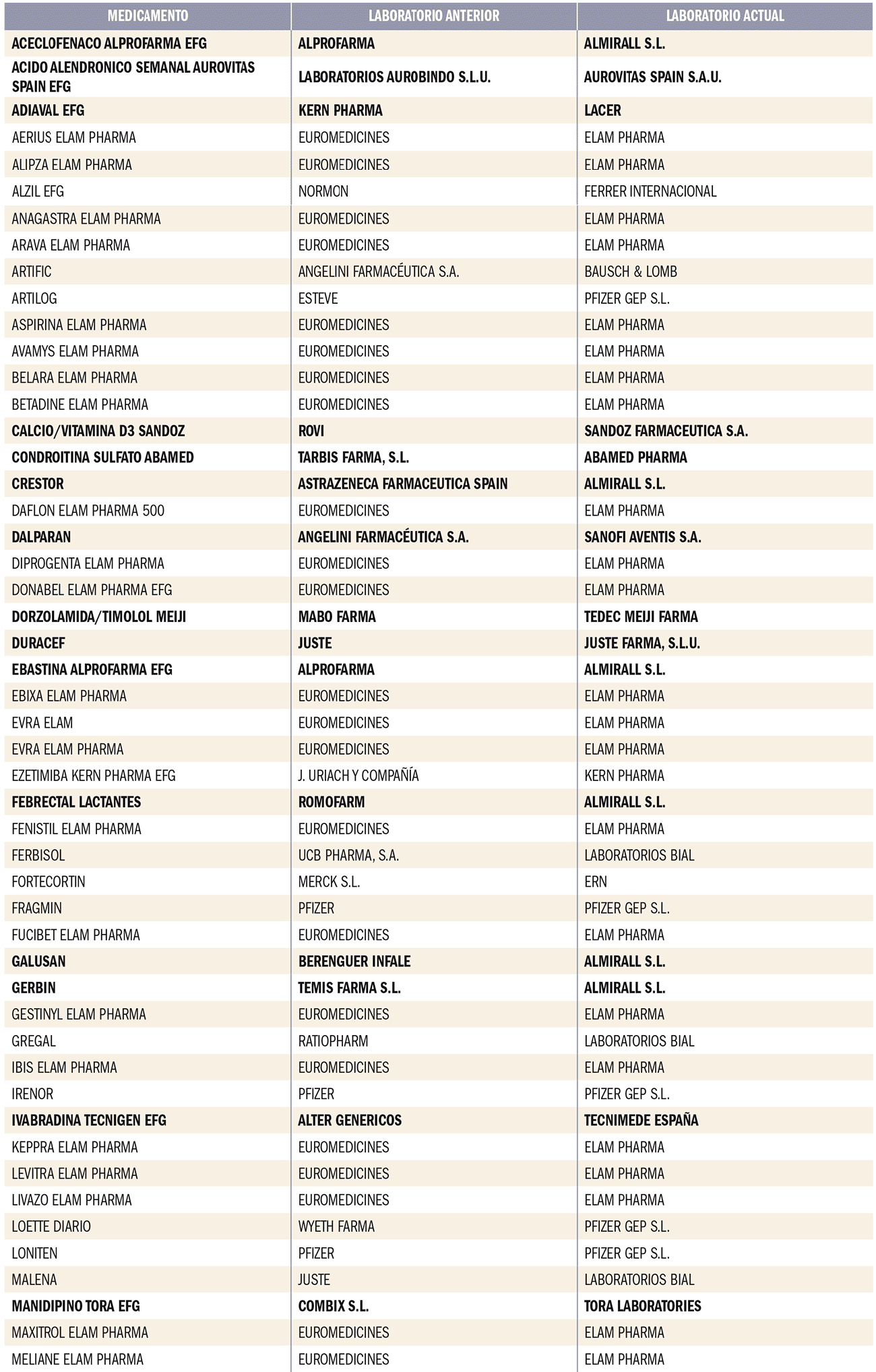
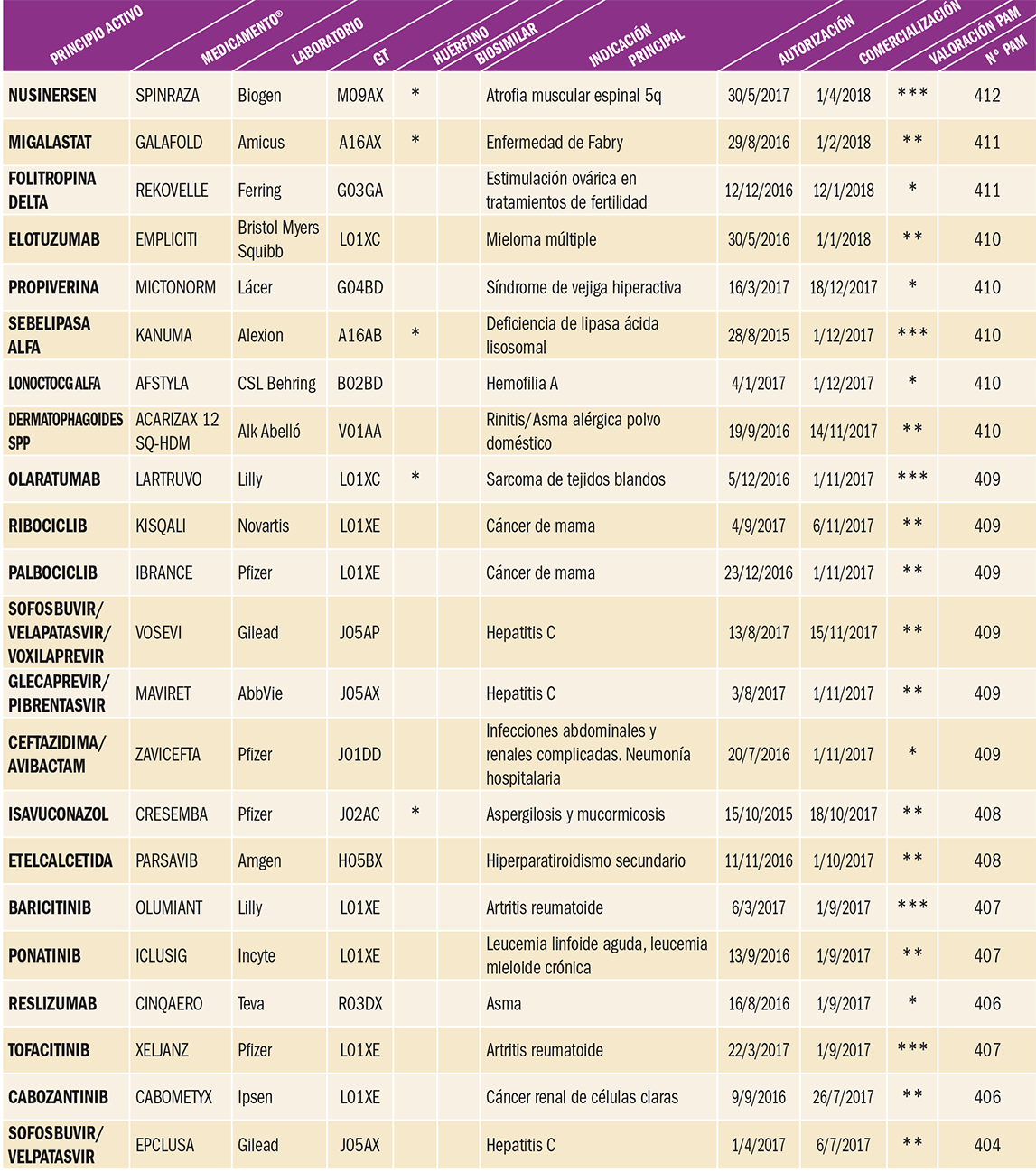
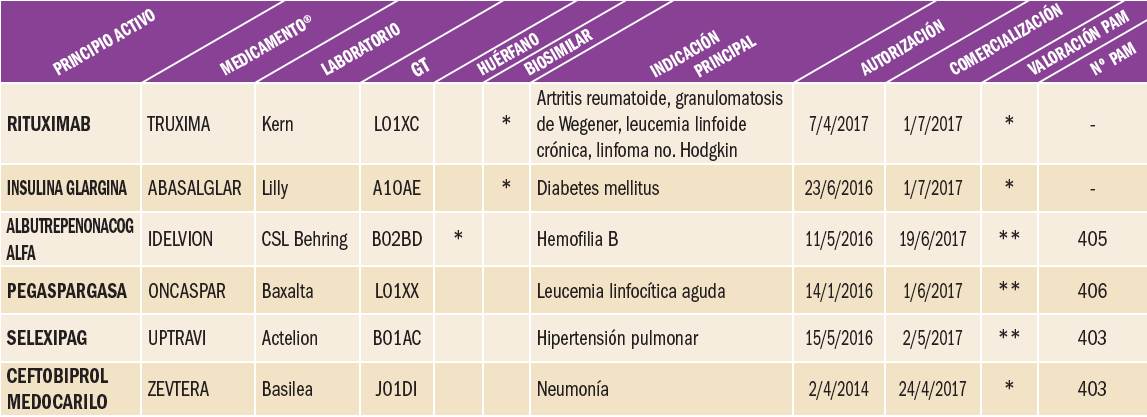
Número 412, Abril 2018
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta ese momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
– SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
– INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
– INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
– Evidencia clínica: mediante estudios controlados, específicamente diseñados y desarrollados para demostrar la eficacia y la seguridad del nuevo medicamento, con demostración fehaciente de lo que puede ser un avance o mejora sobre la terapia estándar hasta ese momento, en el caso de que exista.
– Plausibilidad científica (potencialidad): existencia de aspectos en el medicamento que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostrados mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
Nusinersén es un oligonucleótido antisentido capaz de incrementar la producción de la proteína SMN funcional, cuya deficiencia está relacionada con el origen de la atrofia muscular espinal 5q, indicación para la cual ha recibido autorización, como medicamento huérfano. El fármaco aumenta la proporción de inclusión del exón 7 en los transcritos del ácido ribonucleico mensajero (ARNm) del gen de supervivencia de la neurona motora 2 (SMN2) al unirse a un sitio ISS-N1 (silenciador del proceso de corte y empalme intrónico) localizado en el intrón 7 del precursor del ácido ribonucleico mensajero (pre-ARNm) del SMN2. Al unirse, el nusinersén desplaza los factores de corte y empalme, produciendo la retención del exón 7 en el ARNm del SMN2 y, por consiguiente, cuando se produce el ARNm del SMN2, se puede traducir en su proteína SMN funcional de longitud completa, lo que se relaciona con la eficacia terapéutica. Aunque con numerosas limitaciones, no cabe duda de que nos encontramos ante un medicamento auténticamente innovador, por varios motivos. Es el primer oligonucleótido antisentido (OAS) que permite actuar sobre el fundamento genético de la atrofia muscular espinal 5q y, lo que realmente resulta más relevante, su capacidad para actuar sobre una enfermedad que actualmente carece de tratamiento etiológico. Ciertamente, los resultados no son espectaculares pero abren una puerta a la esperanza, aunque habremos de esperar los datos de los ensayos clínicos en curso para poder establecer con mayor consistencia las conclusiones definitivas.
La atrofia muscular implica el desgaste o pérdida del tejido muscular. El origen del problema puede ser físico o neurológico. La atrofia por desuso ocurre en la mayoría de las personas, especialmente en aquellas con trabajos sedentarios o que padecen afecciones que limitan el movimiento. Este tipo de atrofia se puede contrarrestar con el ejercicio físico adecuado. Sin embargo, el tipo más grave de atrofia muscular es la neurógena, que ocurre cuando hay una lesión o enfermedad de un nervio que controla el movimiento de un músculo o grupo muscular. Este tipo de atrofia muscular tiende a evolucionar más rápidamente que la atrofia por desgaste o por desuso funcional (Cuéllar, 2014).
Las atrofias musculares espinales proximales son un grupo de trastornos musculares caracterizados por una debilidad muscular progresiva resultado de una degeneración y pérdida de las neuronas motoras inferiores en la médula espinal y en los núcleos del tronco encefálico (Topaloglu, 2009). En concreto, la atrofia muscular espinal (AME) implica la degeneración de las neuronas motoras del asta anterior de la médula espinal; se trata de una enfermedad hereditaria de carácter autosómico recesivo, con incidencia global de 8,5-10,3 casos por cada 100,000 recién nacidos vivos, lo que le otorga la condición de enfermedad rara, y representa la causa de muerte de origen hereditaria más común causada por enfermedades infantiles (Parente, 2018).
La AME es un resultado de la reducción de los niveles de la proteína SMN (survival motor neuron 1; supervivencia de la neurona motora-1). Cerca del 98% de los casos están provocados por la herencia de mutaciones, mientras que el restante 2% de los casos están causados por mutaciones de novo (no heredadas). Las mutaciones consisten mayoritariamente en deleciones (pérdida de uno o varios nucleótidos de la secuencia del gen) homocigóticas del exón 7 o de los exones 7 y 8 en el gen SMN1, localizado en el cromosoma 5 (5q12.2-q13.3), responsable de la codificación de la proteína SMN fisiológica.
La falta de proteína SMN causa disfunción y, en última instancia, la muerte de las neuronas motoras. El gen SMN1 se encuentra en una región duplicada e invertida del cromosoma que incluye una copia casi idéntica del gen SMN1, llamado SMN2 (5q13.2). Aunque ambos genes codifican proteínas con idéntica secuencia de aminoácidos, aunque el gen SMN2 difiere del SMN1 en 11 nucleótidos. Una de estas diferencias de nucleótidos, la que supone la sustitución de citosina por timina, se produce en el exón 7 del gen SMN2, lo que da como lugar a un patrón de empalme alternativo que favorece la omisión del exón 7. El 90% de las transcripciones producidas a partir del gen SMN2 carecen de exón 7, lo que da como resultado un producto proteico truncado que es defectuoso e inestable, la proteína SMNΔ7.
Se supone que el aumento de la cantidad de transcripción de longitud completa a partir del gen SMN2 (no de la versión truncada) da como resultado un aumento en la proteína SMN fisiológica en pacientes con SMA. En este sentido, los seres humanos tienen un número variable (de 0 a 8) de copias del gen SMN2. Este número, por lo indicado anteriormente, es considerado como un importante predictor de la gravedad de la enfermedad de SMA, de tal manera que los pacientes con más copias generalmente tienen una forma menos grave de la enfermedad.
Se han definido 5 tipos en función de la edad de aparición y de la gravedad de la enfermedad. Todos ellos se caracterizan por debilidad muscular y atrofia de gravedad variable, que afectan principalmente a los miembros inferiores y a los músculos respiratorios. La debilidad es casi siempre simétrica y progresiva, pudiendo darse retracciones musculares, escoliosis y contracturas articulares; el estreñimiento y el reflujo gastroesofágico son frecuentes.
El tipo 0 o AME prenatal es un tipo raro en el que los bebés nacen con signos clínicos de enfermedad, como graves contracturas articulares y compromiso respiratorio que a menudo conduce a la necesidad de ventilación mecánica al momento o poco después del nacimiento. Estos pacientes generalmente tienen 1 copia del gen SMN2. Muchos de los pacientes mueren o requieren ventilación permanente dentro de las semanas después del nacimiento
El tipo 1 (AME1) o forma infantil representa el 50-60% de casos y se manifiesta durante los primeros seis meses de vida. Los pacientes suelen tener 2 (ocasionalmente, 3) copias del gen SMN2. Los niños presentan debilidad muscular, arreflexia e hipotonía generalizada. No suele haber alteraciones de carácter sensitivo, pero la debilidad es muy grande y algunos pacientes no llegan a sostener la cabeza y nunca consiguen la sedestación, es decir, la capacidad para mantenerse erguido por medios propios al estar sentado. Suelen fallecer antes de cumplir 2 años en más del 80% de los casos. El tipo 1 se subdivide en 1A, 1B y 1C en función del tiempo de aparición de los síntomas; la forma más grave (tipo 1A, que suelen identificarse con el tipo 0), puede manifestarse con graves alteraciones articulares, cardiopatía y trastornos vasculares que conducen a la muerte al cabo de pocas semanas.
El 30% de los pacientes están afectados por el tipo 2 (AME2) o forma intermedia, en la que los síntomas aparecen entre los 6 y los 24 meses, y más del 80% de estos pacientes tienen 3 o 4 copias del gen SMN2. Los niños consiguen la sedestación pasiva, pero no llegan a caminar. Las complicaciones respiratorias y la escoliosis son los problemas más importantes y su expectativa de vida es variable, aunque dos terceras partes de ellos llegan a alcanzar la edad de 25 años.
Por su parte, el 10-20% de los casos corresponden al tipo 3 (AME3), en los que los síntomas suelen manifestarse a partir de los 24 meses. Los pacientes llegan a caminar y en general alcanzan la edad adulta. La gran mayoría de los pacientes presentan 3 o 4 copias del gen SMN2. Aquellos que manifiestan antes de los 3 años la enfermedad (tipo 3a) pierden posteriormente la capacidad de para mantenerse de pie (bipedestación) y caminar (deambulación), mientras que los casos de presentación más tardía (tipo 3b) pueden mantener la deambulación por más tiempo incluso décadas.
Finalmente, la forma más benigna (<5% de los casos), el tipo 4 (AME4), corresponde a la forma adulta, que aparece en la segunda o tercera década de la vida. Estos pacientes tienen capacidad de caminar sin ayuda y en general presentan una afectación clínica menos grave que las tres formas antes descritas. Suelen tener 4 o más copias del gen SMN2.
Hasta el momento, el manejo de los pacientes ha sido meramente sintomático, con un enfoque multidisciplinar y dirigido a aumentar la calidad de vida, a base de terapias respiratorias y ocupacionales, y fisioterapia. También puede llegar a requerirse ventilación no invasiva y gastrostomía; en caso de infección pulmonar se requiere terapia antibiótica, mientras que la escoliosis y las manifestaciones articulares pueden requerir una corrección quirúrgica, pudiendo necesitar silla de ruedas o el uso de un corsé o de una faja lumbar como soporte. El pronóstico depende de la gravedad de la enfermedad, lo que generalmente está asociado con la edad de aparición: las formas de aparición temprana suelen tener un pronóstico peor, mientras que la esperanza de vida puede ser normal en las formas de aparición tardía. La muerte puede ocurrir debido a una insuficiencia respiratoria y a las infecciones.
Nusinersén es un oligonucleótido antisentido capaz de incrementar la producción de la proteína SMS funcional, cuya deficiencia está relacionada con el origen de la atrofia muscular espinal 5q, indicación para la cual ha recibido autorización. El fármaco aumenta la proporción de inclusión del exón 7 en los transcritos del ácido ribonucleico mensajero (ARNm) del gen de supervivencia de la neurona motora 2 (SMN2) al unirse a un sitio ISS-N1 (silenciador del proceso de corte y empalme intrónico) localizado en el intrón 7 del precursor del ácido ribonucleico mensajero (pre-ARNm) del SMN2. Al unirse, el nusinersén desplaza los factores de corte y empalme, produciendo la retención del exón 7 en el ARNm del SMN2 y, por consiguiente, cuando se produce el ARNm del SMN2, se puede traducir en su proteína SMN funcional de longitud completa, lo que se relaciona con la eficacia terapéutica.
La atrofia muscular espinal (AME) 5q es un resultado de la reducción de los niveles de la proteína SMN (survival motor neuron 1; supervivencia de la neurona motora-1), como consecuencia de la existencia de mutaciones (heredadas o de novo), consistentes mayoritariamente en deleciones homocigóticas del exón 7 o de los exones 7 y 8 en el gen SMN1, localizado en el cromosoma 5, responsable de la codificación de la proteína SMN fisiológica. La falta de proteína SMN causa disfunción y, en última instancia, la muerte de las neuronas motoras. El gen SMN1 se encuentra en una región duplicada e invertida del cromosoma que incluye una copia casi idéntica del gen SMN1, el SMN2. Aunque ambos genes codifican proteínas con idéntica secuencia de aminoácidos, el gen SMN2 difiere del SMN1 en unos pocos nucleótidos, lo que da lugar a que el 80-90% de las transcripciones producidas a partir del gen SMN2 carecen de exón 7, dando como resultado un producto proteico truncado que es defectuoso e inestable, la proteína SMNΔ7. Sin embargo, el restante 10-20% producido corresponde a la transcripción de longitud completa a partir del gen SMN2 (no de la versión truncada), lo que da como resultado un aumento en la proteína SMN fisiológica en pacientes con AME.
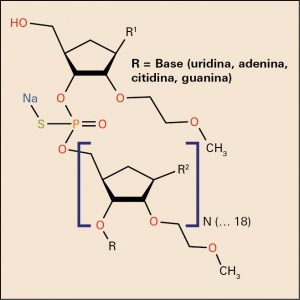
El nusinersén es un oligonucleótido antisentido. Es decir, se trata de un pequeño fragmento de ácido nucleico (un ARN, generalmente) constituido por menos de 30 nucleótidos, cuya secuencia es complementaria (de ahí el término antisentido) a la de un gen específico, con objeto de unirse al mRNA del mismo con la finalidad de silenciar determinados exones que contienen una mutación perjudicial, excluir un pseudoexón – provocado por una mutación en el lugar de empalme exón-intrón en la cadena de ADN – o saltar uno o varios exones de forma que la lectura del ADN evite la zona mutada. En el caso concreto del nusinersén, éste aumenta la proporción de inclusión del exón 7 en los transcritos del ácido ribonucleico mensajero (ARNm) del gen de supervivencia de la neurona motora 2 (SMN2) al unirse a un sitio ISS-N1 (silenciador del proceso de corte y empalme intrónico) localizado en el intrón 7 del precursor del ácido ribonucleico mensajero (pre-ARNm) del SMN2. Al unirse, el nusinersén desplaza los factores de corte y empalme, produciendo la retención del exón 7 en el ARNm del SMN2 y, por consiguiente, cuando se produce el ARNm del SMN2, se puede traducir en su proteína SMN funcional de longitud completa, lo que se relaciona con la eficacia terapéutica.
El nusinersén está formado por 18 nucleótidos uniformemente modificados en la posición 2’ de la molécula de ribosa, donde tienen un grupo 2-metoxietilo: (-OCH2CH2OCH3) de cada uno de los nucleótidos. Los nucleótidos están unidos entre sí por un puente fósforo-tioato sódico (-O-P[O][SNa]-O-) y varios de ellos están metilados (Me). La secuencia del nusinersén es1: 5’-MeUMeCAMeCMeUMeUMeUCAMeUAAMeUGMeCMeUGC-3.
La eficacia y la seguridad clínicas del nusinersén han sido adecuadamente contrastadas en la indicación autorizada mediante dos ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados, doblemente ciegos y controlados con simulacro de administración. El medicamento fue administrado por vía intratecal mediante un bolo lento (1-3 minutos) utilizando una aguja de anestesia espinal, insertándola mayoritariamente en el espacio intervertebral L3/L (o un espacio por encima o por debajo, en algunos casos). La simulación de administración en los pacientes del grupo de control se hizo mediante la punción superficial con la aguja, sin inyectar ningún contenido, en el mismo espacio intervertebral que en los pacientes tratados con el medicamento.
