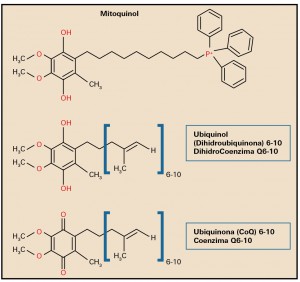
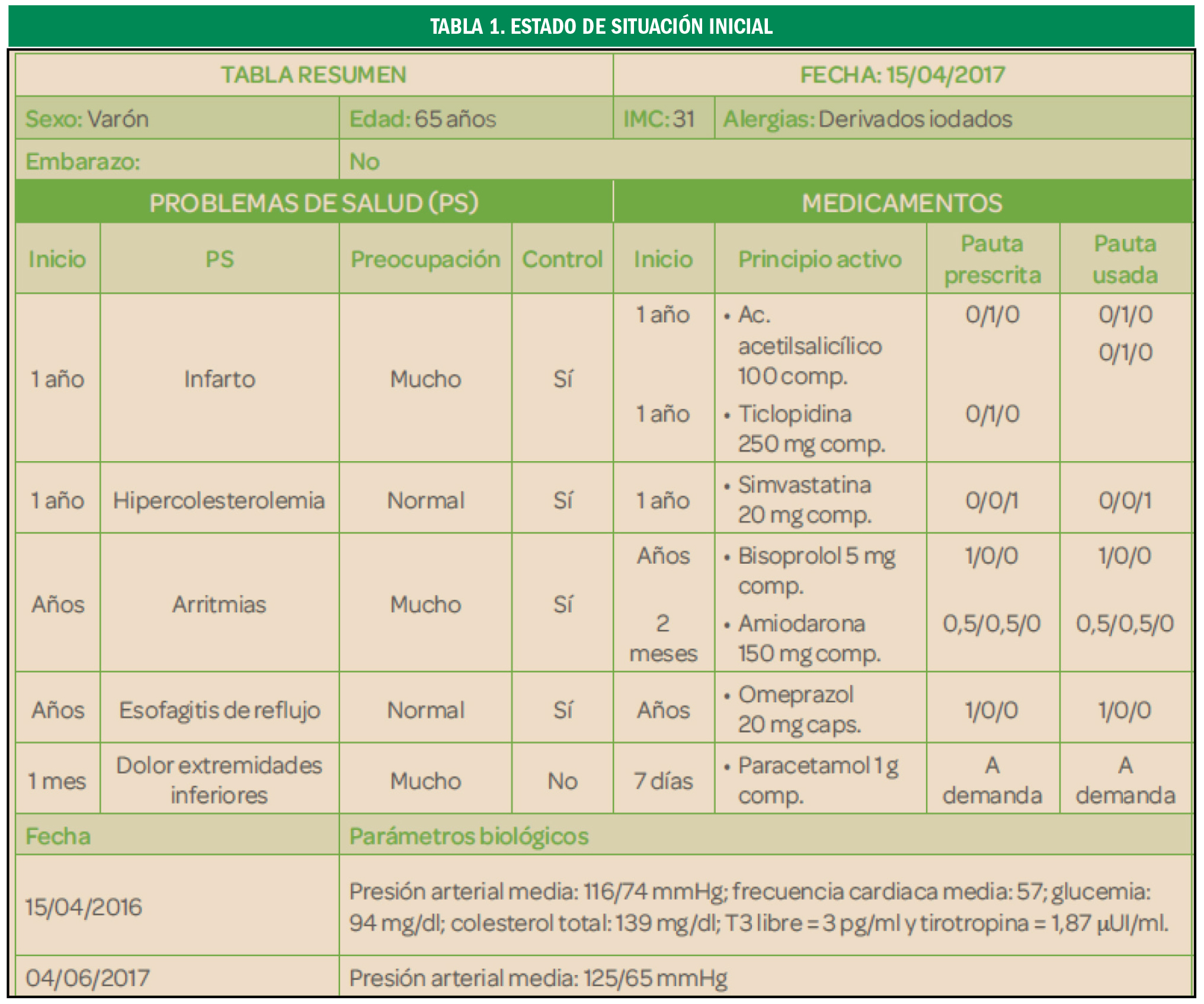
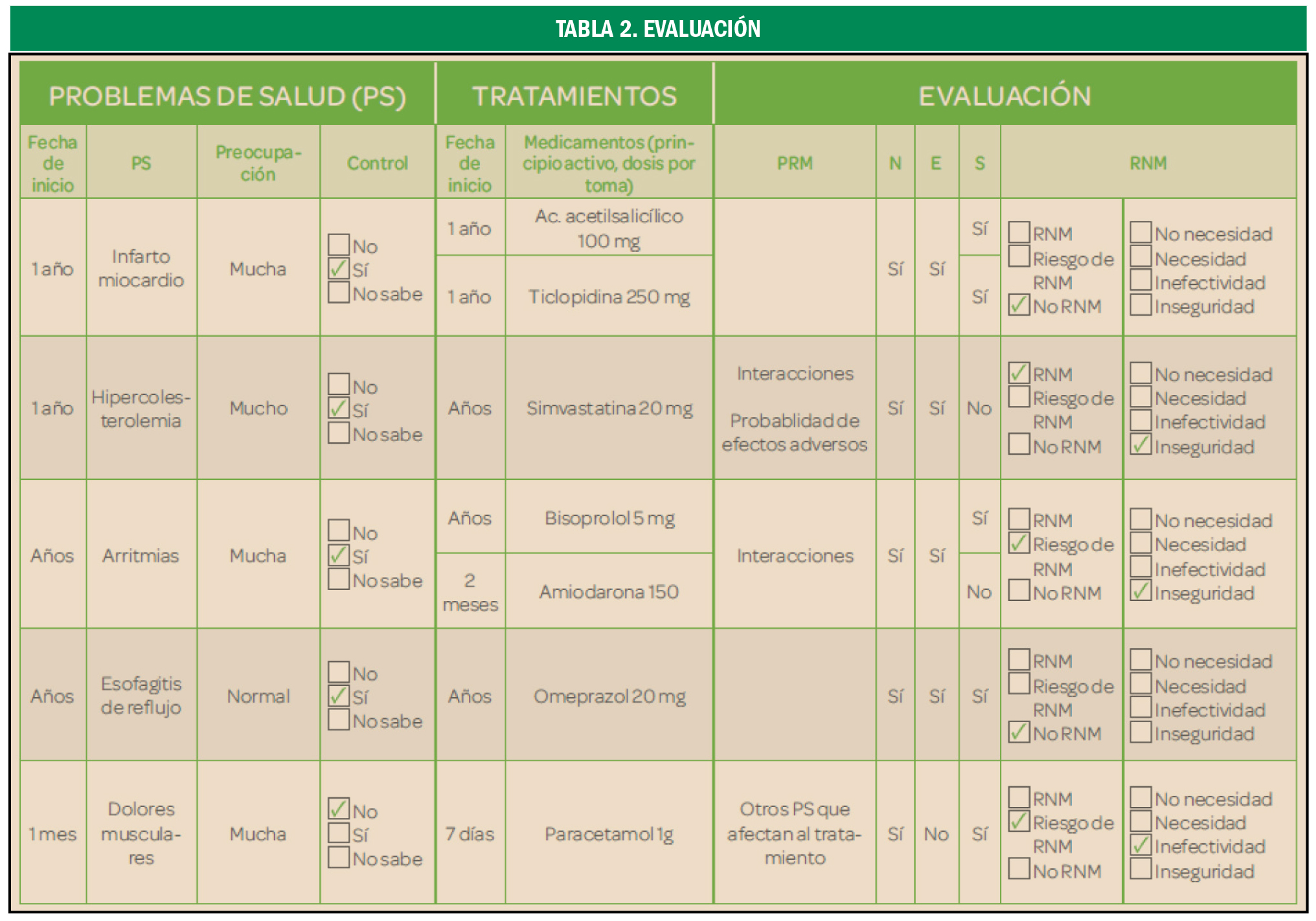
Preeclampsia
El valor preventivo del ácido acetilsalicílico
La administración de ácido acetilsalicílico en embarazadas con alto riesgo de preeclampsia reduce la duración de la estancia en la unidad de cuidados intensivos neonatales en un 68%, fundamentalmente debido a una disminución en la tasa de prematuros con menos de 32 semanas de gestación.
