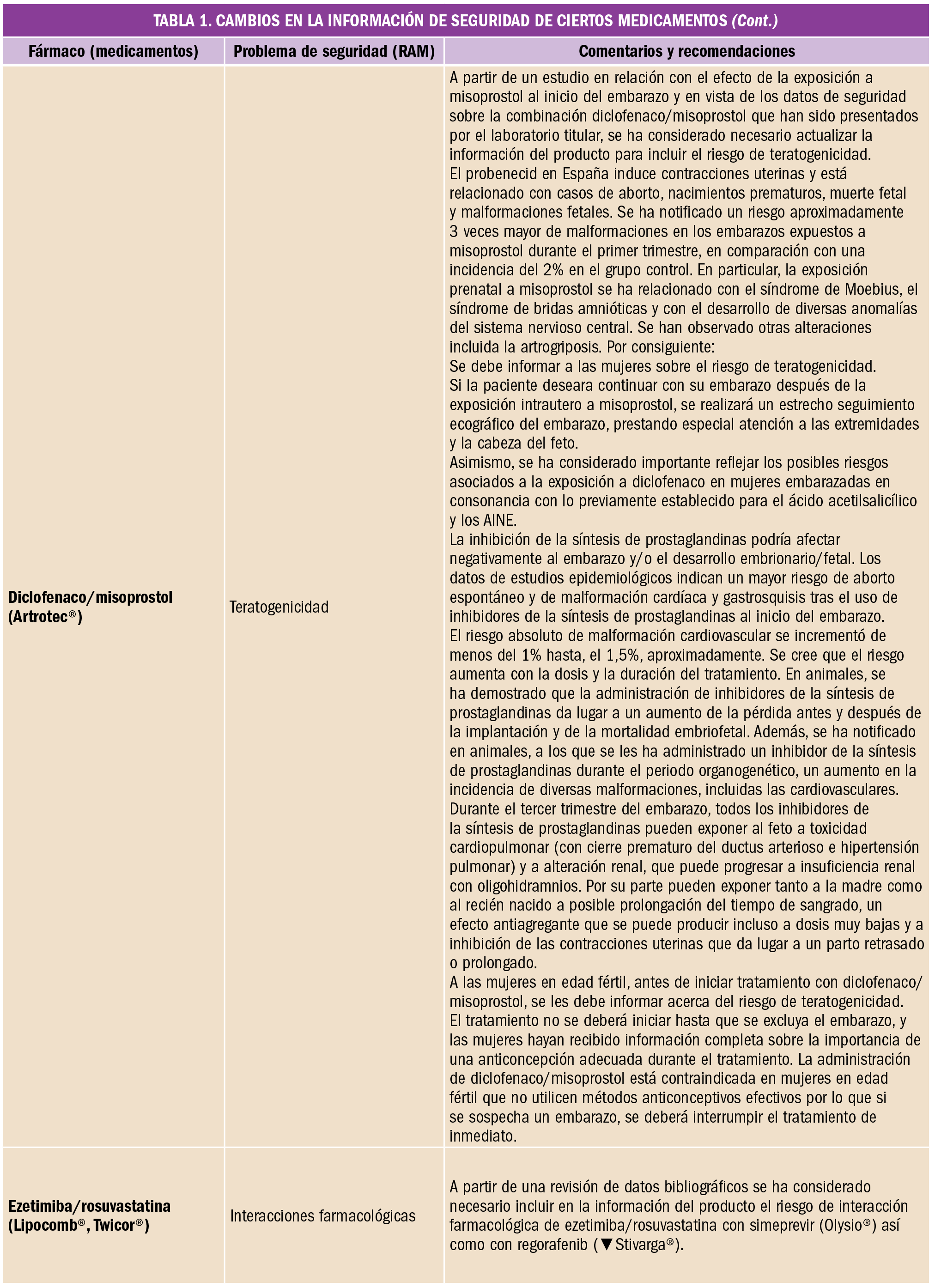
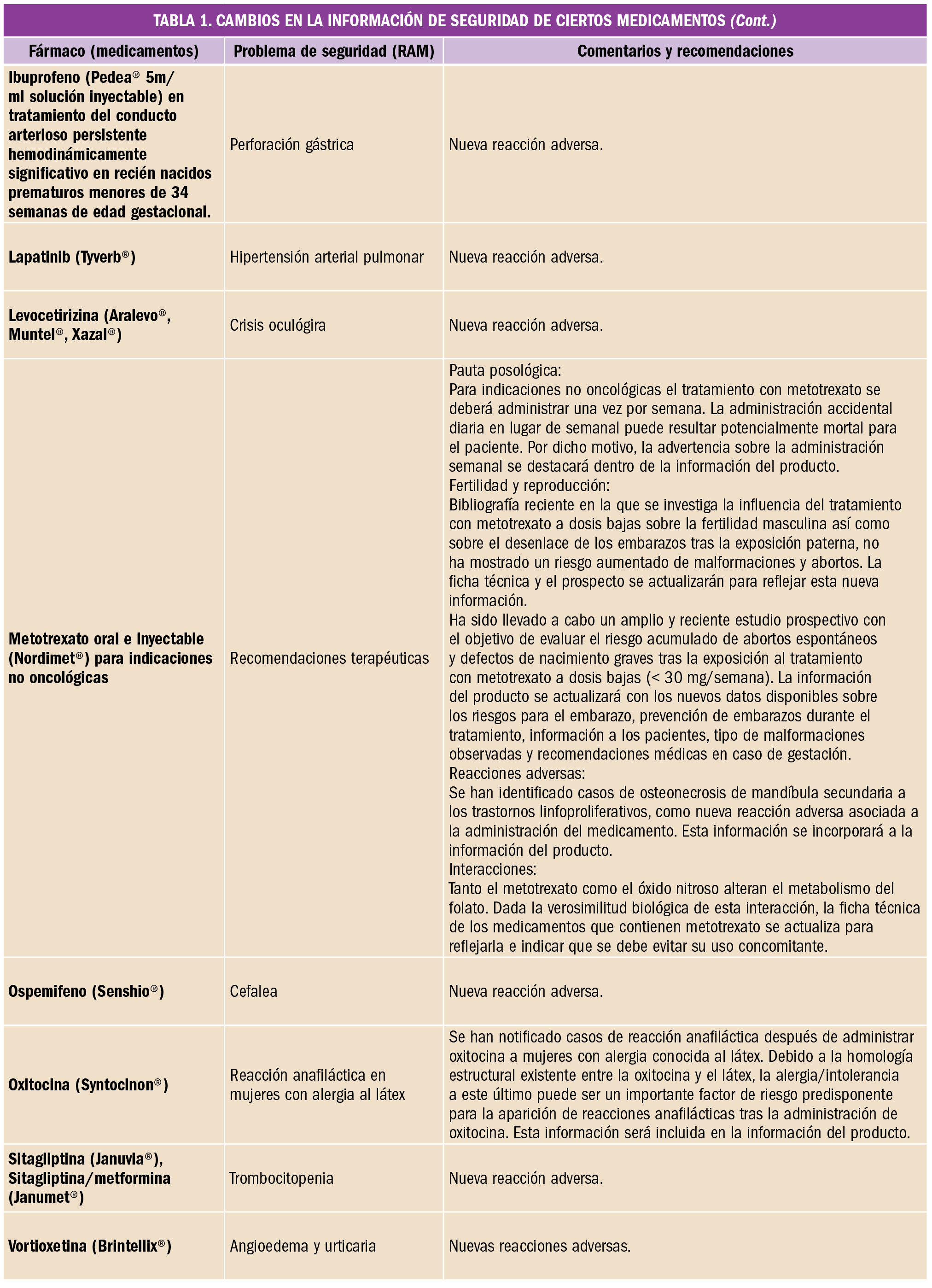
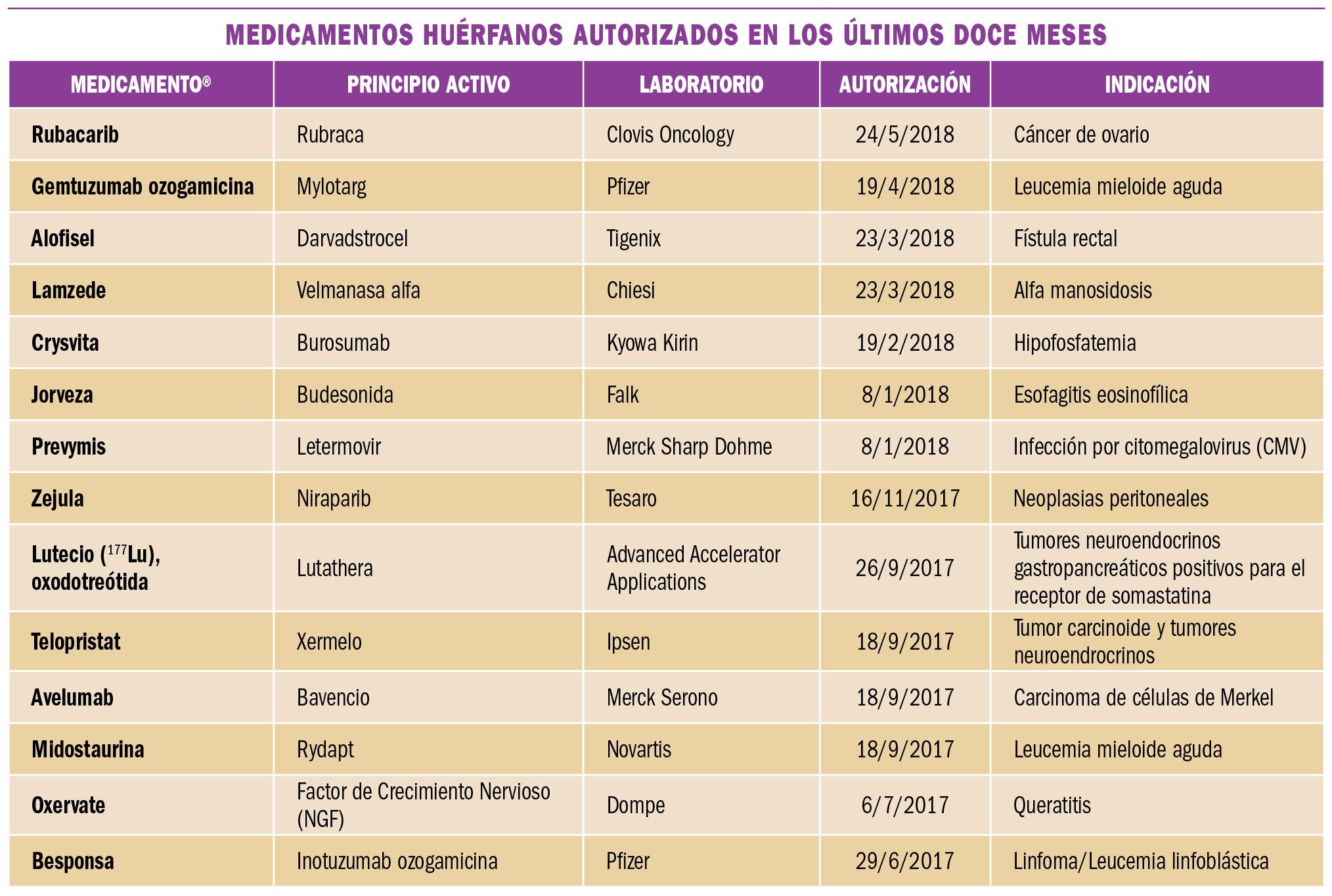
Es frecuente que lleguen a las farmacias prescripciones de vacunas que en ese momento no tenemos en stock o bien que están en desabastecimiento. ¿Cómo debemos proceder para atender a la demanda de los pacientes actuando de acuerdo a las recomendaciones científicas y la normativa legal?
En primer lugar, es importante recordar que las vacunas son medicamentos no sustituibles, por la Orden SCO/2874/2007, de 28 de septiembre, por la que se establecen los medicamentos que constituyen excepción a la posible sustitución por el farmacéutico con arreglo al artículo 86.4 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. En el acto de dispensación no se puede cambiar una marca por otra. Se debe dispensar siempre la marca prescrita; si no la tenemos en ese momento en la farmacia, hemos de pedirla.