El primero de los estudios (ENDEAR; Finkel, 2017) se llevó a cabo en pacientes con AME tipo 1 (recién nacidos). Se incluyeron en el estudio un total de 122 pacientes (de los que 81 recibieron el tratamiento y los otros 41 fueron sometidos a la simulación). La dosis empleada fue de 12 mg, salvo en los niños menores de 2 años, que recibieron dosis ajustadas a su edad. Se realizaron cuatro dosis de carga, los días 1, 15, 29 y 64, seguido de dosis de mantenimiento cada 4 meses, hasta totalizar 10 meses de tratamiento. Las variables principales fueron la tasa de respondedores2 según la escala Hammersmith Infant Neurological Examination (examen neurológico infantil de Hammersmith) y la supervivencia libre de eventos (tiempo hasta la muerte o el uso de ventilación asistida permanente3). Como variables clínicas secundarias se determinaron la supervivencia global, la tasa de respondedores según otras escalas neurológicas (CHOP INTEND) o la de respondedores a la prueba del grado de inervación CMAP (Compound Muscle Action Potential).
En el análisis final, un porcentaje significativamente más alto de recién nacidos en el grupo de nusinersén que en el grupo control tuvo una respuesta motriz (51 vs 0%), y la probabilidad de la supervivencia libre de eventos fue mayor en el grupo nusinersén que en el grupo control (cociente de riesgos instantáneos para la muerte o el uso de ventilación asistida permanente, 0,53; p = 0,005). La probabilidad de supervivencia global fue mayor en el grupo nusinersén que en el grupo control (tasa de riesgo de muerte = 0,37; p = 0,004), la tasa de respondedores CHP INTEND fue del 71 vs. 3%, mientras que en la prueba CMAP fue de 36 vs. 5%. Los lactantes con una duración de enfermedad más corta fueron más propensos a beneficiarse del nusinersén que los que tenían una duración más larga. La incidencia y la gravedad de los eventos adversos fueron similares en los dos grupos.
El segundo estudio (CHERISH; Mercuri, 2018) fue realizado en 126 niños con AME que presentaron síntomas después de los 6 meses de edad (AME-2 o AME-3). Los niños fueron asignados aleatoriamente, en una proporción de 2: 1, para recibir administración intratecal de nusinersén a una dosis de 12 mg (grupo de nusinersén) o un procedimiento simulado (grupo de control) los días 1, 29, 85 y 274. La variable principal fue la variación desde el inicio en la puntuación Hammersmith Functional Motor Scale-Expanded (HFMSE4) a los 15 meses de tratamiento. Las variables secundarias incluyeron el porcentaje de niños con un aumento clínicamente significativo desde el inicio en la puntuación HFMSE (≥3 puntos)5. En el análisis intermedio preespecificado, hubo un aumento medio desde el inicio hasta el mes 15 en la puntuación HFMSE en el grupo nusinersén de 4,0 puntos y una disminución media en el grupo control de –1,9 puntos, con una diferencia significativa entre grupos que favorece a nusinersén (5,9 puntos; IC95% 3,7 a 8,1; p < 0,001). Este resultado provocó la terminación anticipada de la prueba. Los resultados del análisis final fueron consistentes con los resultados del análisis intermedio. En el análisis final, el 57% de los niños en el grupo nusinersén en comparación con el 26% en el grupo control tuvo un aumento desde el inicio hasta el mes 15 en la puntuación HFMSE de al menos 3 puntos (p < 0,001) y la incidencia general de eventos adversos fue similar en el grupo nusinersén y el grupo control (93 vs. 100%).
Desde el punto de vista de la seguridad, la mayoría de los eventos adversos descritos en los pacientes tratados con nusinersén fueron consistentes con la naturaleza y la frecuencia de los eventos típicamente asociados a la propia enfermedad. De hecho, los pacientes pediátricos presintomáticos con nusinersén experimentaron menos eventos adversos que los pacientes sintomáticos. Además, la mortalidad en los grupos de pacientes infantiles tratados fue, aproximadamente, la mitad que en los pacientes sometidos a la simulación (EMA, 2017).
Los eventos adversos reportados en al menos un 20% de los pacientes fueron infecciones del tracto respiratorio superior (30% con nusinersén vs. 22% en los controles), distrés respiratorio (26 vs. 29%), neumonía (29 vs. 17%), insuficiencia respiratoria (25 vs. 39%), atelectasia (23 vs. 29%), insuficiencia respiratoria aguda (14 vs. 24%), infección viral del tracto respiratorio superior (10 vs. 17%), reducción de la tasa de saturación de oxígeno (13 vs. 24%), tos (11 vs. 20%), fiebre (56 vs. 59%), estreñimiento (35 vs. 22%), vómitos (18 vs. 20%), reflujo gastroesofágico (13 vs. 20%) y disfagia (11 vs. 20%).
En torno al 6% de los pacientes desarrollan anticuerpos anti-nusinersén durante el tratamiento, aunque no se relacionó con afectación de la respuesta clínica, en los eventos adversos o en el perfil farmacocinético del nusinersén.
Nusinersén es un oligonucleótido antisentido capaz de incrementar la producción de la proteína SMS funcional, cuya deficiencia está relacionada con el origen de la atrofia muscular espinal 5q, indicación para la cual ha recibido autorización, como medicamento huérfano. El fármaco aumenta la proporción de inclusión del exón 7 en los transcritos del ácido ribonucleico mensajero (ARNm) del gen de supervivencia de la neurona motora 2 (SMN2) al unirse a un sitio ISS-N1 (silenciador del proceso de corte y empalme intrónico) localizado en el intrón 7 del precursor del ácido ribonucleico mensajero (pre-ARNm) del SMN2. Al unirse, el nusinersén desplaza los factores de corte y empalme, produciendo la retención del exón 7 en el ARNm del SMN2 y, por consiguiente, cuando se produce el ARNm del SMN2, se puede traducir en su proteína SMN funcional de longitud completa, lo que se relaciona con la eficacia terapéutica.
En el estudio pivotal ENDEAR realizado en pacientes de inicio infantil, un porcentaje estadísticamente significativamente mayor de sujetos logró la respuesta motriz motora predefinida en el grupo nusinersén (51%), en comparación con el grupo control (0%; p < 0,0001). Además, el tiempo hasta la muerte o la ventilación permanente, la otra variable primaria, se mantuvo estadísticamente significativamente prolongado en los sujetos tratados con nusinersén (HR=0,53) en comparación con los controles. Esto fue aún más prolongado en los sujetos tratados con nusinersén que estaban por debajo de la mediana para la duración de la enfermedad al inicio (HR = 0,21), lo que sugiere que el tratamiento temprano con el fármaco puede conferir una beneficio para la supervivencia libre de eventos. Por su parte, en el estudio CHERISH en pacientes con AME de inicio tardío, se observó un cambio estadísticamente significativo en la puntuación HFMSE desde el inicio a los 15 meses, la variable primaria, en el grupo nusinersén en comparación al grupo de control simulado (+4,0 vs. –1,9; p < 0,001); también se observó una mejoría en la capacidad funcional de la extremidad superior (EMA, 2017).
Como puntualiza la EMA, es importante considerar que no se han estudiado los efectos del nusinersén en pacientes con síntomas muy graves de inicio prenatal (tipo 0), así como tampoco sobre aquellos con un curso leve de inicio en adultos (tipo 4). Otra limitación de los datos actualmente disponibles reside en la falta de datos a largo plazo. Esto implica el desconocimiento de si la magnitud del efecto podría cambiar a medida que la enfermedad progresa y los pacientes crecen o envejecen. Dado que es previsible que el tratamiento pueda aumentar la supervivencia de una parte relevante de la población con AME, el desarrollo físico de estos pacientes probablemente requiere un ajuste de la dosis en función de uno o varios parámetros antropométricos, lo que actualmente es desconocido.
Con todo, no cabe duda de que nos encontramos ante un medicamento auténticamente innovador, por varios motivos. Es el primer oligonucleótido antisentido (OAS) que permite actuar sobre el fundamento genético de la atrofia muscular espinal 5q y, lo que realmente resulta más relevante, su capacidad para actuar sobre una enfermedad que actualmente carece de tratamiento etiológico. Ciertamente, los resultados no son espectaculares pero abren una puerta a la esperanza, aunque habremos de esperar los datos de los ensayos clínicos en curso para poder establecer con mayor consistencia las conclusiones definitivas.

BIBLIOGRAFÍA
La pancitopenia es la disminución simultánea de los valores de las tres series hematológicas por debajo de rangos normales en sangre periférica y puede manifestarse con distinto grado de intensidad. Los síntomas y signos asociados a pancitopenia son habitualmente secundarios a ésta, e independientes de la etiología: astenia, infecciones, hematomas, esplenomegalia, adenopatías. El conjunto de enfermedades que lo producen es muy amplio, pudiendo afectar o no a la médula ósea. Es importante saber enfocar el cuadro de manera inicial, distinguir las situaciones de gravedad, y conocer las indicaciones del estudio de médula ósea.
Todos los huesos que componen nuestro esqueleto tienen una estructura similar; están formados por hueso compacto, hueso esponjoso y médula ósea. El hueso compacto forma la capa exterior, el esponjoso se encuentra, en los extremos y la médula ósea es un tipo de tejido que se encuentra en el interior de los huesos. Puede ser de dos clases: roja y amarilla. En la médula ósea roja se fabrican las células sanguíneas. Este proceso de fabricación se denomina hematopoyesis o hemopoyesis. La médula amarilla se compone de grasa y no participa en la formación de la sangre. Durante la niñez, la mayor parte de la médula es roja, pero con el paso de los años, se convierte en amarilla, aunque puede volverse a convertir en médula roja si fuese necesario. La médula ósea roja, en los adultos, está ubicada en las costillas, el esternón, la columna vertebral, el cráneo, la escápula y la pelvis. Contiene las células madre (o hemoblastos) que originan los tres tipos de células sanguíneas:
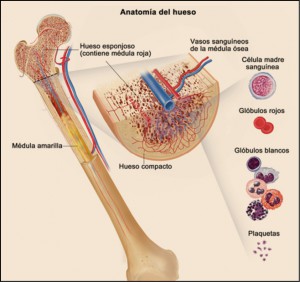
La médula ósea mantiene el número normal de los tres tipos de células sanguíneas, sustituyendo a las antiguas, que sufren muerte natural. Además, si necesitara aumentar el número de ellas, por cualquier motivo, formaría con rapidez nuevas células.
Cuando hablamos de pancitopenia nos referimos a la presencia simultánea de anemia (hemoglobina menor a 13 grs/dL en hombres y menor a 12 grs/dL en mujeres), leucopenia (recuento de leucocitos menor a 4.500/mm3) y trombocitopenia (recuento de plaquetas menor a 150.000/mm3). No es una enfermedad en sí, sino el signo de una enfermedad que necesita ser diagnosticada. Se trata de un problema hematológico bastante frecuente en la práctica clínica y debe sospecharse cuando un paciente presenta palidez, fiebre prolongada y tendencia a los sangrados. Las pancitopenias se clasifican en centrales y periféricas, según exista disminución de las células hematopoyéticas en la médula ósea (MO), o un descenso periférico de los elementos formes de la sangre con MO (médula ósea) normal o no, y en este caso pueden ser por destrucción (pancitopenias autoinmunes) o por secuestro (hiperesplenismo).
Las causas son múltiples y el pronóstico depende de las mismas. En los pacientes sintomáticos, el cuadro clínico de presentación está relacionado con el de las tres citopenias: anemia (palidez y síndrome anémico), leucopenia (fiebre) y plaquetopenia (tendencia a los sangrados cutáneo-mucosos). La aparición simultánea de los tres elementos es un fuerte orientador diagnóstico de pancitopenia. En cambio los pacientes asintomáticos son diagnosticados principalmente por el examen físico y/o la analítica. En ambos casos (sintomáticos y asintomáticos), la esplenomegalia se presenta con gran frecuencia, excepto en aquellos con anemia aplásica. La hepatomegalia también se ve con frecuencia, exceptuando las gammapatías monoclonales y el paludismo donde no suele aparecer. Las adenopatías en cambio son raras de observar, salvo en las neoplasias hematológicas (leucemia aleucémica y linfoma no Hodgkin) y en la tuberculosis diseminada.
En la analíticas realizadas a estos pacientes, podemos observar que la concentración de hemoglobina, el recuento de leucocitos, el número de plaquetas y la velocidad de eritrosedimentación no es diferente entre las diferentes entidades que cursan con pancitopenia, es por esto que los hallazgos obtenidos de los exámenes básicos de laboratorio no ayudan en el diagnóstico etiológico del síndrome, sino en la evaluación de la severidad. Según el hemograma, la pancitopenia se puede categorizar en:
El frotis de sangre periférica tiene un rol fundamental en el estudio del paciente con pancitopenia, existiendo algunas alteraciones morfológicas que caracterizan por sí mismas determinadas entidades. Las técnicas actuales para obtener muestra de tejido medular consisten en punción-aspiración de médula ósea (PAMO), biopsia de hueso con aguja y escisión quirúrgica. La PAMO es la técnica más comúnmente utilizada para este fin y es esencial en el diagnóstico de las pancitopenias. Podremos encontrar alteraciones tanto desde el punto de vista cuantitativo como cualitativo que nos pongan sobre la pista del diagnóstico. Sin embargo, en algunas ocasiones, este procedimiento no proporciona el material adecuado para estudio por ausencia de grumos celulares; a esto se le llama “aspirado seco”. No se trata de un situación frecuente, pero cuando aparece suele estar relacionado con patologías graves, debiendo realizarse biopsia, ya que permite el diagnóstico con mayor eficiencia que las otras técnicas. Las entidades con las que más se asocia son la mielofibrosis idiopática y la mieloptisis, o mielofibrosis secundaria a procesos infiltrantes, como por ejemplo, la tricoleucemia. En pacientes que se presentan con pancitopenia, este hallazgo se asocia en dos de cada tres casos a mieloptisis.

Se trata de un síndrome caracterizado por pancitopenia periferica con MO hipocelular por reemplazo del tejido hematopoyético por tejido adiposo, en ausencia de patología primaria que infiltre, anule o reemplace la hematopoyesis; siendo la única diferencia entre aplasia e hipoplasia medular el grado de severidad de la pancitopenia.
Las manifestaciones clínicas de enfermedad comienzan de forma escalonada. Las primeras en disminuir son las plaquetas, por lo que como primeros signos podrán verse petequias, hemorragias gingivales o retinianas y epistaxis. Los pacientes suelen presentar un síndrome anémico, pero difícilmente el cuadro clínico debuta con una infección, y cuando se produce suele ser bacteriana, debido a la neutropenia. La ausencia de hepato-esplenomegalia y adenopatías es característica; el hallazgo de alguna de ellas debe orientar hacia otras causas de pancitopenia. En el laboratorio se observa pancitopenia con reticulopenia absoluta y células sanguíneas morfológicamente normales. En los eritrotrocitos puede observarse una ligera macrocitosis con anisopoiquilocitosis moderada. Hay por lo general 70 a 90% de linfocitos en el frotis de sangre periférica a pesar de la leucopenia absoluta. La médula ósea es profundamente hipocelular con disminución de todas las series y, con espacios medulares compuestos, fundamentalmente, por grasa y elementos del estroma en ausencia de fibrosis. Las células hematopoyéticas residuales son morfológicamente normales.
Se considera aplasia medular grave cuando la celularidad de la médula ósea es menor del 25% o se encuentra entre el 25 y el 50%, con menos del 30% de células hematopoyéticas residuales y además existan al menos dos de los siguientes hallazgos en sangre periférica: polimorfonucleares (PMN) menores a 500/mm3, plaquetas menores a 20.000/mm3 o reticulocitos menores a 20.000/mm3. Se considera muy grave o severa si los PMN son menores de 200/mm3. La elección del tratamiento más apropiado para la aplasia grave va a depender de diversos factores, que incluyen la edad, la disponibilidad de un donante hermano HLA idéntico y el estado general del paciente. El pronóstico de la aplasia de MO se encuentra determinado por su severidad en el momento del diagnóstico y por la edad de los pacientes.
La franja etaria de comienzo está entre los 20 y 25 años, no existiendo diferencia entre sexos. Aunque entre un 40 y un 70% es considerada idiopática, la aplasia medular se relaciona con diversos agentes etiológicos, principalmente:
La mielofibrosis se define como el depósito de material colágeno en la médula ósea y puede acontecer como una enfermedad hematológica primaria (llamada mielofibrosis con metaplasia mieloide), o como mielofibrosis secundaria o mieloptisis, la cual representa una reacción a diversas situaciones patológicas. El diagnóstico de mielofibrosis secundaria se basa en determinar la existencia de una enfermedad asociada. Cuando la mielofibrosis es secundaria a una infección, ésta es usualmente crónica y de fácil diagnostico. En los casos de mielofibrosis secundaria a neoplasias, ésta última suele encontrarse en un estadio avanzado, con metástasis que permiten reconocer el origen, pero no siempre una neoplasia puede ser demostrable en la MO cuando se desarrolla mielofibrosis.
La forma de presentación puede ser con anemia severa, normocítica normocrómica, con valores normales de leucocitos y plaquetas o como un cuadro de pancitopenia con diferentes grados de severidad. En el frotis de sangre periférica se presentan marcadas alteraciones morfológicas eritrocitarias, como anisocitosis, dacriocitos, eliptocitos y esquistocitos, que son sugestivas de mieloptisis. También la policromatofilia, los eritroblastos y neutrófilos inmaduros (mielocitos, promielocitos, etc) son frecuentes. La punción aspirativa de la medula ósea frecuentemente es “seca”, no se obtiene material por la presencia de fibrosis medular, por lo que es necesario complementar el estudio con la biopsia. La posible fisiopatogenia es la interrupción de la microcirculación de la medula ósea por la infiltración y la fibrosis que generaría la aparición de focos hematopoyéticos extramedulares que determinan la presencia de elementos inmaduros eritroides y granulocíticos en la sangre periférica. El mecanismo más importante de la anemia es la menor actividad eritropoyética por hipoplasia o inhibición eritroide, déficit relativo de eritropoyetina o eritropoyesis ineficaz.
Supone la disminución de la producción de células sanguíneas a pesar de un incremento en los precursores medulares. Se refleja en un aumento de la bilirrubina indirecta y de la LDH, con descenso de la haptoglobina por la hemólisis intramedular acompañado de descenso de los reticulocitos en sangre periférica. Las principales causas son el déficit de factores de maduración (vitamina B12 y ácido fólico) que produce hematopoyesis megaloblástica, y los síndromes mielodisplásicos.
Existe una disminución de la síntesis del ADN que provoca un trastorno madurativo de los precursores eritroides y mieloides lo que da lugar a una hematopoyesis ineficaz con detención de la maduración que compromete las tres líneas celulares de la médula ósea. En más del 95% de los casos se produce por déficit de vitamina B12 y ácido fólico. La etiología de la carencia es multifactorial (ingesta inadecuada, como es el caso de alcohólicos y vegetarianos estrictos, defectos en la absorción entre los que se encuentra la anemia perniciosa (causa más común de deficiencia de vitamina B12), incremento en los requerimientos (embarazo) y fármacos).
Se puede presentar con clínica de síndrome anémico de intensidad variable; piel seca y amarillenta, ictericia leve, glositis atrófica, diarrea y dispepsia; siendo los síntomas específicos de deficiencia de cobalamina: parestesias, disminución de la sensibilidad superficial y profunda, apalestesia, deambulación inestable, incoordinación, signo de Romberg positivo, pérdida de la fuerza muscular, hiperreflexia, espasticidad, clonus y signo de Babinsky bilateral, psicosis franca y demencia de causa reversible con el tratamiento específico. En el hemograma se puede encontrar macrocitosis con VCM > 100 fL y CHCM aumentada, anisocitosis y poiquilocitosis acompañado de grados variables de pancitopenia. Con niveles de cobalamina en suero menor de 200 pg/mL. Otras pruebas más sensibles consisten en la cuantificación de ácido metilmalónico y de homocisteína, ambos se encuentran elevados en la carencia de cobalaminas, mientras que la homocisteína solo está elevada en la carencia de folatos. Los niveles de ácido fólico en sangre deben ser inferiores a 4 ng/mL. Ante la sospecha de anemia perniciosa, el diagnóstico se realiza a través de la prueba de Schilling, En el frotis de sangre periférica es característica la presencia de macroovalocitos y neutrófilos hipersegmentados pudiéndose observar también microcitos y dacriocitos. Son comunes los cuerpos de Howell-Jolly y glóbulos rojos nucleados con cariorrexis. La médula ósea es hipercelular, con aumento relativo de los precursores eritroides, núcleos de aspecto inmaduro y citoplasma hemoglobinizado, metamielocitos gigantes así como megacariocitos.
Son desórdenes hematológicos clonales caracterizados por hematopoyesis ineficaz. Típicamente se manifiestan con citopenias periféricas a pesar de la hipercelularidad de la médula ósea. La historia natural de estos síndromes varía desde un curso crónico que puede durar años, hasta un curso rápido de progresión hacia la leucemización. Se describen cinco categorías basados en criterios morfológicos obtenidos de aspirados de médula: anemia refractaria, anemia refractaria con sideroblastos en anillo, anemia refractaria con exceso de blastos, leucemia mielomonocítica crónica (LMMC) y anemia refractaria con exceso de blastos en transformación. La media de edad de presentación es entre la séptima y la octava década de vida y es aproximadamente tres veces más frecuente en hombres Es importante distinguir entre los SMD de novo y aquellos relacionados con la exposición a agentes tóxicos (quimioterápicos, productos químicos o radiación ionizante), que constituyen aproximadamente el 20%. Se presentan con fallo medular y la evidencia clínica de las diferentes citopenias: infecciones, sangrados y síndrome anémico. Existen pacientes asintomáticos pero con el hallazgo de anemia, trombocitopenia, leucopenia o una combinación de estos en un examen de rutina. En el frotis aparecen una o más citopenias. La anemia es la más comúnmente encontrada y se observa en alrededor del 85 % de los casos, acompañada de una macrocitosis moderada (VCM entre 100-110 fL); puede coexistir ocasionalmente una población normocrómica con otra hipocrómica. Es frecuente encontrar eliptocitos, esquistocitos, punteado basófilo y hasta el 10% de células rojas nucleadas. El recuento de reticulocitos habitualmente es bajo, Las alteraciones granulomonocitarias en el momento del diagnóstico están presentes en alrededor del 50 % de los pacientes. La médula ósea es generalmente hipercelular, aunque se han descrito casos con hipocelularidad. La eritropoyesis es comúnmente hiperplásica con la presencia de vacuolas y la basofilia intensa, alteraciones nucleares e incremento del número de eritroblastos hasta en el 50%. La coloración de azul de prusia es positiva y en ella se puede observar la presencia de sideroblastos anillados debido a las alteraciones en el metabolismo del hierro. Cuando esta tinción es negativa el diagnóstico de SMD suele ser dudoso.