La preeclampsia es la complicación del embarazo más común y peligrosa, que pone en peligro la vida del feto y de la madre, de ahí la importancia de su diagnóstico y tratamiento precoz por lo que debe diagnosticarse y tratarse rápidamente.
La preeclampsia es una patología que ocurre únicamente en los embarazos humanos, afectando al 5-10%. Se diagnostica en la madre al presentarse una presión arterial elevada, junto con edema en las extremidades, después de la semana 20 del embarazo que podría desembocar en una eclampsia, con daño del endotelio materno, riñones e hígado, así como daño neurológico. Algunos de los síntomas más característicos solor dolor de cabeza, dolor abdominal, dificultad respiratoria, sensación de ardor epigástrico, náuseas, vómito, confusión mental, sensación creciente de ansiedad y cambios en la visión tales como sensibilidad excesiva a la luz, visión borrosa, sensación de destellos intermitentes o auras. Aunque tradicionalmente se requería la presencia adicional de proteinuria, cada vez es mayor la tendencia clínica centrada exclusivamente en la hipertensión localizada temporalmente, dado que los problemas renales y hepáticos pueden ocurrir incluso sin proteinuria y que la cantidad de proteína en la orina no predice la severidad de la enfermedad. El único tratamiento de la preeclampsia es la inducción del parto o la cesárea, aunque las complicaciones pueden aparecer hasta seis semanas después del parto.
La detección de preeclampsia a las 11-13 semanas de gestación mediante una combinación de características demográficas y antecedentes médicos con mediciones de biomarcadores puede identificar a aproximadamente el 75% de las mujeres que desarrollan preeclampsia pretérmino con parto a < 37 semanas de gestación y el 90% de las preeclampsia precoz a < 32 semanas.
En un ensayo reciente (Combined Multimarker Screening and Randomized Patient Treatment with Aspirin for Evidence-Based Preeclampsia Prevention) se informó que, en mujeres identificadas por el cribado del primer trimestre como de alto riesgo de preeclampsia, el uso de ácido acetilsalicílico (AAS) en dosis de 150 mg/día del primero al tercer trimestre, redujo la incidencia de preeclampsia prematura en un 62% (IC95% 26 a 80%) y la de preeclampsia temprana en un 89% (IC95% 53 a 97%), en comparación con el placebo. Sin embargo, el sorprendente hallazgo de este ensayo clínico fue que a pesar de la reducción de la preeclampsia, la incidencia de ingreso a la unidad de cuidados intensivos neonatales no se vio afectada significativamente (OR = 0,93; IC95% 0,62 a 1,40).
Con el objetivo de examinar el efecto del uso profiláctico de AAS durante el embarazo en mujeres con alto riesgo de preeclampsia en la duración de la permanencia en la unidad de cuidados intensivos neonatales, se llevó a cabo un análisis secundario de los datos del estudio Aspirin for Evidence-Preeclampsia Prevention, en el que participaron 1.620 pacientes, con un total de 1.571 recién nacidos vivos. La duración total de la permanencia en cuidados intensivos neonatales fue sustancialmente más prolongada en el grupo de placebo que de AAS (1.696 frente a 531 días). Este es un reflejo de la duración media significativamente más corta de la permanencia de bebés ingresados en la unidad de cuidados intensivos neonatales de la AAS que en el grupo placebo (11,1 vs. 31,4 días; una reducción de 20,3 días, IC95% 7,0 38,6; p = 0,008). La terapia intensiva neonatal de bebés nacidos con < 32 semanas de gestación contribuyó en 1.856 (83,3%) del total de 2.227 días en cuidados intensivos en ambos brazos de tratamiento. Estos ocurrieron en 9 (1,2%) de los 777 nacidos vivos en el grupo de AAS y en 23 (2,9%) de 794 en el grupo placebo (OR = 0,42; IC95% 0,19 a 0,93; p = 0,033). En general, en toda la población estudiada la duración media de la permanencia fue más prolongada en el grupo placebo que en el de AAS (2,06 vs. 0,66 días, reducción de 1,4 días; IC95% 0,45 a 2,81; p = 0,014). Esto corresponde a una reducción en la duración del 68% (IC95% 20 a 86%).
{kind=link}