Pero, ¿qué pasa cuando la marca prescrita está en desabastecimiento? ¿Cómo debemos actuar? Veamos cada caso:
- Si es la primera dosis, debemos comprobar que existe otra especialidad con la misma composición y mismas indicaciones, que la edad del paciente está dentro de las edades para los que la vacuna está indicada en ficha técnica y que no es alérgico a ninguno de los componentes. Una vez comprobados estos datos, derivaremos al médico para que prescriba la marca disponible en el mercado.
- Si no es la primera dosis, la regla general es continuar la vacunación con la especialidad con la que se comenzó a vacunar, informarnos si es posible de cuánto va a durar el desabastecimiento y comprobar en función de las dosis que se le han administrado y cuando las recibió si podemos esperar para continuar la vacunación con la misma marca. Cuando esto no es posible, en las fichas técnicas de algunas vacunas se refleja que, aunque no es lo ideal, se puede proceder a intercambiar con el fin de lograr la mejor inmunización posible de nuestros pacientes.
Vacunas que pueden intercambiarse según fichas técnicas (y siempre y cuando todas las demás posibilidades se han agotado) son por ejemplo varicela y hepatitis B, sin embargo, otras vacunas no han demostrado ser intercambiables, como es el caso de las vacunas de virus de papiloma humano, o la de meningococo C; la reciente publicación de un estudio realizado en Reino Unido, en el que se utilizaron de forma indistinta vacunas frente al meningococo C con diferentes carriers (MenC-TT o MenC-CRM), concluye que estas vacunas no son intercambiables.
Ante cualquier duda acerca de intercambiabilidad de vacunas, podemos consultar al CIM (Centro de Información del Medicamento) de cada Colegio Profesional o bien al CAV (Comité Asesor de Vacunas de la Asociación Española de Pediatría), que tiene en su página web un apartado donde se pueden formular preguntas.
Bibliografía