Otras causas de la disminución de la producción de células sanguíneas, a pesar de un incremento en los precursores medulares, es la hipocupremia. Los síntomas de la deficiencia nutricional de cobre son la anemia refractaria (resistente a la terapéutica ferrosa), diarrea, hipotonía, hipotermia, retardo psicomotor y otros trastornos neurológicos, trastornos visuales, despigmentación de la piel y del pelo, trastornos de inmunocompetencia, dermatitis seborreica, ruptura vasculares y osteoporosis.
Diferentes enfermedades autoinmunes pueden potencialmente terminar generando un cuadro de pancitopenia, sin embargo, en ellas es conocido que el ataque inmunológico de las células sanguíneas suele no limitarse a la periferia pudiendo también tener como blanco a los precursores hematopoyéticos en la MO.
El bazo es un órgano reticuloendotelial que entre otras funciones se encarga de mantener el control de calidad de los elementos formes de la sangre, eliminando las células circulantes al terminar su ciclo fisiológico. Cuando aumenta de tamaño (infecciones, infiltración neoplásica, hipertensión portal) aumenta también su actividad, con lo que suele producirse mayor retención y destrucción de células sanguíneas; a este estado se le conoce como hiperesplenismo. Aunque la principal manifestación suele ser la anemia, no es infrecuente observar también pancitopenia.
Bibliografía
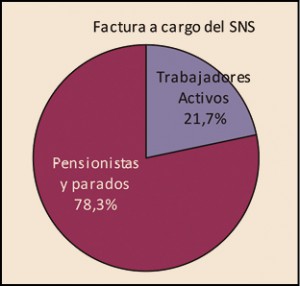
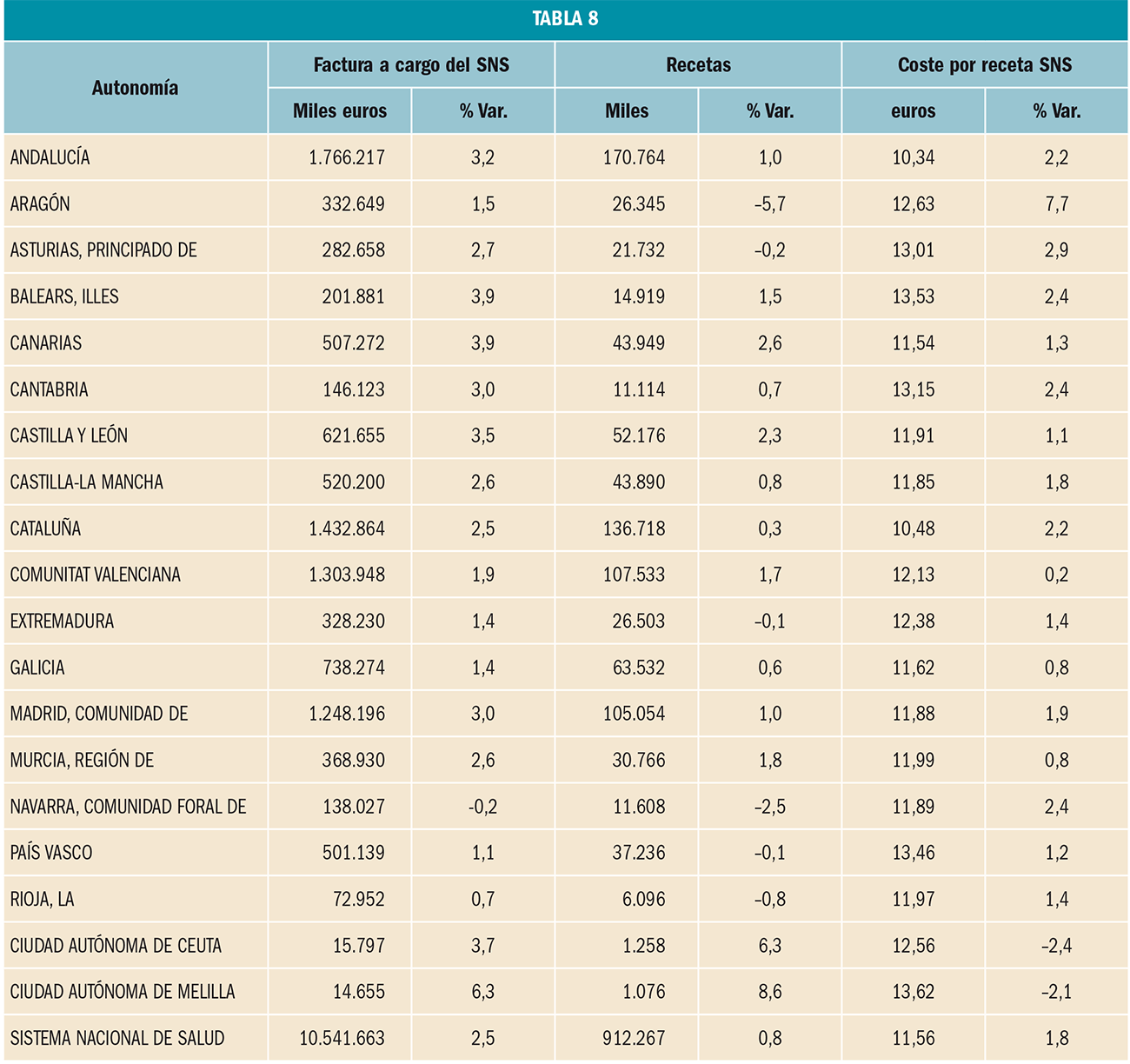
La utilización de medicamentos y productos sanitarios del Sistema Nacional de Salud en 2017 aumentó un 2,1% respecto a 2016, mientras que el importe de la factura a cargo de la Administración se incrementó un 2,5%. El incremento del importe de la factura farmacéutica se explica por el aumento del número de recetas un 0,8% y el incremento del coste por receta un 1,8%. La aportación de los beneficiarios se ha incrementado un 1,5% respecto a la aportación realizada en 2016 y su participación en la financiación de los medicamentos y productos sanitarios utilizados a cargo del SNS se sitúa en el 9,7% del importe de la facturación a PVPIVA. Los agentes del sector farmacéutico han contribuido con 769,2 millones de euros a la financiación de la factura farmacéutica, de los cuales, 432,7 millones de euros, el 56%, han sido aportados por las farmacias. En 2017 el Sistema Nacional de Salud ha pagado de media por la factura farmacéutica unos 878 millones de euros al mes. Mientras que en marzo se registró la liquidación más alta, 936 millones de euros, el mes de febrero registró la liquidación más baja, 809 millones de euros. La factura de medicamentos, que en 2017 ha supuesto el 91% de la factura total a cargo del SNS, se ha incrementado un 2,3% respecto a 2015 mientras que la factura de efectos y accesorios que ha representado el 6,7% de la factura total se ha incrementado un 4,3%. En relación con las recetas dispensadas, las de medicamentos, que han representado el 97,1% del total, han aumentado un 0,6% respecto a 2016, las de efectos y accesorios un 5,2% y las de fórmulas un 8,4%. La factura farmacéutica a cargo del SNS correspondiente a los pensionistas y parados sin derecho a prestación por desempleo se ha incrementado un 2,4% respecto al año anterior y la factura de los trabajadores activos un 3%. En cuanto al número de recetas, han aumentado en ambos colectivos, las de pensionistas y parados un 0,7%, y las de los trabajadores activos un 0,8%. Los pensionistas y parados sin derecho a prestación, que representan el 28,9% de la población protegida, generan el 78,3% de la factura a cargo del SNS, mientras que los trabajadores activos, que representan el 71,1% de los beneficiarios, generan el 21,7% de la factura. A nivel individual, el gasto del SNS en recetas médicas por pensionistas y parados en el año 2017 fue de 644,22 euros, muy por encima del gasto medio en recetas de los trabajadores activos – 72,62 euros –. El gasto medio por persona protegida en el Sistema Nacional de Salud fue de 237,83 euros, un 2,1% más respecto a 2016. Excepto en Navarra, en todas las Comunidades Autónomas se ha incrementado el importe de sus respectivas facturas farmacéuticas respecto a las del año anterior. El mayor crecimiento de la factura se ha registrado en Melilla, un 6,3%, seguida de Baleares y Canarias, un 3,9%. En el extremo opuesto La Rioja y el País Vasco con unos crecimientos del 0,7% y del 1,1% respectivamente.
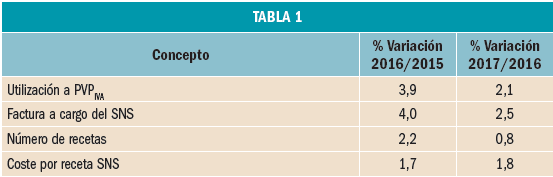
El comportamiento de las principales magnitudes de la prestación Farmacéutica en el Sistema Nacional de Salud a través de recetas médicas oficiales en 2017, comparado con el comportamiento en 2016, se resume en la Tabla 1.
El volumen generado por la utilización de medicamentos y productos sanitarios financiados por el Sistema Nacional de Salud valorado a PVPIVA en 2017 ha aumentado un 2,1% respecto al año anterior, mientras que el importe de la factura final a cargo de la Administración – deducida la aportación de los beneficiarios y de los agentes del sector – se ha incrementado un 2,5% respecto a 2016.
La factura de medicamentos y productos sanitarios o factura farmacéutica a cargo del SNS incluye, además del coste industrial del medicamento (a cargo de los laboratorios), el coste de la distribución mayorista (a cargo de los distribuidores) y el coste de la dispensación (a cargo de las farmacias), por lo que, con el importe total de la factura, se remunera a todos los agentes del sector, a cada uno en proporción a su participación en el precio de los medicamentos, incluidos los descuentos a cargo de cada uno de ellos.
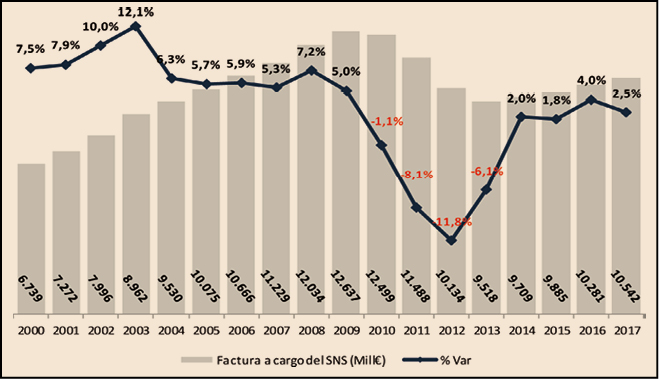
Figura 1.
Como se puede observar en la Figura 1, la factura farmacéutica a cargo del SNS, que registró una importante disminución entre 2010 y 2013 como consecuencia de las medidas de control del gasto que se adoptaron en ese periodo, retomó en 2014 tasas de crecimiento positivas que ha llevado a situar la factura de 2017 ligeramente por debajo de la factura del año 2006: 10.542 millones de euros en 2017 frente a 10.666 millones de euros en 2006.
En la Figura 2 se visualiza la evolución mensual de la factura desde el mes de enero de 2005 y hasta diciembre de 2017.
Como se puede observar en la Figura 2, hay dos periodos claramente diferenciados que marcan el inicio de las medidas que más impacto han tenido sobre la factura:
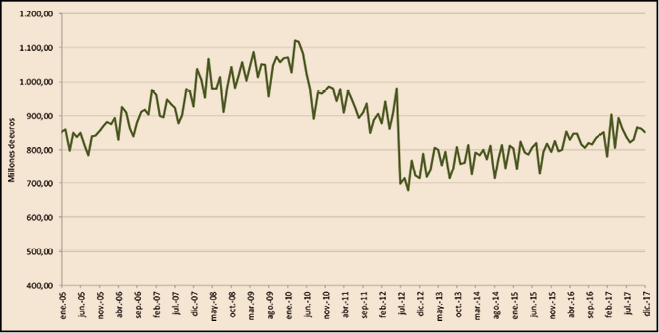
Figura 2.
La gráfica también refleja claramente cómo, tras la brusca caída de la factura por la aplicación del RDL 16/2012, y en concreto por la modificación de las aportaciones de los usuarios del SNS, esta vuelve a aumentar, desde un nivel muy inferior, pero aumenta, lo cual se explica por las características de la demanda de medicamentos, fuertemente influenciada por factores como el envejecimiento de la población, la cronicidad, las pautas de prescripción, etc.
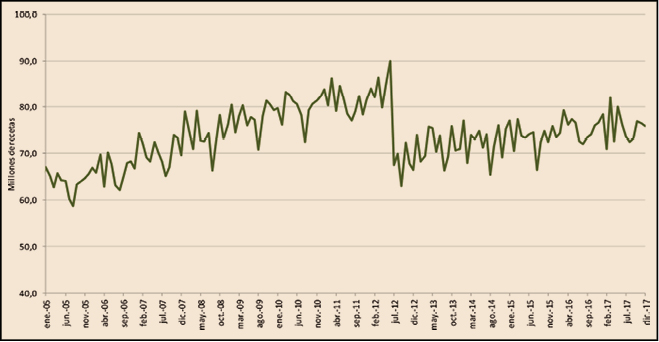
Figura 3.
Como complemento de lo anterior, en la Figura 3 se visualiza la evolución mensual de las recetas dispensadas con cargo al SNS en el mismo periodo. En este caso se distingue claramente la única medida que ha tenido verdadero impacto sobre la demanda de medicamentos, el RDL 16/2012 (julio de 2012) y, también refleja claramente cómo, tras la brusca caída de la demanda, la serie aumenta desde el nuevo nivel de consumo marcado por la aplicación del RDL 16/2012.
La factura de medicamentos y productos sanitarios a cargo del SNS en 2017 se descompone de la forma descrita en la Tabla 2.
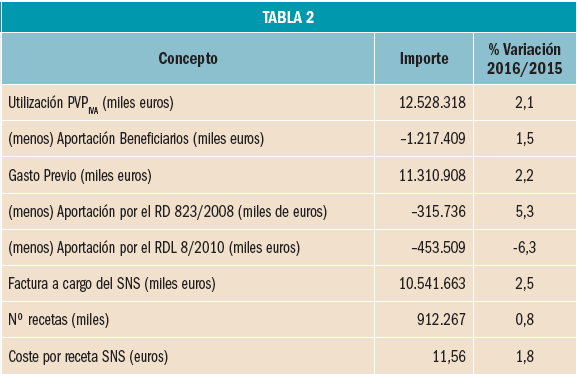
En el año 2017, se ha incrementado el coste medio de la receta pagado por el SNS respecto al coste medio del año anterior un 1,8% situándose en 11,56 € en 2017 frente a 11,36 en 2016. En 2017 se ha consolidado la aplicación de los precios de referencia incluidos en la Orden SSI/1305/2016, de 27 de julio. El retraso en la aprobación de la Orden SSI/1157/2017, de 28 de noviembre, ha supuesto que los nuevos precios de referencia no hayan tenido efecto en ese año al resultar de aplicación efectiva en las dispensaciones realizadas a partir del 1 de enero de 2018.
En cuanto al número de recetas dispensadas en el año 2017, estas han aumentado un 0,8% respecto a 2016.
Como consecuencia de lo anterior, la factura de medicamentos y productos sanitarios a cargo del SNS en 2017, ha sido un 2,5% superior a la factura de 2016, lo que ha provocado, a su vez, el incremento de las aportaciones de las farmacias derivada de la aplicación de la escala de deducciones establecida en el RD 823/2008, de 16 de mayo, por el que se establecen los márgenes, deducciones y descuentos correspondientes a la distribución y dispensación de medicamentos de uso humano, en concreto, un 5,3% más que en 2016.
En total, las farmacias han aportado en 2017, 315,7 millones de euros por la aplicación de la escala de deducciones que representa el 2,5% sobre el importe de la facturación a PVPIVA (Tabla 3).
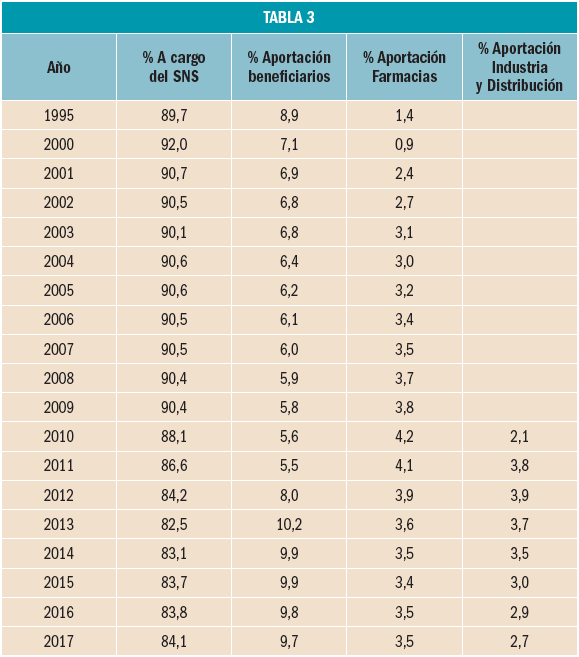
Frente al aumento de las aportaciones de las farmacias por el RD 823/2008, las deducciones sobre el precio de los medicamentos por la aplicación del RDL 8/2010, que se reparten entre todos los agentes de la cadena farmacéutica – Industria, Distribución y Farmacia –, se han reducido un 6,3 % respecto a las deducciones practicadas en 2016, y ello como consecuencia de la entrada en precios de referencia de medicamentos sobre los que dejan de aplicarse las deducciones del RDL 8/2010.
En total, las deducciones por la aplicación del RDL 8/2010 han ascendido a 453,5 millones de euros, que supone el 3,6% del importe de la facturación a PVPIVA (utilización a PVPIVA) que ha sido financiada por todos los agentes del sector. Del importe anterior, 312,5 millones de euros corresponden a la Industria, 24 millones a la Distribución y 117 millones a las Farmacias.
El resultado de añadir a las aportaciones derivadas de la aplicación de la escala del RD 823/2008 – 315,7 millones de euros – 117 millones de euros de las deducciones del RDL 8/2010, es de 432,7 millones de euros que es el importe total con el que las farmacias han contribuido a la financiación del gasto en medicamentos de las CC AA, el 3,5% del importe de la facturación PVPIVA, de medicamentos y productos sanitarios. En total, las aportaciones que han realizado las farmacias se han incrementado un 1,6% respecto a las realizadas en 2016 y representan el 56% de las realizadas por el sector en su conjunto y que han ascendido a 769,2 millones de euros.
En cuanto a la aportación que realizan los beneficiarios, se ha incrementado un 1,5% respecto a la del año anterior. En total, los beneficiarios han aportado 1.217 millones de euros, el 9,7% del importe de la facturación a PVPIVA, ligeramente por debajo de la aportación realizada en 2016 en el que la aportación por este concepto representó el 9,8% de la facturación de ese año.
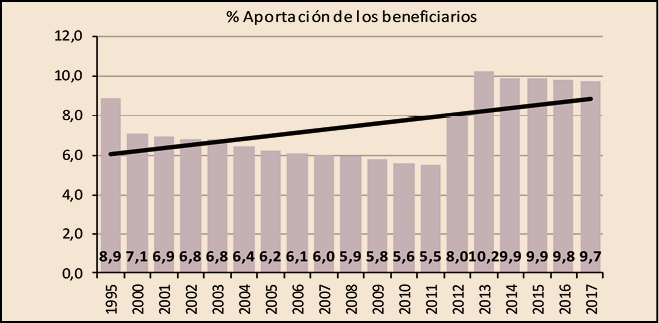
Figura 4.
De cada uno de los dos grandes colectivos en los que se agrupan los beneficiarios del SNS, los trabajadores activos1 habrían aportado 704 millones de euros, el 22,1% de la facturación a PVPIVA de este grupo (3.188 millones de euros), frente a los 514 millones de euros que habrían aportado los pensionistas, el 5,5% de su facturación a PVPIVA (9.340 millones de euros).
Como se puede observar en la Figura 4, en 2012 y 2013 se invirtió la tendencia decreciente que venía registrando la participación de los beneficiarios en la financiación del gasto en medicamentos y productos sanitarios, como también se invirtió la tendencia creciente en las aportaciones realizadas por las farmacias, excepto las de este último año.
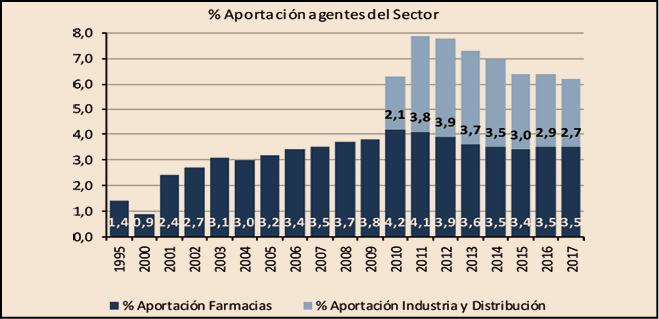
Figura 5.
La utilización de medicamentos y productos sanitarios se caracteriza por la gran variabilidad de las tasas de crecimiento que se registran en los distintos meses del año (Tabla 4). Si en 2016, agosto fue el mes en el que más se incrementó la utilización de medicamentos y productos sanitarios, un 10,9% más que en agosto de 2015, en 2017 el mayor crecimiento se ha registrado en enero, un 6,9% más que en enero de 2016.
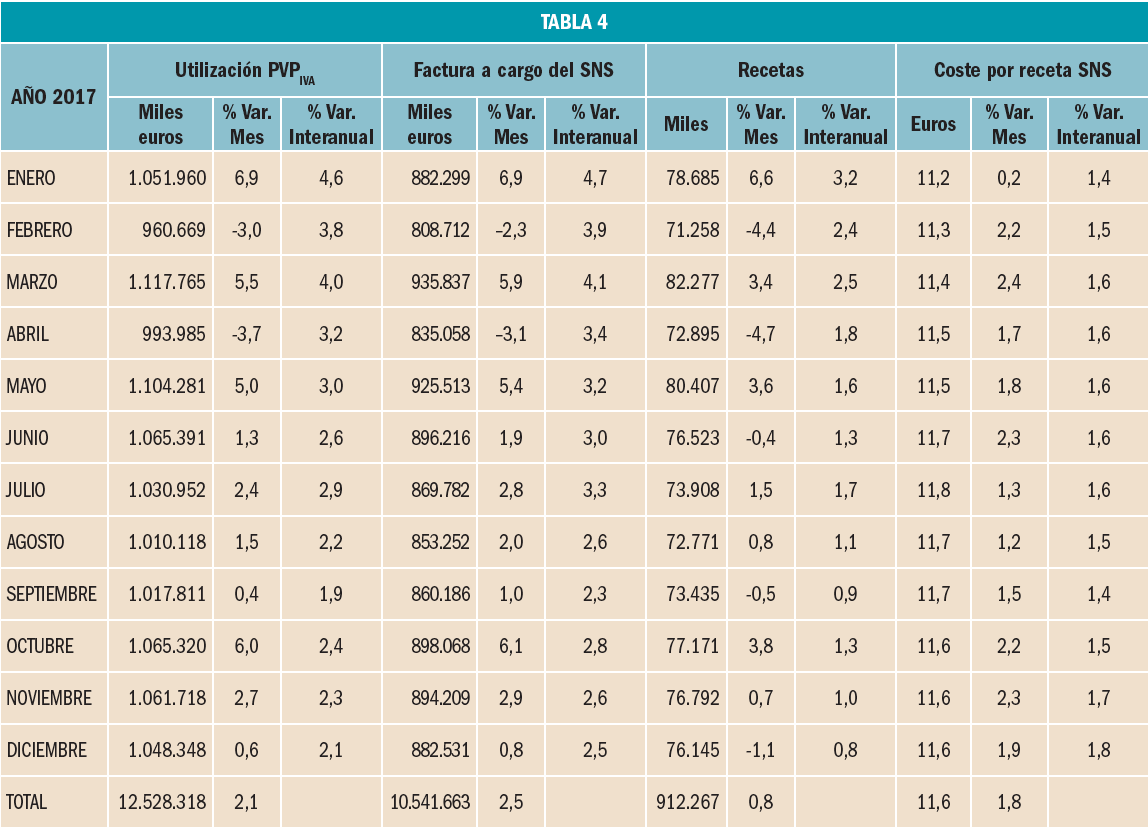
Respecto a la factura a cargo del SNS, en valores absolutos, el Sistema ha pagado de media unos 878 millones de euros al mes, un 2,5% más que el año anterior. Además, mientras que en marzo se registró la liquidación más alta, 936 millones de euros, un 6,5% por encima de la media, el mes de febrero registró la liquidación más baja, 809 millones de euros, un 7,9% por debajo de la media.
En cuanto a las recetas dispensadas y su coste medio, que son las variables que determinan el comportamiento de la factura, destacar que en 2017 se dispensaron en promedio unos 76 millones de recetas mensuales, siendo marzo y febrero los meses con mayor y menor número de dispensaciones respectivamente, y además, que el coste medio por el coste medio por receta ha registrado, al finalizar el año, un crecimiento del 1,8% respecto a 2016.
En 2017, la factura de medicamentos que ha supuesto el 91% de la factura total a cargo del SNS, se ha incrementado un 2,3% respecto a 2016 mientras que la factura de efectos y accesorios que ha representado el 6,7% de la factura total se ha incrementado un 4,3%. En cuanto a la factura de fórmulas, con un peso del 2,4% de la factura total, se ha incrementado un 6,6% respecto a 2016.
En cuanto al comportamiento del número de recetas dispensadas, las de medicamentos, que han representado el 97,1% de las recetas que se dispensan a través de farmacia, han aumentado un 0,6% respecto a 2016, las de efectos y accesorios un 5,2% y las de fórmulas un 8,4%.
Respecto al coste medio de las recetas, el de los medicamentos se ha incrementado un 1,7% respecto a 2016, mientras que el de los efectos y accesorios y el de las fórmulas se han reducido un 0,8% y un 1,7% cada uno de ellos respecto al año anterior.
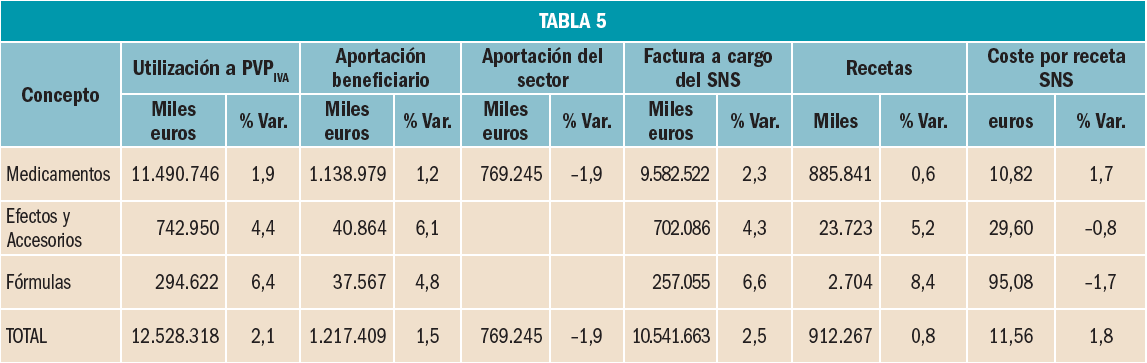
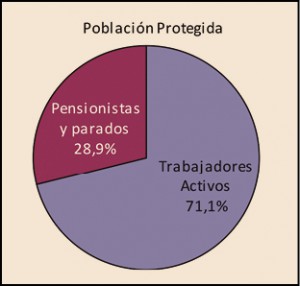
Desde la entrada en vigor del RDL 16/2012, de 20 de abril, los beneficiarios del Sistema Nacional de Salud (CC AA e Ingesa), se agrupan en dos grandes colectivos: el de los trabajadores activos, que representan el 71,1% de la población protegida y del de los pensionistas y parados sin prestación de desempleo, que representan el 28,9%2. Sin embargo, el nivel de gasto de cada uno de estos colectivos es muy diferente. Mientras que el gasto para el SNS de los trabajadores activos3 supone el 21,7% del gasto total del SNS, el de los pensionistas es el 78,3%, tres veces superior al de los trabajadores activos, a pesar de que éstos son más del doble en número que los pensionistas y parados.
En 2017, la factura farmacéutica correspondiente a los pensionistas y parados se ha incrementado un 2,4% respecto al año anterior, por debajo de lo que se ha incrementado la factura de los trabajadores activos, un 3%:
A nivel individual, el gasto del SNS en recetas médicas por pensionistas y parados en el año 2017 fue de 644,22 euros, muy por encima del gasto medio en recetas de los trabajadores activos – 72,62 euros –. El gasto medio por persona protegida en el Sistema Nacional de Salud fue de 237,83 euros, un 2,1% más respecto a 2016.
En 2017, con la única excepción de Navarra, en todas las Comunidades Autónomas se ha incrementado el importe de sus respectivas facturas farmacéuticas respecto a las del año anterior. El mayor crecimiento de la factura se ha registrado en la Ciudad Autónoma de Melilla, un 6,3% más que en 2016, seguida de Baleares y Canarias con un crecimiento del 3,9%.
En el extremo opuesto, La Rioja y el País Vasco son las Comunidades en las que menos se han incrementado sus respectivas facturas en 2017, un 0,7% y un 1,1%, respectivamente y sólo en Navarra se ha reducido la factura, en concreto un –0,2% respecto a 2016.
En cuanto a las recetas dispensadas en 2017, en cinco CC AA se han reducido las dispensaciones, en concreto en Aragón
(–5,7%), Navarra (–2,5%), La Rioja (–0,8%), Asturias (–0,2%) y en Extremadura y País Vasco (–0,1%). En las demás CC AA, los crecimientos han ido desde el 8,6% registrado en Melilla y el 6,3% en Ceuta, hasta 0,6% de Galicia y el 0,3% de Cataluña.
Respecto al coste por receta, los de Melilla, Baleares y el País Vasco son los más elevados superando en más de un 16% el coste medio del SNS (11,56 €). Por el contrario, el coste por receta en Andalucía y Cataluña es de los más bajos, en concreto un 10,5% y un 9,3% menos respectivamente y en relación al coste medio del SNS.
Como ocurre con las células normales, las células tumorales expresan proteínas con carácter antigénico – antígenos tumorales – en su membrana que pueden ser reconocidos como extraños por el sistema inmune. Esto puede ayudar a entender cómo ciertos tumores experimentan una regresión espontánea sin tratamiento y, al mismo tiempo, está permitiendo desarrollar estrategias para modular las respuestas inmunes antitumorales. Las inmunoterapias contra el cáncer que explotan esta capacidad constituyen un cambio de paradigma en el tratamiento del cáncer, produciendo éxitos donde las terapias convencionales contra el cáncer – basadas en la destrucción no selectiva de células con alta tasa de proliferación – fracasan. Los avances en este campo son continuos, centrándose fundamentalmente en la caracterización de los antígenos dirigidos por la inmunidad antitumoral y el aprendizaje correspondiente para diseñar inmunoterapias específicas de mayor beneficio clínico. Los mecanismos por los que el sistema inmune reacciona frente a las células tumorales suponen una intrincada red de eventos que implican tanto el sistema inmune innato como el adaptativo. Sin embargo, también las células tumorales son capaces de desarrollar mecanismos de escape o evasión – inmunoevasión – frente a la acción del sistema inmune; para paliarlos, se han ido desarrollando diversos enfoques terapéuticos que actúan en diferentes etapas de la cascada del proceso inmune antitumoral. Básicamente, pueden dividirse en dos grandes grupos; por un lado, la inmunoterapia a base de citocinas u otras moléculas inmunomoduladoras, capaces de potenciar la actividad general del sistema inmune; por otro, aquellas terapias que provocan una respuesta inmune específica in vivo o el empleo de células inmunitarias del propio paciente estimuladas y cultivadas (expandidas) ex vivo que posteriormente son reintroducidas en el paciente.
Aunque pueden aparecer células tumorales en cualquier tejido de nuestro organismo, éstas no proceden de cualquier célula sino de unas denominadas células madre tumorales o (cancer stem cells, CSC), que se caracterizan por tener la capacidad de dividirse de forma autónoma escapando de los mecanismos de control de la división celular y de los mecanismos de muerte celular programada (apoptosis). Al mismo tiempo, las CSC tienen pierden su diferenciación celular, posiblemente como una estrategia biológica para poder invadir el estroma extracelular adquiriendo algunas la competencia acceder a los vasos linfáticos y sanguíneos, y extenderse a otros órganos para formar áreas de crecimiento tumoral alejadas del tumor primario (metástasis distales).
La adquisición de la capacidad metastásica por las células tumorales es un aspecto de enorme interés, como resulta obvio. En este sentido, datos experimentales recientes sugieren que el incremento de la expresión de CD361, también conocido como glicoproteína plaquetaria 4 (platelet glycoprotein 4) o translocasa de ácidos grasos (fatty acid translocase; FAT), podría facilitar la transformación de una célula normal en metastásica; por el contrario la administración de anticuerpos bloqueadores de CD36 a animales con metástasis ya establecidas, parece conducir a la eliminación total o a una drástica reducción de dichas metástasis. Además, se ha demostrado que el efecto ejercido por CD36 sobre la metástasis es común para las células tumorales de melanoma y de cáncer de mama, y asimismo se ha confirmado las metástasis de cáncer de ovario, vejiga y pulmón también son dependientes de CD36.
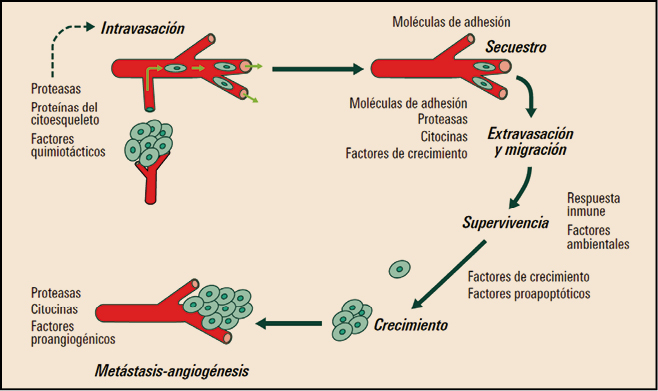
Figura 1. Diseminación metastásica y factores implicados.
Por otro lado, el estado madurativo de las CSC no es uniforme y sus capacidades proliferativas tampoco lo son, lo que explica la existencia habitual de diferentes subclones celulares dentro de un mismo tumor y justifica su heterogeneidad tanto en su comportamiento patológico como en la respuesta a fármacos; asimismo, la capacidad de proliferación de estas células condiciona el crecimiento del tumor y su capacidad de diseminarse.
Por último, estas células deben adaptar su metabolismo y, especialmente, escapar del reconocimiento por parte del sistema inmunitario. Así pues, la respuesta inmune – o, más bien, su ausencia o deficiencia – resulta determinante para la supervivencia de las células tumorales.
Las células de cada individuo expresan proteínas específicas en su superficie que actúan como marcas de identidad; esto permite que las células de una persona sean reconocidas como propias por su sistema inmunitario, evitando que sean atacadas y eliminadas. Dichas proteínas específicas tienen un carácter de antígeno en tanto que son capaces de ser detectadas y formar una biblioteca genética en las células del sistema inmune, lo que permitirá que sean reconocidas como propias: tolerancia inmune. Por el contrario, cuando el sistema inmune detecta antígenos que no reconoce (bacterias, virus, células tumorales, etc.), pone en marcha una serie de mecanismos contra estos antígenos extraños y sus estructuras portadoras, con el objetivo de eliminarlos.
Como ocurre con las células normales, las células tumorales expresan proteínas con carácter antigénico – antígenos tumorales – en su membrana, lo que supone la superficie celular de contacto con el entorno tisular, que pueden ser reconocidos como extraños por el sistema inmune si dichas proteínas han experimentado algún cambio o mutación con respecto a su origen fisiológico. Se han identificado diversos tipos de antígenos tumorales que pueden ser reconocidos por las células del sistema inmune, principalmente por linfocitos T, linfocitos B y células natural killers. Esto puede ayudar a entender cómo ciertos tumores experimentan una regresión espontánea (sin tratamiento) y, al mismo tiempo, está permitiendo desarrollar estrategias para modular las respuestas inmunes antitumorales.
En definitiva, todo ello supone que las células T (linfocitos T) endógenas son capaces de reconocer y destruir las células cancerosas. En este sentido, las inmunoterapias contra el cáncer que explotan esta capacidad constituyen un cambio de paradigma en el tratamiento del cáncer, produciendo éxitos en los que las terapias convencionales contra el cáncer – basadas en la destrucción poco selectiva de células con alta tasa de proliferación – fracasan. Los avances en este campo son continuos, centrándose fundamentalmente en la caracterización de los antígenos dirigidos por la inmunidad antitumoral y el aprendizaje correspondiente para diseñar inmunoterapias específicas de mayor beneficio clínico.
La expresión de un único tipo de receptor en cada célula T (T cell receptor; TCR) permite a ésta escanear selectivamente antígenos presentados en las moléculas del complejo principal de histocompatibilidad (MHC) en la superficie de la célula tumoral. En términos generales, estos antígenos tumorales se agrupan en dos familias:
Los primeros pueden ser compartidos por múltiples cánceres, pero también se puede expresar en los tejidos normales, mientras que los neoantígenos no solo son específicos de cada tipo de tumor, sino incluso de cada paciente.
La exclusividad de la expresión de los neoantígenos por las células tumorales – por tanto, no son expresados por las células normales – les convierte en evidentes dianas terapéuticas en los pacientes con cáncer. En unos casos, estos antígenos corresponden a proteínas que han experimentado cambios como consecuencia de mutaciones acumuladas en ADN de la célula tumoral; en otros, en cambio, se trata proteínas normales pero que solo se expresan en determinados momentos o etapas vitales, especialmente durante el desarrollo embrionario, y por tanto están habitualmente ausentes el resto de la vida. Lamentablemente, la mayoría de las mutaciones que experimenta el material genético de las células tumorales no crea neoantígenos y las que lo hacen son generalmente mutaciones que solo se observan de forma puntual durante la progresión del cáncer y únicas para cada paciente, lo que dificulta notablemente su detección y caracterización.
Por su parte, los denominados antígenos asociados a tumores se expresan tanto en tumores como en las células normales, y de hecho en su mayoría son componentes normales de las células pero su expresión por células tumoral es aberrante, excesiva o presenta una regulación anómala durante los procesos tumorales. Un ejemplo de esto último es la mucina 1 (MUC1), una proteína transmembrana expresada en células de pulmón, mama, páncreas, riñón, ovario, colon y otros tejidos. Tiene un dominio extracelular N-terminal que contiene un número variable de unidades de repetición en tándem de 20 aminoácidos (variable number tandem repeat; VNTR) y la región C-terminal transmembrana e intracelular. En la porción de péptido central de MUC1, cada región de repetición en tándem contiene cinco sitios potenciales de O-glicosilación en residuos de serina o treonina. La mucina 1 está intensamente glicosilada en células normales, mientras que en células cancerosas la glicosilación es baja o aberrante, es decir, muy diferente del patrón fisiológico. Esta diferencia estructural en MUC1 entre tejidos normales y cancerosos le convierte en un objetivo atractivo para la inmunoterapia contra el cáncer.
Otro ejemplo interesante de antígeno asociado a tumores es el sulfato de condroitina proteoglicano-4 (CSPG4), un proteoglicano de la superficie celular sobreexpresado en un amplio rango de lesiones neoplásicas humanas y caninas, afectando al microambiente tumoral, y de células iniciadoras de cáncer. El CSPG4 juega un papel central en las vías oncogénicas necesarias para la progresión tumoral y la formación de metástasis. Gracias a estas características y a su escasa expresión en las células de los tejidos sanos adultos, el CSPG4 representa un oncoantígeno ideal y, por tanto, otro objetivo atractivo para la inmunoterapia antitumoral.
Las mutaciones impulsan el crecimiento sin control, la heterogeneidad y la capacidad adaptativa de las células cancerosas, haciendo que muchos cánceres sean en origen o se vuelvan refractarios a los tratamientos convencionales. Sin embargo, lo que dificulta el tratamiento también, curiosamente, constituye el talón de Aquiles del cáncer al facilitar y señalizar el ataque inmunitario específico; de hecho, las diferentes formas de inmunoterapia están teniendo ya excelentes resultados explotando esta vulnerabilidad. Sea cual sea el tipo de antígeno tumoral, el sistema inmune tiene la capacidad de reconocer a las células cancerosas a través de la detección de estos antígenos, lo que permite que el sistema mantenga una constantemente vigilancia para detectar la aparición de células tumorales, para destruirlas y eliminarlas antes de que se acumulen formando un tumor o emigren para incorporarse y proliferar en otras zonas distales del organismo (metástasis).
No obstante, aún queda mucho espacio por recorrer. Básicamente, los principales desafíos relacionados con el descubrimiento de objetivos y la implementación terapéutica pueden agruparse en tres:
Tradicionalmente, la inmunidad se cataloga en dos gran apartados en función de la especialización de sus cometidos: innata y adaptativa.
La inmunidad innata corresponde a células y moléculas que actúan de manera rápida contra agentes extraños; se trata de la primera barrera defensiva, pero no genera memoria inmunológica. Forman parte de ésta forma defensiva las barreras físicas como la piel y mucosas, las secreciones antimicrobianas de estos tejidos, las moléculas que favorecen la inflamación y las células especializadas. De otro lado, está la inmunidad adaptativa, que actúa después de un primer contacto con un agente extraño, y es mediada por receptores específicos en la membrana de los linfocitos, fruto de lo que se conoce como memoria inmunitaria. Tras este primer contacto, algunas células se especializan en reconocer y reaccionar rápidamente ante un nuevo contacto con el mismo agente extraño. Las células principales que componen este sistema son los linfocitos y, por ello, son los principales responsables de la respuesta inmune antitumoral.
Las respuestas inmunes innatas están mediadas por macrófagos, granulocitos, mastocitos, células dendríticas (DC; dendritic cells) y células asesinas naturales (NK; natural killers), mientras que las respuestas adaptativas están mediadas por linfocitos T y B, que no solo pueden desencadenar una respuesta inmune antitumoral específica, sino también pueden promover una inflamación crónica, condicionar el escape de la acción del sistema inmune y el desarrollo neoplásico. Además de estas células, el balance entre la respuesta inmune antitumoral y la inflamación crónica está regulado por una compleja red de citocinas y mediadores solubles, orientados por quimocinas y moléculas de adhesión, que actúan en el entorno del microambiente del tumor y de los ganglios linfáticos.
La respuesta inmune innata representa la primera línea de defensa cuando los mecanismos de homeostasis celular se alteran por los procesos iniciales de la carcinogénesis. En este sentido, los macrófagos presentes en el tejido, los mastocitos y otras células secretan mediadores solubles como citocinas y quimocinas que reclutan leucocitos polimorfonucleares (PMN), otros monocitos, células NK y células provenientes de la circulación. Al mismo tiempo las células dendríticas reconocen los antígenos tumorales y migran a los ganglios linfáticos cercanos, lo que desencadena el procesamiento y presentación de antígenos a las células del sistema inmune adaptativo (linfocitos T y B), lo cual origina activación, proliferación, diferenciación y funciones efectoras antitumorales.
La activación inicial del sistema inmune innato puede asociarse a mecanismos de inflamación aguda y desencadenar mecanismos inmunes adaptativos, que pueden ser efectivos en la etapa de eliminación de las células tumorales. En este escenario, la respuesta se polariza a macrófagos asociados al tumor (TAM; tumor-associated macrophages) tipo 1 y a linfocitos facilitadores o cooperadores (TH; T helper) CD4+ del patrón TH1 (productores de interleucina-12 e interferón γ), además hay predominio de respuesta citotóxica por linfocitos T citotóxicos (CTL; citolytic T lymphocyte) CD8+ específicos y por células inespecíficas NK. También los linfocitos B participan en las respuestas a través de la producción de anticuerpos que reconocen antígenos tumorales.
Por otro lado, la perpetuación de las células tumorales puede originar cambios inflamatorios crónicos que se asocian a la presencia de células supresoras de origen mieloide (MDSC; myeloid-derived suppressor cells), patrones de linfocitos CD4+ Th2 y linfocitos T reguladores (Treg), las que inhiben la citotoxicidad de los CTL CD8+ y polarizan la respuesta a TAM tipo 2, con la secreción de citocinas supresoras como IL-4, IL-13, IL-10, IL-6, el factor de crecimiento transformante β (TGF-β; transforming growth factor β) y la secreción de factores angiogénicos como el factor de crecimiento del endotelio vascular (VGEF; vascular endotelial growth factor). Todos estos factores a nivel del microambiente condicionan la promoción y progresión del tumor.
Como se ha visto, en las respuestas inmunitarias antitumorales participan tanto la inmunidad innata como la adaptativa. En una respuesta inmune antitumoral, las células presentadoras de antígenos (APC; antigen-presenting cells) presentes en todo el organismo como células dendríticas y macrófagos, son capaces de detectar y eliminar a células tumorales, mediante la expresión de fragmentos de los antígenos de las células tumorales capturadas, presentándolos a los linfocitos T, las auténticas células efectoras (destructoras) del sistema inmune. En este proceso, el linfocito T se activa con el contacto con la célula presentadora (APC), y adquiere la capacidad de reconocer cualquier elemento (célula o fragmento) que contenga el antígeno presentado por la APC como extraño; por este motivo, el antígeno en cuestión pasa a convertirse en un auténtico marcador biológico. Una vez activado el linfocito T, viaja por todo el cuerpo y cada vez que encuentra una célula que tenga sobre superficie o secrete el antígeno tumoral, la destruye. Se trata de una respuesta muy específica, dado que ese linfocito T sólo puede reconocer un sólo antígeno y no a otros.
Aunque la descripción de este mecanismo sugiere que el proceso de vigilancia y actuación antitumoral natural es muy eficiente, en muchas ocasiones no es así. El motivo de ello es que muchas de las células tumorales expresan una baja cantidad de antígenos reconocibles como extraños, de modo que la capacidad de inducir una respuesta inmune es demasiado leve como para ser eficaz. Por otro lado, la rapidez con que muchas veces proliferan y se propagan las células tumorales sobrepasa la capacidad de respuesta del sistema inmune para erradicarlas.
Además, muchas células tumorales disponen de mecanismos especializados para evitar las respuestas inmunes del organismo, lo que se conoce como inmunoevasión tumoral. Entre ellos cabe destacar la capacidad de algunas células tumorales para modificar, reducir o incluso anular la expresión de antígenos tumorales en su superficie, impidiendo el reconocimiento por parte de las células del sistema inmune; también pueden secretar sustancias que disminuyen el funcionamiento del sistema inmune, incapacitando o dificultando la labor de los linfocitos. Asimismo, algunos tumores sólidos son capaces de desarrollarse sin provocar ninguna reacción inflamatoria, lo que limita drásticamente su detección. En general, un deficiente funcionamiento del sistema inmune favorece el desarrollo tumoral; de hecho, algunos cuadros de inmunodeficiencia generalizada son relacionados con cuadros neoplásicos específicos, tal como ocurre con el sarcoma de Kaposi ligado al SIDA.
Las células presentadoras de antígeno (APC) son capaces de entrar en contacto, endocitar, procesar y presentar un antígeno promoviendo una respuesta inmunitaria específica. Aunque los macrófagos y linfocitos B también pueden cumplir esta función, en realidad las únicas células con capacidad de estimular linfocitos T que no han tenido contacto previo con el antígeno (linfocitos T naïve) son las células dendríticas (CD), un tipo de glóbulos blancos cuyo nombre deriva de la presencia de prolongaciones celulares similares a las dendritas neuronales. Precisamente, el descubrimiento de la capacidad de las células dendríticas para activar linfocitos T que no habían tenido una experiencia previa con un antígeno le valieron a Ralph Steinman el premio Nobel de medicina en el año 2011.
Las células dendríticas tienen su origen en las células hematopoiéticas de la médula ósea y básicamente pueden distinguirse dos tipos, las mieloides y las plasmocitoides. Las primeras proceden desde la médula ósea, distribuyéndose a través del torrente sanguíneo – mayoritariamente en forma de monocitos CD14+ – hacia los tejidos donde se diferencian y residen como células dendríticas inmaduras, caracterizadas por disponer de una gran superficie de contacto debida a la existencia de las prolongaciones citoplasmáticas (dendritas), que les facilitan el proceso de captación de antígenos presentes en el medio. Aun en estado de inmadurez, se especializan en internalizar material antigénico mediante endocitosis a través de diversos tipos de receptores de superficie: receptores de complemento, receptores para la porción Fc de las inmunoglobulinas (CD32), receptores tipo lectina C (CD209, CD205, BDCA, langerina, receptores de manosa) y receptores tipo scavenger (LOX-1, CD91, CD36). Paralelamente, expresan en baja intensidad las moléculas presentadoras de antígeno del complejo principal de histocompatibilidad de clase I y II (MCH-I y MHC-II) y de moléculas co-estimuladoras (CD40, CD80 y CD86).
En este estado inmaduro, las células dendríticas inducen y mantienen la tolerancia frente a los antígenos propios, provocando que los linfocitos T que reciben esta información y que reconocen los autoantígenos entran en muerte celular, lo que elimina la capacidad de autorreactividad. Es decir, si un antígeno es presentado a los linfocitos por células dendríticas inmaduras puede generar la deleción o anergia clonal de dichos linfocitos o inducir la producción de linfocitos T reguladores (Treg). Por el contrario, cuando el antígeno es endocitado en un contexto proinflamatorio, las células dendríticas inmaduras experimentan un profundo cambio que supone, de hecho, su maduración, durante la cual reducen su capacidad de endocitosis, cambian el patrón de expresión de moléculas de adhesión y adquieren mayor movilidad y sensibilidad a las quimiocinas presentes en el ganglio linfático, aumentando la expresión de moléculas implicadas en el proceso de presentación antigénica y estimulación linfocitaria, como lo son el complejo principal de histocompatibilidad de tipo II (CMH-II) y las moléculas co-estimulatorias CD40, CD83, CD80, CD86. Estas glicoproteínas de membrana permiten estimular linfocitos T que reconocen sobre las células dendríticas su antígeno específico. Asimismo, la célula dendrítica madura incrementa su capacidad para secretar diversas citocinas como IL-12, IL-15, interferón alfa. En concreto, la IL-12 induce una respuesta de linfocito Th1 (facilitador), con capacidad de secretar interferón gamma, y la activación de linfocitos Tc (citotóxicos) efectores. Todos estos cambios adaptativos facilitan su migración al ganglio linfático regional y su capacidad para realizar una presentación antigénica eficaz a los linfocitos T.
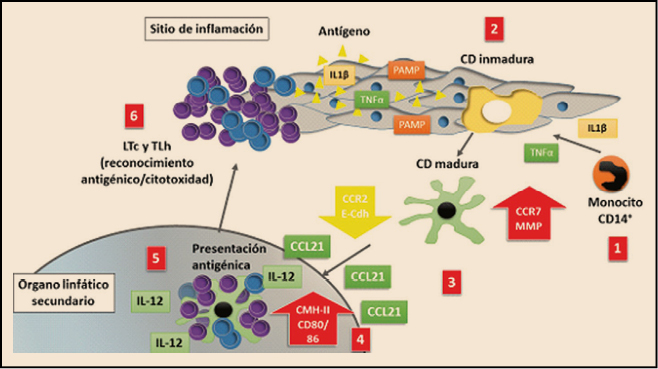
Figura 2.
Están presentes en la mayoría de los sitios del cuerpo, especialmente las superficies corporales, con la finalidad de captar antígenos; además, son capaces de migrar a los órganos linfoides para seleccionar clones de linfocitos T reactivos al antígeno e iniciar la inmunidad. Es importante destacar que las células dendríticas son capaces de responder a un amplio espectro de estímulos mediante una extensa capacidad de diferenciación o maduración, para convertirse en células inmunoactivas. Aunque hay diferentes tipos o subconjuntos de células dendríticas, todos ellos son capaces de experimentar un proceso de maduración apropiada para mejorar la inmunidad. Dicha maduración debe ir acompañada por la captación, procesamiento y presentación del antígeno para que la célula dendrítica sea capaz de actuar como agente inmunoactivo; de lo contrario, la célula dendrítica podría inducir tolerancia o silenciamiento de células T específicas de antígeno.
El estado de la inmunidad antitumoral natural está determinado por muchos factores y es prácticamente exclusivo para cada paciente. Por ejemplo, un paciente con inmunidad antitumoral adecuadamente activa antes de la extirpación quirúrgica de un tumor tiene un buen pronóstico de protección mediada por inmunidad posquirúrgica contra futuras metástasis; de hecho, en algunos tipos de cáncer de mama una mastectomía total puede no requerir quimioterapia posquirúrgica. Por el contrario, los pacientes sin inmunidad antitumoral suficiente antes de la cirugía deben recibir quimioterapia para prevenir o reducir el riesgo de metástasis inducidas por la cirugía.
Por consiguiente, es evidente la importancia de determinar el estado de la inmunidad antitumoral de cada paciente con cáncer. Lamentablemente, el estado actual de las pruebas clínicas no permite medir la inmunidad antitumoral de forma específica y ese es, precisamente, uno de los principales objetivos en el tratamiento del cáncer: el desarrollo de biomarcadores selectivos que determinen con precisión el estado específico de inmunidad antitumoral de cada paciente.
Por el momento, los oncólogos no tienen más remedio que recurrir a signos y síntomas clínicos para hacerse una idea del estatus inmunológico del paciente canceroso. Por ejemplo, un paciente con síntomas inflamatorios que son capaces de ser controlados con procedimientos estándar sugiere la existencia de una activación de la inmunidad innata en el paciente, con posible establecimiento de inmunidad antitumoral adaptativa. Esta observación puede ser corroborada mediante diversas pruebas, tales como cambios en la concentración de determinados marcadores tumorales, como el activador del plasminógeno urocinasa (uPA) e inhibidor del activador del plasminógeno (PAI-1) en cáncer de mama; alfa-fetoproteína (AFP) en cáncer de hígado y tumores de células germinativas; antígeno carcinoembrionario (CEA) en cáncer colorrectal y algunos otros cánceres; antígeno prostático específico (PSA) en cáncer de próstata; calcitonina en cáncer de tiroides; cromograninaA (CgA) en tumores neuroendocrinos; enolasa neuronal específica (NSE) en cáncer de pulmón microcítico y neuroblastoma, etc.
Todos estos marcadores deben ser valorados conjuntamente con pruebas de imagen metabólicas del sitio tumoral mediante PET (tomografía por emisión de positrones) o TC (tomografía computadorizada). En este sentido, la aparición de ganglios linfáticos agrandados sin actividad metabólica detectada por PET/TC puede indicar un historial de metástasis y posterior control (erradicación o supresión) por inmunidad antitumoral del paciente, lo cual sugiere que la activación de esta inmunidad por quimioterapia o radiación seguida de cirugía puede proporcionar una cura clínica. Por el contrario, un paciente que presenta un tumor primario único muy activo descubierto durante un chequeo regular sin ningún signo o síntomas (inflamación), probablemente no ha establecido una inmunidad antitumoral relevante; aunque el paciente pudiera ser un buen candidato para la cirugía, la deficiencia de inmunidad antitumoral no permitiría una protección posquirúrgica contra futuras metástasis.
Los mecanismos por los que el sistema inmune reacciona frente a las células tumorales – que hemos descrito de forma muy sucinta anteriormente – suponen, en realidad, una intrincada red de eventos que implican tanto el sistema inmune innato como el adaptativo, iniciado por la captación, procesamiento y presentación de antígenos tumorales por células presentadoras de antígeno (APC), seguido por cebado y activación de linfocitos T y concluyendo con la infiltración de linfocitos T efectores al sitio del tumor donde ejercen su actividad citotóxica; todo lo cual desemboca potencialmente en la eliminación del tumor. Pero también hemos visto cómo las células tumorales son capaces, eventualmente, de desarrollar mecanismos de escape o evasión – inmunoevasión – frente a la acción del sistema inmune.
Para paliar dichos mecanismos de inmunoevasión tumoral, se han ido desarrollando diversos enfoques terapéuticos que actúan en diferentes etapas de la cascada del proceso inmune antitumoral. Básicamente, pueden dividirse en dos grandes grupos; por un lado, la inmunoterapia a base de citocinas u otras moléculas inmunomoduladoras, capaces de potenciar la actividad general del sistema inmune; por otro, aquellas terapias que provocan una respuesta inmune específica in vivo o el empleo de células inmunitarias del propio paciente estimuladas y cultivadas (expandidas) ex vivo que posteriormente son reintroducidas en el paciente.
El ciclo inmune antitumoral ideal supone:
Desde su primera aplicación en 1890 por William Coley, que trató a los pacientes con cáncer con una mezcla de bacterias muertas observando remisión completa en el 10% de los casos, la inmunoterapia contra el cáncer ha recorrido un largo camino, culminando de forma práctica en 2010 con la primera inmunoterapia personalizada aprobada por la FDA contra el cáncer de próstata. Sin embargo, aún estamos lejos de conseguir una plena utilización de la inmunología en el tratamiento del cáncer en sus múltiples variedades y, más aún, en sus particularidades personales, el auténtico caballo de Troya del cáncer: ni todas las células tumorales son iguales, aunque procedan de un mismo origen, ni las características fisiopatológicas de los pacientes son necesariamente superponibles. Todo lo cual nos lleva, inevitablemente, a que el objetivo final de la terapéutica es actuar selectivamente sobre los tipos tumorales específicos presentes en cada paciente, sin afectar significativamente a las células sanas.
Aunque tradicionalmente se piensa que el uso de fármacos citotóxicos – quimioterapia – tiene efectos fundamentalmente inmunosupresores, este punto de vista está siendo progresivamente modificado. El motivo es que el sistema inmune innato discrimina entre la apoptosis y la necrosis. La muerte celular inocua – apoptosis o muerte celular programada – podríamos considerarla como fisiológica, ya que forma parte de la homeostasis general, mientras que la necrosis supone la muerte celular como consecuencia de un daño infligido al margen de la regulación general del organismo. Asimismo, desde hace algún tiempo se viene considerando un nuevo tipo de apoptosis, denominada apoptosis inmunogénica o muerte celular inmunogénica (immunogenic cell death; ICD), observada tras el empleo de radioterapia o quimioterapia muy citotóxica en el tratamiento del cáncer. A diferencia de la apoptosis, la muerte celular inmunogénica expone extracelularmente ciertas moléculas intracitoplasmáticas y nucleares que, en globalmente, se han denominado patrones moleculares asociados al daño (danger-associated molecular patterns; DAMP) o señales de peligro. Los DAMP alertan al organismo y participan colaborando en el reconocimiento de antígenos tumorales y en la inducción de una eficiente respuesta inmunitaria antitumoral.
La liberación de estas moléculas inmunoestimuladoras (DAMP) por células cancerosas tras su apoptosis inmunogénica, puede contribuir a una mayor captación de neoantígenos por las células dendríticas (células APC) y al ciclo inmunitario antitumoral; es decir, la muerte celular inmunogénica podría alertar al sistema inmunitario de la presencia de células cancerosas moribundas y convertirlas en auténticas vacunas para estimular la inmunidad contra el cáncer a través de la maduración de las células dendríticas y la activación de los linfocitos T citotóxicos, así como la potenciación de la actividad citotóxica de las células NK. Las DAMP más comúnmente asociados con la apoptosis inmunogénica son la calreticulina unida a membrana (CRT) y la proteína de secreción de HGMB1 (high mobility group box 1) del núcleo. Las proteínas de choque térmico (heat shock protein; HSP) 70 y 90 también se han encontrado en la superficie celular durante la apoptosis inmunogénica. Todas ellas funcionan literalmente como señales de “comedme” para los fagocitos, mejorando la captación de antígeno y maduración de células dendríticas.
Las citocinas – especialmente, determinadas interleucinas – juegan un papel crucial en la estimulación y regulación de la respuesta inmunitaria frente a los antígenos, pero su uso directo en clínica es muy limitado debido a los graves efectos tóxicos relacionados con su naturaleza pleiotrópica3 y a menudo doble función al estimular y suprimir simultáneamente la respuesta inmune a diferentes niveles, tal como ocurre con la interleucina 2 (IL-2), empleada en clínica como aldesleukina (Proleukin®) para el tratamiento del carcinoma metastásico de células renales. También se ha estudiado la administración directa de interleucina 12 (IL-12) mediante nanopartículas de quitosano, dada la condición de la IL-12 de ser una potente citocina proinflamatoria que potencia la diferenciación de los linfocitos facilitadores Th1, la proliferación de linfocitos T activadas y de células NK y la inmunidad mediada por células.
Los virus oncolíticos son virus modificados genéticamente con capacidad para infectar a todas las células pero que solo se replican en las células tumorales, acumulándose en gran cantidad dentro de éstas y se liberan de manera masiva, provocando la muerte selectiva de la célula tumoral. La oncolisis puede ser una propiedad natural del virus, como ocurre con algunos Reovirus, o una consecuencia de la manipulación del genoma viral, para lo que suele echarse mano habitualmente de los Adenovirus. Esta propiedad ha permitido el desarrollo de la viroterapia oncolítica – el uso de virus activos con capacidad replicante – para el tratamiento de determinadas formas de cáncer.
Aunque el concepto puede parecer revolucionario, en realidad, la viroterapia oncolítica no es nueva. De hecho, los primeros informes que refieren una drástica regresión tumoral datan de hace más de un siglo (1910); en concreto, el caso de una paciente con cáncer de cuello uterino que recibió la vacuna viva atenuada Pasteur-Roux contra la rabia. Ya en la década de 1940 se iniciaron los primeros estudios sistemáticos en seres humanos con diferentes tipos de virus, aunque la era de la viroterapia oncolítica moderna se inició realmente a principios de los 90, cuando una cepa de virus del Herpes simple (HSV) modificada genéticamente y atenuada por timidina cinasa (TK) se inyectó localmente en modelos de xenoinjerto de glioma humano, mostrando resultados prometedores.
El talimogene laherparepvec (Imlygic®; T-VEC) es una variante del virus del Herpes simplex de tipo 1 (HSV-1) genéticamente modificada, que fue autorizado en los Estados Unidos (FDA) y la Unión Europea (EMA) en 2015 para tratar a adultos con melanoma que no se puede extirpar quirúrgicamente y que se ha extendido a otras partes del cuerpo (pero no a los huesos, los pulmones, el cerebro u otros órganos internos). La cepa herpética fue modificada para replicarse selectivamente dentro de los tumores y producir, dentro de estos, factor estimulante de colonias de granulocitos y monocitos (GM-CSF; Granulocyte Macrophage Colony-Stimulating Factor), provocando la muerte de las células tumorales y la liberación de antígenos tumorales, lo que a su vez promueve una respuesta antitumoral inmune sistémica y una respuesta de células T efectoras.
Las modificaciones introducidas en talimogene laherparepvec de HSV-1 incluyen deleción de los genes ICP34.5 e ICP47. La deleción de ICP47 previene la regulación negativa de las moléculas de presentación de antígenos e incrementa la expresión del gen US11 de HSV, mejorando así la replicación viral en células tumorales. Además, se ha insertado la secuencia de codificación para el factor estimulante de colonias de macrófagos de granulocitos humanos (GM-CSF), en lugar de ICP34.5 original. El GM-CSF es una citocina implicada en la estimulación de respuestas inmunes a través de su efecto sobre las células presentadoras de antígeno; puede activar las células dendríticas para aumentar la presentación del antígeno y puede potenciar tanto las respuestas inmunitarias mediadas por células como humorales.
Imlygic® se ha investigado en un ensayo clínicos en el que participaron 436 pacientes con melanoma metastásico (salvo en huesos o cerebro) que era inoperable. El estudio, que duró 24 meses, comparó Imlygic® con GM-GSF administrados mediante inyección subcutánea. La variable primaria de la eficacia fue el porcentaje de pacientes que respondieron al tratamiento y en los que la respuesta duró al menos seis meses, antes de que la salud del paciente empeorase o necesitase otro tratamiento; la respuesta al tratamiento se definió como un descenso de al menos el 50% en los signos de melanoma. Al considerar el subgrupo de pacientes en el estudio (249 pacientes) cuya enfermedad no se había extendido al pulmón ni a otros órganos internos, el 25% de los pacientes tratados con Imlygic® mostraron una respuesta sostenida al tratamiento en comparación con aproximadamente el 1 % de los pacientes tratados con GM-CSF. Los efectos adversos más frecuentes (≥25%) con Imlygic® fueron fatiga, escalofríos, fiebre, náuseas, síndrome pseudogripal y dolor en el lugar de la inyección, la mayoría de carácter leve o moderado; el efecto grave más frecuente (2%) fue celulitis. Es importante tener en cuenta que Imlygic® contiene un virus herpético, el cual podría reactivarse posteriormente causando infecciones como el herpes labial; incluso, en pacientes cuyo sistema inmunitario está debilitado (VIH, uso crónico o con elevadas dosis de corticosteroides, etc.), podría provocar una infección herpética generalizada.
Algunos virus oncolíticos han sido diseñados o combinados con otros inductores de la muerte celular inmunogénica para promover un cebado cruzado de células T más eficaz y, en muchos casos, la ruptura de la tolerancia inmunológica funcional. También se ha intentado incorporar material genético que permita la expresión de citocinas/quimiocinas estimuladoras de linfocitos T para reclutar y sostener la potente inmunidad antitumoral en el microambiente tumoral para enfocar su actividad terapéutica dentro de las localizaciones tumorales. Asimismo, las combinaciones de virus oncolíticos con fármacos inmunomoduladores o anticuerpos que reacondicionan el microambiente tumoral han demostrado ser muy prometedoras, así como las combinaciones con otros regímenes inmunoterapéuticos, como las células T CAR, que veremos posteriormente. Finalmente, los virus oncolíticos se han combinado con agentes activos sobre señalización bioquímica (inhibidores de tirosina cinasa, etc.), mostrando una notable eficacia antitumoral.
Las bacterias presentan características muy útiles para ser utilizadas como terapia antitumoral. Por un lado, la relativamente fácil manipulación de su material genético permite realizar modificaciones para que puedan producir toxinas que destruyan una células tumorales o producir factores que aumenten la actividad del sistema inmune antitumoral del paciente, así como también pueden actuar como transporte de otras estructuras antitumorales, como los ácidos ribonucleicos sintéticos de interferencia(siRNA) o los virus oncolíticos ya mencionados. Por otro lado, muchas bacterias tienen una elevada movilidad gracias a la presencia de flagelos, presentes tanto en bacterias Gram-positivas como Gram-negativas, generalmente en bacilos y raramente en cocos. Esta movilidad facilita la difusión bacteriana desde el torrente sanguíneo a los tejidos, lo que permitiría una mayor penetración dentro de un tumor.
Se está estudiando actualmente una terapia personalizada conocida como pLADD, consistente en una cepa de Listeria monocytogenes doblemente suprimida (LADD, Listeria monocytogenes doubled-deleted), viva y atenuada, que ha sido diseñada para codificar múltiples neoantígenos específicos de tumores. La plataforma LADD es un enfoque atractivo para la inmunoterapia personalizada debido a la rápida modificación y liberación de cepas clínicas. Además, se ha establecido su perfil clínico de seguridad y eficacia en numerosos pacientes, y se ha mostrado una robusta activación de la inmunidad innata y el remodelado del microambiente tumoral en modelos preclínicos y en pacientes.
Los ácidos ribonucleicos sintéticos de interferencia (synthetic interference RNA; siRNA) son pequeños fragmentos sintéticos de ARN que se unen selectivamente al ARN mensajero (ARNm) que porta la información genética (procedente del ADN) de un oncogén determinado, impidiendo la traducción de éste y, por lo tanto, interfiriendo con la expresión de dicho oncogén. Esta técnica ha demostrado ser muy efectiva en ensayos in vitro; sin embargo, el factor que aún limita su uso clínico es conseguir que estas moléculas, relativamente pequeñas, accedan selectivamente a las células tumorales e ingresen a ellas, manteniendo la integridad.
Los siRNA están formados por moléculas de ARN de doble cadena con apenas 20-25 nucleótidos complementarios, producidos a partir de un ARN largo de doble cadena (double strand RNA, dsRNA), que pueden tener una procedencia endógena o exógena (virus, transgenes, etc.). El enzima responsable la fragmentación selectiva de los dsRNA en siRNAs es el Dicer, una ribonucleasa de la familia de las ARNasa III, presente en el citoplasma celular.
Los siRNA suprimen la expresión de los genes diana mediante el corte del ARN mensajero (ARNm) complementario en dos mitades, a través de la interacción de la cadena antisentido del siRNA con el complejo RISC (RNA-induced silencing complex). Las dos mitades del ARNm son posteriormente degradadas por la maquinaria celular, lo que impide la expresión del gen. Asimismo, los siRNA promueven la modificación del ADN, facilitando el silenciamiento de la cromatina, ya que favorecen la expansión de los segmentos de heterocromatina, a través del complejo RITS (RNA-induced transcriptional silencing).
Actualmente, la principal barrera para implementar terapias basadas en siRNA en la práctica clínica es la falta de un sistema de administración eficaz que pueda proteger las moléculas de ARN de la degradación de la nucleasa, administrarlas al tejido tumoral y liberarlas en el citoplasma para atacar las células cancerosas, todo ello sin inducir efectos adversos.
No obstante, ha habido un progreso considerable en el empleo de siRNA en el cáncer de mama a través de varios sistemas de administración. Se ha optimizado el diseño modificando el núcleo y la superficie de los vehículos de administración para abordar los diversos desafíos actuales. Hay varios estudios en los que se ha empleado la pegilación (incorporación de cadenas largas de polietilenglicol, PEG) para proteger el sistema de administración de la degradación por nucleasas; también se ha modificado para mejorar su capacidad de escapar del endosoma, mediante la incorporación de polímeros como la polietilenimina (PEI). En todos los casos, el tamaño de casi todos los vehículos de administración de nanopartículas es menor de 200 nm, lo que hace compleja la producción de nanopartículas utilizables en clínica.
La doble utilidad de los anticuerpos selectivos frente a antígenos tumorales o antígenos asociados a tumores reside en que pueden ser empleados directamente como agentes terapéuticos, bloqueando la actividad de proteínas vitales para la célula tumoral, “marcando” a las células tumorales para que las células citotóxicas del sistema inmune (NK) las destruyan selectivamente, actuando como elementos transportadores que permiten trasladar sustancias tóxicas que actúen exclusivamente en el ambiente local tumoral, o bien permite localizar anatómicamente a las células tumorales, tanto en el tumor primario como en otras localizaciones (metástasis). En la Tabla 1 están indicados los anticuerpos monoclonales antineoplásicos actualmente comercializados en España (febrero 2018).
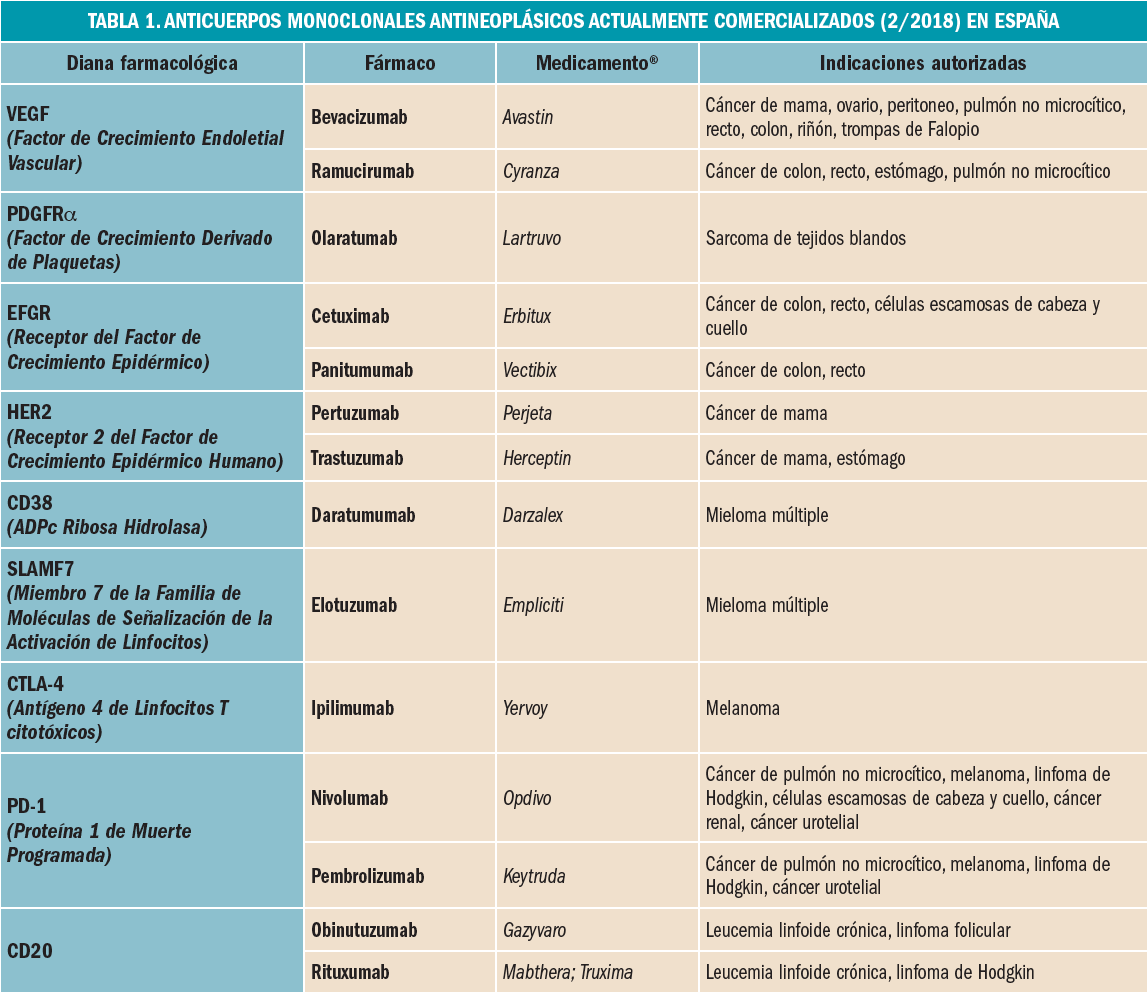
También se ha desarrollado anticuerpos portadores de toxinas que inducen la muerte celular, donde el anticuerpo tiene como misión favorecer el ingreso selectivo de la toxina a la célula tumoral para ejercer su efecto. Por ejemplo, el brentuximab vedotina (Adcetris®) es un anticuerpo monoclonal conjugado con un agente citotóxico (vedotina) que es capaz de provocar la apoptosis específicamente de células que presenten la proteína CD30 en su superficie. Ha sido autorizado para el tratamiento de pacientes adultos con linfoma de Hodgkin CD30+ en recaída o refractario; también está indicado para el tratamiento de pacientes adultos con linfoma anaplásico de células grandes sistémico en recaída o refractario. El anticuerpo se une selectivamente a la parte extracelular del CD30 presente en la superficie de la membrana, formando un complejo que es internalizado mediante endocitosis. Una vez en el interior celular, el fármaco accede al interior de los lisosomas donde sufre un proceso de escisión proteolítica liberando la vedotina (monometil auristatina E). La vedotina es un potente agente citotóxico que actúan interfiriendo con la polimerización de la tubulina; por ello, impide la formación del huso mitótico durante la división celular bloqueando el ciclo celular en fase G2/M, lo que provoca la activación de los mecanismos de apoptosis (muerte celular programada) y, en definitiva, la muerte celular.
Por su parte, el trastuzumab emtansina (Kadcyla®) es un conjugado de un anticuerpo dirigido contra HER2 (trastuzumab), unido mediante enlace covalente al inhibidor microtubular DM1 (un derivado de maytansina) a través del enlace tioéter estable (MCC4). La emtansina representa el complejo MCC-DM1. Cada molécula de trastuzumab está conjugada con una media de 3,5 moléculas de DM1. El medicamento está indicado para el tratamiento como agente único de pacientes adultos con cáncer de mama HER2 positivo localmente avanzado irresecable o metastásico, que han recibido previamente trastuzumab y un taxano por separado o en combinación.
La conjugación de DM1 a trastuzumab confiere selectividad al agente citotóxico por las células de tumores que sobreexpresan HER2, lo que potencia el transporte intracelular de DM1 directamente hacia el interior de las células malignas. La unión a HER2 causa la internalización de trastuzumab emtansina mediada por el receptor y la consiguiente degradación en lisosomas, lo que da lugar a la liberación de catabolitos citotóxicos que contienen DM1 (principalmente lisina-MCC-DM1). El DM1 es un agente citotóxico que actúa uniéndose a la tubulina, de forma similar a los taxanos y los alcaloides citotóxicos de la Vinca rosea, impidiendo su polimerización, lo que se traduce en la detención del ciclo celular en la fase G2/M, provocando la muerte celular por apoptosis. DM1 es, en términos equimoleculares, entre 20 y 200 veces más potente como citotóxico que los taxanos y los alcaloides de la vinca.
La radioinmunoterapia (RAIT) consiste en incorporar un radionúclido emisor de radiación ionizante letal pero de corto radio de influencia (milímetros o centímetros) a un anticuerpo dirigido contra un antígeno específico de un tumor. El efecto tumoricida se consigue por una baja pero continua dosis de radiación. Existan ya fármacos de este tipo autorizados, como el ibritumumab tiuxetan itrio (90Y) (Zevalin®), un anticuerpo monoclonal recombinante murino tipo IgG1 kappa específico para el antígeno CD20 de las células B, ligado a itrio radiactivo; está indicado para el tratamiento de consolidación después de la inducción de la remisión en pacientes con linfoma folicular no tratados anteriormente; asimismo, está indicado para el tratamiento de pacientes adultos con linfoma no Hodgkin (LNH) folicular de células B CD20+ en recaída o refractario a rituximab.
El anticuerpo (ibritumumab) se une específicamente al antígeno CD20, que se localiza en la superficie de los linfocitos B malignos y normales. Durante la maduración de las células B, el CD20 se expresa por primera vez en el estadio medio del linfoblasto B (prolinfocito B) y desaparece durante el estadio final de maduración de las células B hacia células plasmáticas. No se desprende de la superficie celular ni se interna en la célula al unirse al anticuerpo. El medicamento marcado con itrio-90 se une específicamente a los linfocitos B, incluyendo las células malignas que expresan el CD20. El isótopo itrio-90 es un emisor beta puro con un alcance medio de 5 mm aproximadamente. Esto le otorga su capacidad de destruir las células diana y las células vecinas. El tratamiento previo con rituximab es necesario para eliminar las células B circulantes, permitiendo que ibritumomab tiuxetan libere la radiación a los linfomas de un modo más específico. El tratamiento con ibritumomab tiuxetan marcado con itrio-90 (90Y) también produce depleción de las células B CD20+ normales, aunque se trata de un efecto temporal; la recuperación de las células B normales suele empezar en un plazo de 6 meses, normalizándose en los 9 meses siguientes al tratamiento.
Un concepto muy similar al anterior es el del tositumomab Iodo (131I), autorizado en Estados Unidos (Bexxar®), pero no en la Unión Europea. Como en el caso anterior, está indicado para el tratamiento de linfomas no-Hodgkin CD20+ en recaída o refractarios, de bajo grado, que hayan progresado durante o después de un tratamiento con rituximab.
Las células tumorales circulantes (circulating tumor cells; CTC) son células cancerosas que se desprenden del tumor primario y después de entrar en el torrente sanguíneo se detienen en sitios distales para iniciar la metástasis del cáncer. A pesar de que su primera noticia se remonta a 1869, en realidad el interés derivado del aislamiento de estas células solo se ha reactivado en el siglo XXI. Los motivos de este relativo abandono están claramente relacionados con los desafíos técnicos que suponen la detección y el aislamiento de estas células, ligada a su extraordinaria heterogeneidad y a su no menos excepcional escasez: una CTD entre uno y mil millones de células sanguíneas normales (1:106 -109). En este sentido, se han ido desarrollando – todavía con carácter preliminar – algunas estrategias como aislar CTC de los productos de leucoféresis, con el fin de poder utilizar volúmenes de sangre mucho mayores (en torno a 10 L) que el comúnmente utilizado para el análisis de CTC (5-10 mL) o, alternativamente, otros grupos están desarrollando productos de ingeniería tisular basados en andamios implantables (implantable scaffolds) que son capaces de capturar y atrapar CTC, con ayuda de siembras celulares o adyuvantes para modular el entorno inmune dentro del andamio.
Las células madres o pluripotenciales (CM, stem cells) son células relativamente indiferenciadas que pueden encontrarse en embriones (CME, embrionarias), algunos tejidos fetales, cordón umbilical, placenta (CMF, fetales) y en tejidos adultos (CMA, adultas). Son células pluri o multipotentes (en algunos casos totipotentes), según su grado de indiferenciación, que pueden dar lugar a distintos tipos celulares, dependiendo de su origen y plasticidad.
Las células madre embrionarias (CME) proceden de la masa celular interna de embriones en estadio de blastocisto; se trata de embriones de 5-6 días con 150-200 células, aproximadamente. La masa celular interna, origen de las CME en condiciones de cultivo in vitro, es la que daría lugar al feto in vivo, si el embrión se implantara definitivamente en la pared uterina y la gestación llegase a término.
Ha sido ampliamente demostrada la posibilidad de convertir células madre adultas (CMA) en células con alto grado de indiferenciación – pluripotentes – con las mismas características que las CME, mediante la transferencia de determinados genes implicados en la pluripotencia. Estas células reprogramadas llamadas iPS (induced Pluripotent Stem Cells) han supuesto una revolución en el campo de la pluripotencia y podrían ser de gran utilidad en la aplicación clínica si se demuestra que es una técnica eficaz y segura. Asimismo, se han desarrollado líneas de investigación que permiten la obtención de CM pluripotentes a través de otros tipos celulares o metodologías.
La utilización de terapia celular somática en oncología ya era objeto de investigación intensiva hace más de una década. En concreto, la capacidad de renovación de las células madres ha sido utilizada ampliamente en el tratamiento de leucemias y linfomas. Células madres hematopoyéticas con capacidad para diferenciarse en los diferentes tipos celulares sanguíneos, han sido utilizadas en conjunto con quimioterapia con el fin de reducir hematotoxicidad de esta esta última. Por otra parte, hay células madres con una especial atracción hacia tejidos alterados y, en concreto, para migrar hacia tumores, por lo cual se ha intentado utilizarlas como vehículo para transportar virus oncolíticos o producir sustancias tumoricidas.
La importancia de la terapia antitumoral celular somática ha adquirido un inusitado protagonismo terapéutico, dado que los espectaculares avances producidos en la farmacología antineoplásica se han visto frenados por el hecho de que frecuentemente es difícil obtener una alta concentración intratumoral de los fármacos, debido a la falta de selectividad, que deriva en la aparición de efectos adversos inaceptables. Ciertamente, se han producido avances notables en el desarrollo de terapias dirigidas específicamente al tumor, como son el uso de anticuerpos monoclonales o de terapia génica. Sin embargo, todavía no se ha llegado a alcanzar un nivel de señalización específica y presentan problemas como una escasa duración en la circulación sanguínea, una adherencia inespecífica a otros tejidos, incapacidad para salir del torrente circulatorio hacia las células diana o la propia activación del sistema inmune contra el fármaco.
Un fármaco antineoplásico ideal debería disponer de un vehículo terapéutico que le permitiese llegar específicamente al tumor, tras salir fácil y rápidamente del torrente sanguíneo, y no presentar problemas de inmunidad. Estas condiciones pueden ser cumplidas satisfactoriamente por algunos tipos celulares del propio paciente. En este sentido, ciertos tipos celulares (linfocitos, progenitores endoteliales, macrófagos, etc.) parecen ser reclutados selectivamente por el tumor durante su desarrollo. Por ejemplo, los tumores, durante su etapa proliferativa, inducen al tejido circundante a la formación de nuevos vasos sanguíneos al secretar factores de crecimiento como el VEGF (factor de crecimiento endotelial vascular; Vascular Endothelial Growth Factor) o el FGF (factor de crecimiento de fibroblastos; fibroblast growth factor). Otros factores secretados por las células tumorales, como el SDF-1 (factor 1 derivado de células estromales; stromal cell-derived factor 1), inducen la migración de ciertas células inmunes.
Las células del sistema inmune como los linfocitos, los macrófagos, las células NK – natural killers – y los eosinófilos, o las células relacionados con neoangiogénesis tumoral, son las opciones más obvias para ser utilizadas como vehículos celulares, pero otros tipos de células también se podrían utilizar, cómo por ejemplo las propias células tumorales o las células madre adultas. En cuanto a la capacidad de las células del sistema inmune para ser utilizadas como vehículos celulares de agentes antitumorales. Así se han utilizado leucocitos de sangre periférica, cultivados in vitro en presencia de diversas citocinas, en especial IL-2, para obtener linfocitos activados. Se han realizado ensayos clínicos combinando la administración de interleucina 2 (IL-2; aldesleukina) y de linfocitos activados, observándose que los efectos antitumorales se han correlacionado con la dosis de IL-2 y el número de células administradas.
Por otro lado, los linfocitos que infiltran los tumores poseen una actividad única antitumoral y pueden ser expandidos ex vivo también con IL-2. Estas células se han utilizado ya en terapias inmunomoduladoras, especialmente con melanomas, obteniéndose respuestas parciales clínicas en los pacientes tratados con la infusión de estos linfocitos. También se ha pensado en utilizar células NK, con la ventaja de que pueden ser obtenidas fácilmente de la sangre periférica de los pacientes; sin embargo, no se ha encontrado evidencia de beneficios en ensayos clínicos con las células NK, a pesar de haberse demostrado su acumulación dentro de las metástasis de los pacientes. Algo similar ocurre con los macrófagos, los cuales parecen ser eficaces a la hora de localizar las metástasis y acumularse alrededor de éstas pero no parecen afectar a los tumores primarios.
La utilización de células tumorales como vehículos terapéuticos podría parecer paradójica. Y, sin embargo, existen datos que indican que puede ser efectiva. Esta aplicación se basa en la observación en modelos animales de que la administración de determinadas células tumorales hacía que éstas se localizasen preferentemente en las áreas tumorales. Adicionalmente, cuando estas células malignas son dopadas con agentes terapéuticos se han obtenido reducciones significativas del tamaño del tumor.
La idea de utilizar las propias células tumorales como vehículos terapéuticos se basa en el conocimiento de que, en general, las metástasis se presentan en órganos determinados según el tipo de tumor. Esta localización preferencial se debe al hecho de que las células tumorales que viajan en la circulación sanguínea responden a los factores producidos en los diversos órganos, a las señales del endotelio y a su capacidad de anidar en sitios específicos. En modelos preclínicos se ha llegado a demostrar que las células tumorales infundidas se localizan en las lesiones metastásicas preexistentes y que, además, transduciendo estas células con genes suicidas o virus oncolíticos, es posible obtener una remisión significativa de las lesiones tumorales.
Hasta hace algunos años se consideraba que las células precursoras adultas específicas de un órgano se hallaban restringidas a ese linaje celular. Hoy se sabe que esto no es así, al menos no lo es siempre y para todos los tejidos; en efecto, hay células precursoras adultas que pueden diferenciarse hacia multitud de tejidos y tipos celulares. Aprovechando esta excepcional pluripotencialidad, se ha comprobado experimentalmente que algunas células madre podrían localizarse en los tumores, en especial las células madre endoteliales, ya que el tumor al crecer necesita vascularización, promoviendo el reclutamiento de células progenitoras endoteliales. La utilidad potencial en oncología es obvia.
Las células endoteliales recubren normalmente la superficie interna de los vasos sanguíneos y constituyen una barrera selectiva entre la sangre y el resto de los tejidos. A pesar de tener un fenotipo muy simple, las células endoteliales desempeñan un papel crítico en una amplia variedad de procesos fisiológicos, incluyendo el mantenimiento de la fluidez de la sangre, el tráfico de células sanguíneas, la inmunidad innata y adaptativa, y la coagulación. Atendiendo a estas características, no es extraño que el potencial interés de la investigación de las células endoteliales en el contexto del cáncer.
La observación de que los tumores trasplantados en ratones eran capaces de captar capilares del huésped data de 1945. A principios de los años 70 se consiguió aislar un factor asociado al tumor que estimula la formación de nuevos vasos sanguíneos tumorales y se propuso una intervención terapéutica para bloquear este factor. El paradigma basado en el bloqueo de la formación de nuevos vasos sanguíneos tumorales (neovasos), para bloquear la progresión del tumor fue el punto de partida de una investigación extensa que todavía está progresando actualmente. De hecho, durante las últimas décadas se han aprobado varios fármacos que actúan alterando el endotelio vascular tumoral principalmente mediante el bloqueo del factor de crecimiento endotelial vascular (VEGF) y sus receptores para el tratamiento de varios cánceres avanzados y en otras condiciones patológicas con predominio neovascular (bevacizumab, aflibercept, ramucirumab, ranibizumab, etc.).
Sin embargo, a pesar de los estudios preclínicos muy exitosos, los tratamientos antiangiogénicos han proporcionado sólo beneficios limitados en pacientes con cáncer, debido a la existencia de mecanismos de resistencia a los tratamientos anti-VEGF, tales como la activación de otras vías angiogénicas o el empleo de modos alternativos de vascularización tumoral. Aunque que una parte de la investigación en curso se centra en eludir estos mecanismos de resistencia, cada vez es mayor el esfuerzo investigador para el diseño de terapéuticas basadas en las características específicas del endotelio vascular tumoral.
Las células endoteliales tumorales influyen en la respuesta inmunitaria del huésped controlando la penetración de las células inmunes en el tumor y modulando su actividad. En consecuencia, los enfoques que estimulan el reclutamiento y la activación de linfocitos por las células endoteliales tumorales están siendo objeto de investigación, principalmente a través de la promoción del reclutamiento/activación de linfocitos (incrementando la expresión de moléculas de adhesión y co-estimuladoras, y la destrucción de células tumorales mediada por linfocitos) y reduciendo la capacidad de penetración y extravasación de las células tumorales, con reduciría su potencial metastásico.
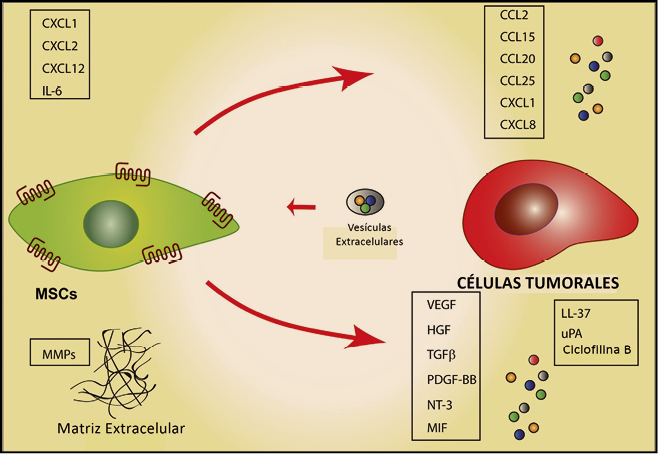
Figura 3. Factores que favorecen la aproximación de células mesenquimatosas (MSC) a las células tumorales. Las células cancerosas secretan una serie de quimiocinas (CCL2, CCL15, CCL20, CCL25, CXCL1, CXCL8) que atraen las MSC a través de los receptores de quimiocinas específicos en su superficie. Las quimiocinas pueden ser liberadas directamente en el medio extracelular o incorporadas en vesículas. Otras citocinas, incluyendo VEGF, HGF, TGF, PDGFF-BB, NT-3, MIF, y factores como la LL-37, UPA y ciclofilina B liberadas por las células tumorales afectan el tropismo de las MSC. Por otra parte, en interacción con las células cancerosas, las MSC secretan citocinas tales como CXCL1, CXCL2, CXCL12 o IL-6 y metaloproteinasas (MMPs); estas últimas tienen la capacidad de facilitar la degrdación de la matriz extracelular y la migración.
Las células madre mesenquimales (mesenchymal stem cells, MSC) o células estromales mesenquimales representan un amplio y diverso conjunto de células. De hecho, las MSC se han aislado a partir de diferentes tipos de tejidos, incluyendo la médula ósea, la sangre del cordón umbilical, el tejido adiposo e incluso también sangre periférica, hígado fetal, pulmón, líquido amniótico o placenta. Todos estos tipos de MSC conservan características similares en lo que se refiere a la capacidad de adherirse fuertemente a las superficies de plástico, se caracterizan por la presencia de marcadores de la superficie (CD14, CD11b, CD19, CD79α, CD34, CD45, HLA-DR, CD73+, CD90+, CD105+) y mantienen el potencial de diferenciarse en condrocitos, osteoblastos y adipocitos bajo condiciones estándar in vitro. A pesar de algunas de las características comunes con respecto al potencial de inmunofenotipo y la diferenciación de las MSC, estas células varían en función de su interacción directa con otras células o a través de una manera paracrina según el microambiente circundante.
Las quimiocinas, o más generalmente las citocinas, se encuentran entre los principales actores responsables de la migración de MSC a los tumores (Figura 3), lo cual no es sorprendente, ya que las quimiocinas se producen en abundancia en los sitios del tumor. Además, el tratamiento de tumor también podría promover la migración de las MSC hacia tumores. En este sentido, se ha comprobado que la irradiación de células de tumor de mama potencia la liberación de TGFb1, VEGF y el factor de crecimiento derivado de plaquetas BB (PDGF-BB) por las células tumorales, lo que mejora la migración de las MSC hacia las células tumorales.
De igual manera, las células madre mesenquimales se pueden alojar selectivamente en los tumores. De hecho, se ha demostrado la presencia de estas células en el interior de los tumores después de que fueran administradas en animales con melanoma. Además, se obtuvo una prolongación de la supervivencia cuando se indujo la secreción de interferón gamma por las células madre mesenquimatosas. En la misma línea, se está trabajando con células precursoras nerviosas, capaces de anidar en los tumores primarios del cerebro. Hay también datos demostrando una reducción de tumores establecidos cuando se usaron estás células transducidas con genes suicidas y virus oncolíticos.
En la Tabla 2 se recogen los medicamentos que han sido clasificados por la EMA5 como de terapia celular somática en indicaciones anticancerosas hasta febrero de 2018, aunque esto no equivale a ningún aval de la plausibilidad del producto, incluido el modo de acción o indicaciones terapéuticas alegadas por el solicitante.
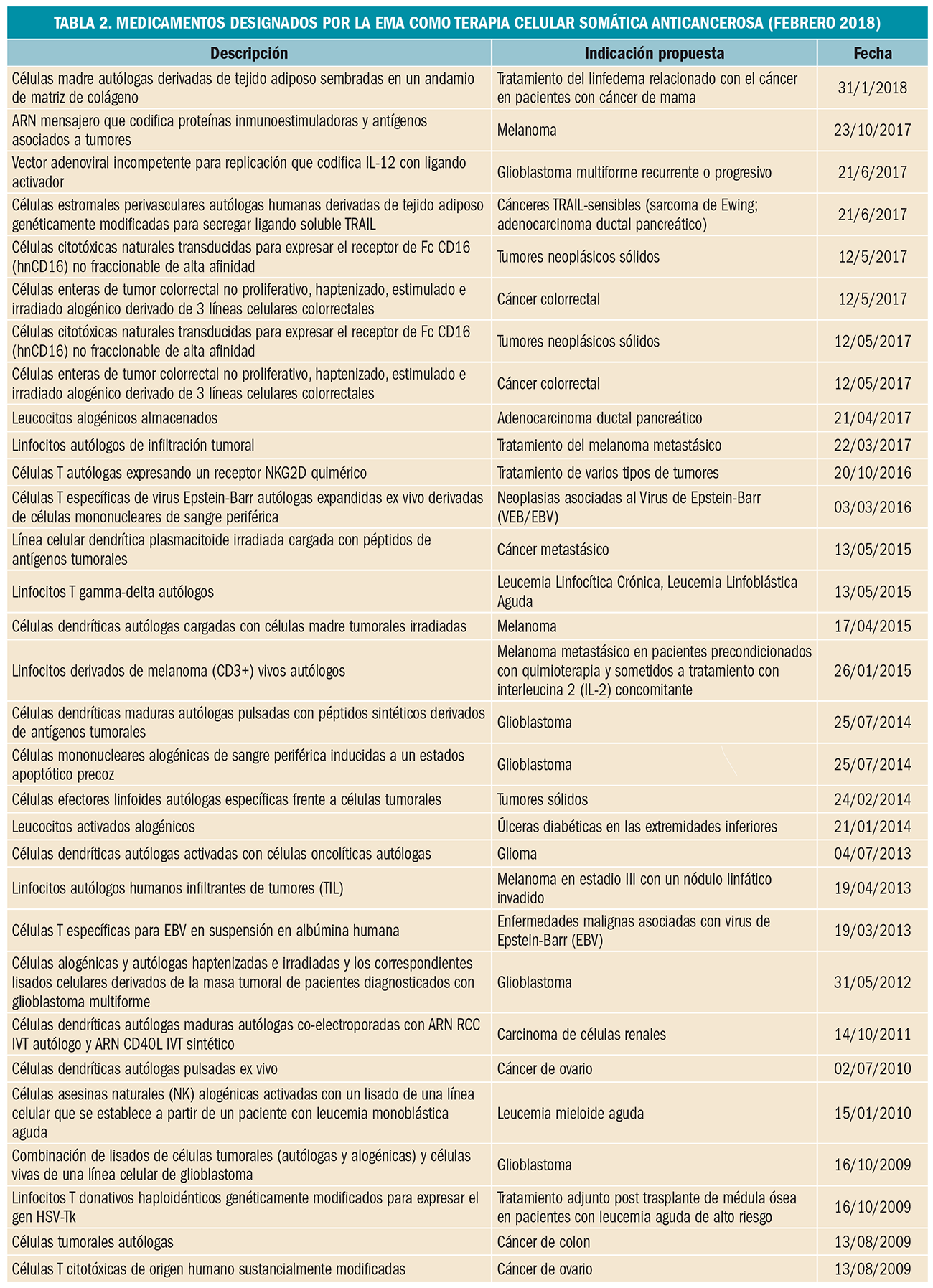
La transferencia o terapia celular adaptativa(adoptive T Cell therapy; ACT) es un tipo de inmunoterapia experimental. Uno de sus métodos consiste en extraer las células T citotóxicas que han invadido el tumor del paciente, conocidas como linfocitos infiltrados en el tumor. Se seleccionan las células con mayor actividad antitumoral, se cultivan – expanden – grandes poblaciones de estas células en el laboratorio y se activan con citocinas. El siguiente paso es volver a administrar las células al paciente. El evidente objetivo del procedimiento es que, si las células tumorales suprimen la actividad de los linfocitos infiltrados en el tumor que son generados de forma natural, sería posible contrarrestar esa supresión exponiendo al tumor a cantidades masivas de linfocitos infiltrantes activados
Alternativamente, especialmente para aquellos tipos de cánceres en los que linfocitos T específicos antitumorales son menos espontáneos, los linfocitos T pueden expandirse a partir de células T modificadas genéticamente por el paciente que expresan un receptor de células T específico de tumor (T cell receptor; TCR) o un TCR quimérico (híbrido) compuesto de una fracción de inmunoglobulina (Ig) sintética fusionada con los componentes de señalización TCR, llamado receptor CAR (chimeric antigen receptor; CAR). Las proteínas CAR facilitan la unión de las células a unas proteínas específicas en la superficie de las células cancerosas, lo cual activa a las células T para atacarlas. Y este es el fundamento de la terapia con células T y CAR (TCAR).
A pesar del entusiasmo inicialmente despertado por la inmunoterapia celular adaptativa, han surgido muchos obstáculos que deben ser abordados antes de que la TCAR se una al arsenal terapéutico disponible para el tratamiento de tumores sólidos. En los tipos de tumores que tienen más de un antígeno asociado al tumor (TAA), existe el problema de determinar cuál es el objetivo óptimo para minimizar la inmunoevasión tumoral. Cuando se expresa más de un TAA, ¿podrían las fracciones variables de inmunoglobulina contra varios antígenos ser manipulados en una configuración de “activación y/o” para enfrentarse a la heterogeneidad del tumor o para mejorar la seguridad? El requisito previo para el preacondicionamiento no mieloablativo también debe ser rigurosamente evaluado. Asimismo, deben explorarse modelos preclínicos apropiados y mecanismos de eficacia y resistencia a la terapia con células T y CAR, idealmente antes de plantearse un desarrollo clínico completo.
En definitiva, la terapia con células T y CAR ha surgido como una prometedora opción inmunoterapéutica para tumores sólidos y, de hecho, varios candidatos prometedores están en fase inicial de ensayos clínicos. A pesar de la heterogeneidad tumoral y antigénica, ya se han identificado varios antígenos asociados a tumores (TAA), tales como MUC-16, GD2, EGFRIII, mesotelina y PSMA, como objetivos para la terapia. En concreto, se han notificado respuestas clínicas en un pequeño subgrupo de tumores sólidos, aunque se requieren mayores tasas de respuesta ya que la eficacia está limitada por varios factores intrínsecos y extrínsecos, incluyendo el tráfico deficiente al sitio del tumor y un microambiente de tumor inmunosupresor. Por ello, se están investigando opciones de ingeniería genética para optimizar el diseño de CAR, como las células T CAR blindadas, o enfoques combinatorios con terapia citotóxica, terapia dirigida e inmunomoduladores.
Las vacunas anticancerosas son sustancias capaces de estimular o restaurar la capacidad del sistema inmunitario para prevenir, detectar y eliminar células tumorales, de igual manera que las vacunas tradicionales ayudan a prevenir las infecciones producidas por microoganismos. De acuerdo con ello, pueden distinguirse dos tipos esenciales de vacunas anticancerosas:
El virus del papiloma humano (VPH) constituye un amplio grupo viral (se conocen cerca de 200 genotipos diferentes) que infecta de fundamentalmente a células epiteliales humanas; cerca de 40 de estos genotipos. El ADN del VPH se detecta en el 99,7 % de los cánceres cervicales y las mujeres infectadas tienen un riesgo de padecer este cáncer entre 50 y más de 150 veces mayor que el de las no infectadas, hasta el punto de actualmente se acepta que el cáncer de cérvix es una enfermedad de transmisión sexual. No se conoce ninguna otra asociación más fuerte entre una infección vírica y un cáncer humano que la que se da entre la infección cervical por el VPH y el cáncer de cérvix uterino.
Entre el 60% y el 80% de los cánceres cervicales en todo el mundo están asociados con dos tipos de VPH de alto riesgo (16/18) y el resto mayoritariamente a los tipos 32 y 51; por su parte, casi el 90% de las verrugas genitales están asociados con dos tipos de VPH de bajo riesgo oncogénico (6/11). Cada año alrededor de 500.000 mujeres desarrollan cáncer cervical invasivo en todo el mundo. Los factores de riesgo asociados con la infección por VPH son una edad temprana en el primer coito, mayor número de parejas sexuales, tabaquismo e historial de infección por virus del herpes simple (VHS). Alrededor del 75% de las mujeres sexualmente activas desarrollan una infección por el VPH, más frecuentemente poco después de su debut sexual, aunque la mayoría de estas infecciones (hasta un 90%) desaparecen espontáneamente por acción del sistema inmune. La minoría restante puede progresar desde la infección aguda hasta el cáncer de cuello uterino, un proceso que requiere habitualmente varias décadas y pasa por estadios intermedios: cáncer in situ y lesiones precancerosas denominadas neoplasia cervical intraepitelial (cervical intraepithelial neoplasia; CIN) de creciente gravedad, de CIN1 a CIN3, aunque es posible una regresión espontánea de las lesiones en cualquier nivel.
En España, la vacunación frente al VPH está incluida en los calendarios vacunales oficiales. Existen tres vacunas frente al VPH, Gardasil®, Gardasil-9® y Cervarix®, constituidas por diferentes combinaciones de formas recombinante de la partícula L1 de la cápside de los diversos tipos del virus del papiloma humano (VPH) potencialmente relacionados con el cáncer de útero. La partícula L1 está normalmente formando complejos de cinco unidades – pentámeros – y es la principal proteína constituyente de la cápside o cubierta viral del VPH y como tal puede mimetizar el comportamiento antigénico del virus completo. La cápside del VPH tiene un tamaño de aproximadamente 60 nm de diámetro, con simetría icosaédrica, y está formada por el ensamblaje de 72 pentámeros de L1.
Gardasil® contiene proteína L1 procedente de VPH de los tipos 6 (20 µg), 11 (40 µg), 16 (40 µg) y 18 (20 µg). Está indicada a partir de los 9 años de edad para la prevención de lesiones genitales precancerosas (cervicales, vulvares y vaginales), lesiones anales precancerosas, cáncer cervical y cáncer anal relacionados causalmente con ciertos tipos oncogénicos del virus del papiloma humano (VPH); asimismo está indicado en la prevención de verrugas genitales (condiloma acuminata) relacionadas causalmente con tipos específicos del VPH. La posología recomendada es, en administración intramuscular, en individuos de 9-13 años de dos dosis (0,5 mL) separadas por dos 6 meses (si la segunda dosis se administra antes, deberá emplearse una tercera dosis; como alternativa, puede seguirse una pauta de tres dosis (0,5 mL a los 0, 2, 6 meses). En individuos de 14 años en adelante se debe utilizar siempre la pauta de tres dosis. Esta pauta requiere que la segunda dosis se administre al menos un mes después de la primera, la tercera se administre al menos tres meses después de la segunda y que las tres dosis se administren dentro del periodo de 1 año.
Gardasil-9® contiene proteína L1 procedente de VPH de los tipos 6 (30 µg), 11 (40 µg), 16 (60 µg) y 18 (40 µg), 31 (20 µg), 33 (20 µg), 45 (20 µg), 52 (20 µg) y 58 (20 µg). Está indicada para la inmunización activa de individuos a partir de los 9 años de edad frente a las lesiones precancerosas y canceres que afectan al cuello de útero, vulva, vagina y ano causados por los tipos del VPH de la vacuna, y las verrugas genitales (condiloma acuminata) causados por tipos específicos del VPH.
La posología recomendada es en individuos de 9 a 14 años de edad (inclusive) en el momento de la primera inyección: Dos dosis, la segunda de las cuales se debe administrar entre los 5 y 13 meses después de la administración de la primera; si la segunda se administra antes de 5 meses, se debe administrar siempre una tercera dosis. También puede administrarse siguiendo una pauta de tres dosis (0, 2, 6 meses), en la que la segunda se debe administrar al menos un mes después de la primera y la tercera se debe administrar al menos tres meses después de la segunda; las tres dosis se deben administrar dentro del periodo de 1 año. En individuos de 15 años en adelante en el momento de la primera inyección: Tres dosis (0, 2, 6 meses), la segunda de las cuales se debe administrar al menos un mes después de la primera y la tercera al menos tres meses después de la segunda; las tres dosis se deben administrar dentro del periodo de 1 año.
Cervarix® contiene proteína L1 procedente de VPH de los tipos 16 (20 µg) y 18 (20 µg). Está indicada a partir de los 9 años de edad para la prevención de lesiones ano-genitales premalignas (cervicales, vulvares, vaginales y anales) y cáncer de cérvix y ano causados por determinados tipos oncogénicos del virus del papiloma humano (VPH). La posología recomendada es, en administración intramuscular, en individuos de 9-14 años de dos dosis (0,5 ml) separadas por entre 5 y 13 meses, si se administrase antes de 5 meses, se requeriría una tercera dosis. En individuos de 15 años en adelante se debe utilizar siempre la pauta de tres dosis (0, 1 y 6 meses). La segunda dosis se puede administrar entre1 mes y 2,5 meses después de la primera dosis y la tercera dosis entre 5 y 12 meses después de la primera dosis.
Los ensayos clínicos con las vacunas bivalente y tetravalente demuestran la seguridad, inmunogenicidad y eficacia de ambas vacunas en la prevención de infecciones por VPH y lesiones precancerosas, especialmente si se administran en adolescentes antes de la exposición al virus. Los ensayos clínicos con la vacuna nonavalente también muestran su seguridad, inmunogenicidad y eficacia en la prevención de infección y enfermedad asociada con los tipos vacunales, y sugieren el potencial de la vacuna para reducir la carga de enfermedad asociada al VPH. Informes poscomercialización en países con programas de vacunación sistemática y altas coberturas sugieren una efectividad muy alta a nivel poblacional, con descensos en la prevalencia de los VPH relacionados con la vacuna, y en la incidencia de verrugas genitales y lesiones cervicales de alto grado.
Tanto la vacuna tetravalente (Gardasil®) como la bivalente (Cervarix®) han demostrado evidencia de diversos grados de protección cruzada frente a genotipos de VPH no vacunales. La protección cruzada es de especial importancia, puesto que los tipos de VPH no incluidos en estas vacunas se asocian aproximadamente con el 30% de los cánceres cervicales a nivel mundial.
Dado que el riesgo de exposición al VPH persiste durante toda la vida sexual activa, la duración de la protección a largo plazo de las vacunas VPH se convierte en uno de los tema clave. En el caso de Gardasil®, el seguimiento hasta 5 años de los participantes en los estudios de eficacia revela la no aparición de lesiones cervicales precancerosas ni verrugas genitales en los vacunados. Asimismo, la respuesta inmune frente a VPH16 se mantenía, mientras el título de anticuerpos frente a VPH18 caía con el paso del tiempo, asimilándose a la infección natural. En el seguimiento de los estudios en niños y niñas de 9-15 años tampoco aparecen casos de enfermedad a los 8 años de la vacunación, manteniéndose el título de anticuerpos entre el 64% y el 100% según el tipo de VPH. La monitorización indica que a los 8 años de seguimiento en países nórdicos (Dinamarca, Suecia, Noruega e Islandia) no ha habido ningún caso de CIN2+, ni de cáncer de vulva o vagina relacionado con los tipos vacunales entre las mujeres vacunadas, y que la respuesta inmune se mantiene por encima del 90% para los cuatro tipos de VPH más oncogénicos.
Resultados similares se han publicado para Cervarix®. El seguimiento a largo plazo refleja que a los 6,4 años de seguimiento la eficacia de la vacuna frente a CIN2+ se mantiene en cifras del 100% para lesiones relacionadas con los tipos vacunales, y que los títulos de anticuerpos frente a VPH16 y 18 se mantienen en niveles más de 12 veces superiores a los inducidos por la infección natural. Un estudio refleja que, hasta los 9,4 años de seguimiento, los niveles de anticuerpos inducidos por la vacuna frente a VPH16 y 18 se mantienen en títulos varias veces superiores a los alcanzados con la infección natural
Aunque algunos resultados podrían sugerir que la vacuna bivalente (Cervarix®) genera una mayor respuesta inmune que la vacuna tetravalente (Gardasil®), en general parece ser que estas diferencias podrían ser debidas a la diversidad de las pruebas de inmunoanálisis utilizadas, y que a pesar del descenso en los títulos de anticuerpos, la eficacia profiláctica de la vacuna tetravalente frente a lesiones anogenitales asociadas con HPV18 se mantiene en el tiempo.
Los resultados de seguridad de los ensayos clínicos de ambas vacunas respaldan que estas vacunas son generalmente bien toleradas y tienen un adecuado perfil de seguridad, siendo muy pocos los abandonos. Los efectos adversos locales más comunes relacionados con las vacunas fueron el dolor transitorio de intensidad leve a moderada, el eritema y la inflamación en el lugar de inyección. Los síntomas sistémicos más comunes potencialmente relacionados con la vacuna fueron fiebre, fatiga, dolor de cabeza y dolor muscular.
Desde su comercialización en 2007, más de 50 países han incluido las vacunas VPH en sus programas nacionales de inmunización. Sin embargo, el impacto poblacional respecto al cáncer de cuello de útero y otros cánceres relacionados con el VPH no se observará en términos reales hasta 30-50 años después del inicio del programa de vacunación, debido al largo intervalo de tiempo que transcurre entre la infección y el cáncer. No obstante, los resultados de un metaanálisis reflejan que, en países con coberturas vacunales de más del 50%, la prevalencia de infección por los VPH16 y 18 en niñas de 13 a 19 años ha descendido en un 68% en el periodo posvacunal en comparación con el periodo prevacunal. La incidencia de verrugas genitales ha descendido el 61% en niñas de 13 a 19años y se ha observado un efecto de protección de rebaño en mujeres de 20 a 39 años y en niños menores de 20 años. En países con coberturas inferiores al 50% también se han registrado descensos significativos en la infección y verrugas genitales debidas a los tipos vacunales, pero no se acompañan de efectos de protección cruzada, ni de inmunidad de grupo.
Cabe destacar que ambas son vacunas profilácticas sin eficacia terapéutica. En mujeres jóvenes, las dos vacunas (bi y tetravalente) han demostrado ser altamente eficaces en la prevención de CIN2+ (neoplasia intraepitelial cervical) relacionados con los VPH de alto riesgo (VPH16 y 18); adicionalmente, ambas vacunas, pero en especial la bivalente (Cervarix®), han demostrado cierto grado de protección cruzada frente a otros tipos de VPH. Las vacunas también son eficaces en la prevención de VIN2+ (neoplasia intraepitelial vulvar) y VaIN2+ (neoplasia intraepitelial vaginal) relacionados con los VPH16 y 18. La vacuna tetravalente (Gardasil®) es eficaz en la prevención de verrugas genitales relacionadas con los VPH6 y 11, tanto en mujeres como en hombres. La vacuna bivalente protege frente a infección anal y de cavidad oral relacionada con los tipos vacunales. Finalmente, los datos disponibles para la vacuna nonavalente (Gardasil-9®) confirman el satisfactorio perfil de seguridad e inmunogenicidad de la vacuna en mujeres jóvenes y una alta eficacia de la vacuna frente a CIN2+, VIN2+ y VaIN2+ relacionado con los VPH31, 33, 45, 52 y 58, y una respuesta de anticuerpos frente a VPH6, 11, 16 y 18 no inferior a la generada por la vacuna tetravalente (Gardasil®).
Según la Organización Mundial de la Salud (OMS), en 2015 había alrededor de 257 millones de personas infectadas con el virus de la hepatitis B y otros 71 millones con el de la hepatitis C, causando 1,34 millones de muertes, casi el mismo número que el VIH-Sida o la tuberculosis, pero mientras el número de fallecimientos por estas dos últimas enfermedades ha caído en los últimos años, el de la hepatitis sigue creciendo (un 22% entre 2000 y 2015); de estos 1,34 millones de muertes, 720.000 fueron causadas por cirrosis y 470.000 por cáncer de hígado.
La evolución crónica de las hepatitis virales B y C provocan paulatinamente lesiones en el hígado que se traducen en la formación de tejido cicatricial. En una primera etapa, el tejido con núcleos cicatriciales evoluciona hacia una fibrosis que impide el desarrollo normal de sus funciones metabólicas; posteriormente, la fibrosis evoluciona a cirrosis y la secuencia evolutiva acaba frecuentemente en un hepatocarcinona. Actualmente, se estima que entre el 20 y el 35% de los infectados crónicamente por el virus de la hepatitis C desarrollará un cáncer de hígado.
En España se diagnostican cada año unos 4.300 nuevos casos de cáncer de hígado, afectando 3.000 a varones y 1.300 a mujeres. Es significativo que hasta un 75% de las personas que desarrollan un hepatocarcinoma dan positivo en el virus de la hepatitis C, lo que enfatiza la importancia de prevenir la infección crónica por los virus de la hepatitis B y C, en tanto que constituyen los factores de riesgo más comunes del cáncer hepático; por consiguiente, la prevención o, en su caso, el tratamiento de la hepatitis asociados a estos tipos virales es fundamental para prevenir el cáncer de hígado.
La vacunación es la mejor forma de prevenir el VHB, aunque la inmunización sólo es eficaz en los individuos que no han estado expuestos previamente al virus; todavía no existe una vacuna contra el VHC, aunque la quimioterapia antiviral actualmente disponible (sofosbuvir, etc.) permite la erradicación viral y la curación clínica de más del 95% de los pacientes e incluso es prácticamente del 100% en determinadas subpoblaciones.
En un reciente (2017) y amplio estudio realizado en Taiwán se estudió específicamente la importancia de la inmunización de los lactantes contra el VHB para impedir el desarrollo de carcinoma hepatocelular (CHC) en su vida de adulto. Para ello, se recogieron datos de registro de CHC correspondientes a 1.509 pacientes (6-26 años de edad) diagnosticados entre 1983 y 2011. De los 1.509 pacientes con CHC, 1.343 nacieron antes y 166 nacieron después de iniciarse el programa de vacunación sistemática contra el VHB.
Los datos epidemiológicos mostraron que la incidencia global de CHC fue de 0,92 casos por 100.000 años-persona en la cohorte no vacunada y de 0,23 en la vacunada. En concreto, el riesgo relativo de contraer un carcinoma hepatocelular (vacunados vs. no vacunados) en pacientes de 6-9 años de edad se redujo en un 74% (RR=0,26; IC95% 0,17 a 0,40), en un 66% en pacientes de 10-14 años de edad (0,34; IC95% 0,25 a 0,48), en un 63% para los de 15-19 años (0,37; IC95% 0,25 a 0,51) y en un 58% para los de 20-26 años (0,42; IC95% 0,32 a 0,56). El riesgo relativo fue significativamente menor en las cohortes más modernas que en las anteriores (nacidos en 1992-2005 vs. 1986-1992 y 1986-1992 vs. 1984-1986).
En España, la vacunación sistemática de la hepatitis B está contemplada en los calendarios vacunales oficiales. Están comercializadas las vacunas Engerix B®, Fendrix®, HBVaxPro®. Adicionalmente, Twinrix®, incluye los antígenos de superficie de los virus de la hepatitis A (VHA) además de los del VHB; asimismo, algunas vacunas hexavalentes (hepatitis B, difteria, tétanos, tos ferina, Haemophilus influenzae y poliomielitis) también contienen antígenos de superficie de VHB: Infanrix Hexa® y Hexyon®.
Uno de los motivos por los que la inmunoterapia es considerada como la principal esperanza del tratamiento del cáncer es el éxito de cosechado por los anticuerpos monoclonales específicos, es decir, la inmunoterapia específica pasiva. Se espera que las vacunas terapéuticas contra el cáncer lleguen a tener incluso un mayor impacto sobre el sistema inmunitario como inmunoterapia específica activa, lo que incrementaría la seguridad y la eficacia de los tratamientos, especialmente en comparación con la quimioterapia anticancerosa. Lamentablemente, sin embargo, por el momento las pruebas clínicas de eficacia para la mayoría de las vacunas terapéuticas contra el cáncer actualmente desarrolladas todavía son muy limitadas, cuando no abiertamente desalentadoras; en este sentido, una reciente revisión retrospectiva mostró que el 74% (17/23) de los estudios de fase 3 (confirmatorios de eficacia y seguridad) sobre vacunas terapéuticas contra el cáncer no mostraron estadísticamente ningún efecto significativo, aplicando los criterios o variables de valoración convencionales, aunque se espera que la pormenorización inmunológica ayudará a mejorar la probabilidad de éxito en ensayos de fase 3, al reducir considerablemente la heterogeneidad de la población de estudio.
De hecho, la heterogeneidad de la población estudiada es considerada como el principal factor que contribuyó al fracaso de las vacunas terapéuticas contra el cáncer en los estudios de fase 3 realizados hasta ahora. La utilización de criterios de inclusión en los ensayos clínicos que son demasiado amplios o que carecen de estratificación, puede impedir la detección de respuestas de eficacia significativa con este tipo de vacunas terapéuticas. Por ello, la restricción de la población de pacientes exclusivamente a aquellos para quienes se puede predecir la eficacia con anticipación, mejorará la probabilidad de éxito de estos estudios.
Por lo tanto, la identificación de factores pronósticos para las vacunas terapéuticas contra el cáncer es fundamental para su éxito futuro. Entre esos factores pronósticos cabe destacar la categorización detallada en estadios tumorales, como un factor pronóstico importante. Asimismo, las características de cualquier pretratamiento o combinación de fármacos deben tenerse en cuenta, ya que pueden tener un impacto no sólo sobre la carga tumoral, sino también sobre el sistema inmunitario. Por último, la correlación entre la respuesta inmune y el resultado clínico debe ser reevaluada; actualmente, la respuesta inmune se utiliza principalmente como un marcador subrogado para predecir el resultado clínico, sin embargo la respuesta inmune también debería ser considerado como un factor pronóstico y la condición inmune del paciente antes del tratamiento debe evaluarse para reducir o estratificar la población de pacientes y reducir la heterogeneidad del paciente en un estudio.
Los primeros intentos de realizar una vacuna anticancerosa de tipo terapéutico se realizaron mediante inyecciones de células tumorales autólogas – provenientes del propio paciente – o de proteínas específicas de tumores administradas solas o con un adyuvante, mientras que más recientemente se ha desarrollado una estrategia alternativa mediante la estimulación directa de células dendríticas ex vivo con antígenos tumorales específicos (neoantígenos), con antígenos asociados a tumores (TAA) o incluso usando un lisado celular tumoral completo, que se vuelven a infundir en los pacientes. Las células dendríticas pueden expandirse o cultivarse ex vivo mediante el aislamiento de precursores de monocitos a partir de sangre periférica, seguida de incubación con factores de crecimiento específicos y citocinas tales como factores estimulantes de colonias como el GM-CSF, interleucinas (IL-4, IL-3), ligando Flt3, c-Kit, etc.
Actualmente, las formulaciones de vacunas anticancerosas terapéuticas se dividan en tres grandes grupos:
Se considera que la forma más eficaz de inmunidad antitumoral en seres humanos debe estar asociada con la presencia de linfocitos T dirigidos contra los neoantígenos, es decir, los antígenos tumorales específicos. Como ya se ha mencionado, los neoantígenos son intensamente inmunogénicos porque no están presentes en los tejidos normales y, por tanto, evitan la tolerancia tímica central. Aunque los neoantígenos fueron concebidos como objetivos óptimos para una respuesta inmune antitumoral, su descubrimiento y evaluación sistemática sólo se hizo factible con la reciente disponibilidad de técnicas de secuenciación masiva paralela para la detección de todas las mutaciones codificantes dentro de los tumores y de los métodos de aprendizaje automático para predecir de forma fiable aquellos péptidos mutados con unión de alta afinidad de moléculas autólogas de antígeno leucocitario humano (HLA). Así pues, la vacunación con neoantígenos debería expandir las poblaciones de células T neoantigénicas preexistentes e inducir un repertorio más amplio de nuevas especificidades de células T en pacientes con cáncer, permitiendo un mayor control antitumoral.
Las vacunas terapéuticas anticancerosas basadas en células dendríticas han sido las primeras en alcanzar el estatus de medicamento autorizado oficialmente, al menos en algunos países, entre los que cabe incluir Estados Unidos, Suiza, Australia, Cuba, Perú y Rusia. Aunque la inmunoterapia del cáncer con células dendríticas está en pleno desarrollo, todavía quedan por resolver importantes cuestiones de eficacia, seguridad, producción, etc.
Básicamente, consiste en extraer células progenitoras de células dendríticas del paciente (a partir de muestras tumorales o por leucoféresis), inducir in vitro (fuera del organismo) su diferenciación y posterior proliferación, activando su carga antigénica específica, para volver a administrarlas al paciente. Existen diferentes procedimientos de diferenciación, activación y dirección de la especificidad de la respuesta inmunitaria, lo que determina una amplia diversidad en los resultados.
El modo de obtención de precursores de células dendríticas más utilizado actualmente para la fabricación de vacunas antitumorales son las células mononucleares de sangre periférica del propio paciente (peripheral blood mononuclear cells; PBMC), constituidas por linfocitos (T, B, NK) y monocitos; a diferencia de las PBMC, los eritrocitos carecen de núcleo y los leucocitos de tipo granulocito (neutrófilos, basófilos y eosinófilos) presentan un núcleo multilobulado.
Estas células se pueden extraer de sangre entera usando determinados productos a base de polisacáridos hidrófilos neutros de alto peso molecular y muy ramificados (como Ficoll®), empleándose para la diferenciación y maduración a partir de PBMC diferentes combinaciones de citocinas, frecuentemente GM-CSF e IL-4. Este procedimiento permite obtener células dendríticas inmaduras, que no son adecuadas como vacuna terapéutica, ya que su inoculación podría inhibir la respuesta inmunitaria mediada por linfocitos T CD8+ e inducir la aparición de linfocitos T específicos productores de IL-10 (citocina inmunosupresora), justo lo contrario de lo que hacen las células dendríticas maduras y activadas. En definitiva, las vacunas terapéuticas precisan asegurar el estado madurativo y de activación de las células dendríticas utilizadas, que es determinante para su eficacia.
Existe una amplia variedad de métodos de activación de las células dendríticas, principalmente con IL-1β, IL-6, factor de necrosis tumoral (TNF), interferones alfa y gamma (IFN-α, IFN-γ), prostaglandinas (PGE2) y ácido poliinosínico: policitidílico (poly I: C)6. Es común también la utilización de productos de origen microbiano que actúan a través de receptores tipo Toll (TLR7), lectinas tipo C y receptores tipo NOD (NLR8); así como la de subpoblaciones de linfocitos T, NK y γδ; proteínas co-estimuladoras como CD40 y citocinas proinflamatorias como IL1β, factor de necrosis tumoral (TNF), IL-6 y prostaglandina E2.
Los antígenos empleados para el cargado de las células dendríticas son diversos: proteínas totales, lisado tumoral, péptidos restringidos por HLA, ARN, híbridos o fusiones celulares, etc. En general, se considera que las células dendríticas cargadas con antígeno tumoral total podrían tener un mayor beneficio que el resto de opciones, permitiendo el procesamiento y selección de epítopos (determinantes antigénicos) de forma natural, que permiten inducir una respuesta inmunitaria policlonal. Hay datos clínicos que justifican una respuesta inmunitaria más eficaz con el uso de células dendríticas cargadas con mezclas antigénicas complejas (lisado de células tumorales, exosomas, células tumorales apoptóticas-necróticas, transfección con ARN total proveniente de células tumorales, hibridomas entre células tumorales y células dendríticas), aunque éstas implican problemas de reproducibilidad de los resultados dada la heterogeneidad en la población de células dendríticas empleada, las variadas vías de administración y esquemas terapéuticos puestos en práctica. Además, la utilización de antígenos tumorales totales expone a las células dendríticas a un gran número de antígenos desconocidos, algunos de los cuales podría inhibir su maduración e incluso generar una respuesta autoinmune.
Tras la reincorporación de las células dendríticas activadas al paciente, éstas deben migrar al ganglio linfático donde harán la presentación antigénica a los linfocitos. El procedimiento más habitual de reincorporación es la vía subcutánea y la inoculación periganglionar, ocasionalmente asociadas con PGE2 o TNF-α para incrementar el porcentaje de células inoculadas que migran al ganglio linfático regional.
En abril de 2010, la FDA de Estados Unidos aprobó la primera vacuna de tratamiento de cáncer. En concreto, fue aprobada para varones con cáncer metastásico de próstata para estimular una respuesta inmunitaria a la fosfatasa ácida prostática (PAP), un antígeno que se encuentra en la mayoría de las células tumorales prostáticas.
Provenge® recibió también autorización de comercialización en la Unión Europea (EMA), el 6 de septiembre de 2013, para tratar a los hombres con cáncer de próstata metastásico (no visceral) resistente a castración asintomático o mínimamente sintomático en los que la quimioterapia no está todavía indicada clínicamente. Sin embargo, el 6 de mayo de 2015 la Comisión Europea retiró la autorización de comercialización de Provenge® en la Unión Europea, a petición del titular de la autorización de comercialización, Dendreon UK Ltd, que notificó a la Comisión Europea su decisión de interrumpir definitivamente la distribución del producto por motivos comerciales.
Provenge® se produce en varios pasos. En primer lugar, se practica una leucoféresis con la sangre del paciente, en la que se recogen algunas células inmunes del paciente que posteriormente son expuestas a una proteína destinada a estimularlas y dirigirlas contra el cáncer de próstata. Después de esta exposición, las células inmunes activadas son infundidas al paciente.
Dado el carácter estrictamente personal de cada vacuna Provenge®, su composición celular variará, dependiendo de las células obtenidas del paciente individual durante la leucoféresis. Además de las células presentadoras de antígenos (APC, mayoritariamente células dendríticas), el producto también contiene linfocitos T, B, NK (natural killers; células citotóxicas naturales) y otros tipos de células. En concreto, los componentes activos de Provenge® son células autólogas presentadoras de antígenos (APC) y la proteína llamada PAP-GM-CSF. Las APC se activan durante un período de cultivo definido con una proteína humana recombinante, PAP-GM-CSF, que consiste en fosfatasa ácida prostática (PAP), un antígeno expresado en tejido de cáncer de próstata, unido al factor estimulante de colonias de granulocitos-macrófagos (GM-CSF ), un activador fisiológico de las células del sistema inmune y mejora la presentación de antígenos. El medicamento se administra por vía intravenosa en un esquema de tres dosis, a intervalos de dos semanas. Cada dosis está precedida por el procedimiento de leucoféresis aproximadamente tres días antes del tratamiento programado.
Los datos procedentes de tres ensayos clínicos de fase 3, doblemente ciegos y controlado con placebo, en pacientes con cáncer de próstata metastásico resistente a castración que recibieron sipuleucel-T o placebo, administrados por vía intravenosa cada 2 semanas en un total de tres infusiones, indican una mejora modesta pero significativa de 4,1-4,3 meses en la supervivencia media (23-25,8 meses en el grupo sipuleucel-T vs. 21,7-18,9 en el grupo placebo), lo que supone un aumento en torno al 40% en la probabilidad de supervivencia a los 36 meses (32 vs. 23%) respecto del grupo placebo. Sin embargo, el tiempo transcurrido hasta la progresión objetiva de la enfermedad no fue estadísticamente diferente con la vacuna y el placebo.
A continuación se indican algunos ejemplos de vacunas terapéuticas cancerosas que están actualmente en fase de investigación clínica. La relación no es exhaustiva, sino que tan solo se pretende ofrecer una panorámica representativa de este amplio grupo de medicamentos, orientando sobre las tendencias actualmente en desarrollo.
Se está estudiando actualmente una terapia personalizada conocida como pLADD, mencionada anteriormente y consistente en una cepa de Listeria monocytogenes doblemente suprimida, viva y atenuada (LADD, Listeria monocytogenes doubled-deleted), diseñada para codificar múltiples neoantígenos específicos de tumores. La plataforma LADD es un enfoque atractivo para la inmunoterapia personalizada debido a la rápida construcción, fabricación y liberación de cepas clínicas. Además, se ha establecido su perfil clínico de seguridad y eficacia en más de 400 pacientes y se ha demostrado la robusta activación de la inmunidad innata y el remodelado del microambiente tumoral en modelos preclínicos y en pacientes.
Algunos estudios que utilizaron ratones portadores de tumores demostraron que una cepa pLADD que expresaba neoepítopos específicos de tumores de células tumorales murinas MC38 podría inducir respuestas robustas de células T CD8+ específicas para los neoepítopos codificados, pero no contra secuencias nativas. Este enfoque personalizado ha demostrado una elevada eficacia en combinación con el bloqueo PD-1 (nivolumab, pembrolizumab); en concreto, se acaba de iniciar un ensayo clínico para evaluar la seguridad y la inmunogenicidad del pLADD en pacientes con cánceres del tracto gastrointestinal, especialmente en cáncer colorrectal con microsatélites estables, una indicación en la cual las respuestas terapéuticas actuales son muy pobres.
GVAX colorrectal (autologous GM-CSF-secreting lethally irradiated colorectal cancer cell vaccine) es una vacuna de cáncer colorrectal autóloga irradiada letalmente y consistente en células de cáncer colorrectal específicas de pacientes modificadas genéticamente para secretar el factor de estimulación de colonias de granulocitos-macrófagos (GM-CSF), con potenciales actividades inmunoestimulantes y antineoplásicas.
La vacuna de células dendríticas autólogas con adenovirus HER2-transducido (AdHER2) / neuexpresa los dominios extracelular y transmembrana de la proteínas HER2 humana.
INO-1400 (synthetic hTERT DNA vaccine INO-1400) es una vacuna de ADN consistente en un plásmido que codifica la secuencia completa del antígeno asociado al tumor, la telomerasa humana, la transcriptasa inversa (hTERT) – subunidad catalítica de la telomerasa humana y sintetiza el ADN telomérico en los extremos del cromosoma – conteniendo dos mutaciones con potenciales actividades inmunoestimulantes y antineoplásicas. Tras la vacunación intradérmica de la vacuna en combinación con electroporación, se expresa la proteína hTERT y activa el sistema inmune para montar una respuesta de células T citotóxicas frente a células tumorales que expresan telomerasa. La telomerasa prolonga la vida funcional de las células mediante la restauración y el mantenimiento de la longitud de los telómeros. Anormalmente activada en la tumorigénesis, la telomerasa se expresa en la mayoría de las células cancerosas humanas, pero su expresión es baja o inexistente en las células normales.
Finalmente, la GI-4000 contiene Saccharomyces cerevisiae recombinante muerta por calor y previamente transfectada con formas mutadas de Ras, un oncogén frecuentemente encontrado en tumores sólidos.
ADXS11-001 es una vacuna contra el cáncer que contiene una cepa atenuada de la bacteria Listeria monocytogenes que codifica la proteína E7 del papilomavirus humano (HPV) 16 fusionado a una proteína de listeriolisina O no hemolítica. Tras la vacunación, Listeria expresa el antígeno HPV 16 E7 y activa el sistema inmunológico para montar una respuesta de linfocitos T citotóxicos (CTL) contra las células cancerosas que expresan HPV 16 E7. Esto puede resultar en lisis de células tumorales. Además, el propio vector de Listeria puede inducir una potente respuesta inmune. La VPH 16 E7 es una glicoproteína de la superficie celular y antígeno asociado al tumor, que se sobreexpresa en la mayoría de las células cancerosas cervicales
DCVAC/OvCa (ovarian tumor antigen-activated autologous dendritic cell vaccine) es una vacuna compuesta de células dendríticas autólogas activadas con un lisado de células tumorales ováricas que contienen antígenos asociados a tumores. Tras su administración, la vacuna puede estimular una respuesta antitumoral de linfocitos T citotóxicos contra células de cáncer ovárico que expresan antígenos específicos de células tumorales ováricas, lo que puede dar lugar a lisis de células tumorales ováricas.
HER-2/neu peptide vaccine es una vacuna formada a partir de péptidos derivados del domino extracelular del antígeno asociado a tumores HER-2/neu. También se está estudiando en pacientes con cáncer de mama.
GVAX pancreatic cancer vaccine es una vacuna de células enteras que expresa el factor estimulante de colonias de macrófagos de granulocitos humanos (GM-CSF). Las células tumorales de pacientes con cáncer de páncreas se cosechan y luego se modifican genéticamente para secretar GM-CSF.
PANC 10.05 pcDNA-1/GM-Neo es una vacuna alogénica compuesta de células de cáncer de páncreas del propio paciente enteramente irradiadas letalmente y previamente transfectadas con un plásmido que porta el gen para el factor estimulante de colonias de granulocitos-macrófagos (GM-CSF). Las células tumorales pancreáticas se derivan de la línea de células tumorales PANC 10.05. Otra vacuna similar es la PANC 6.03 pcDNA-1/GM-Neo, que deriva de una línea de células tumorales PANC 6.03.
p53MVA (modified vaccinia virus ankara vaccine expressing p53) es una vacuna terapéutica compuesta por un vector viral Ankara modificado (MVA) recombinante que codifica la forma de tipo salvaje de la proteína tumoral p53 (wt p53). Tras la vacunación subcutánea con la vacuna MVA que expresa p53, la p53 expresada puede estimular el sistema inmune del huésped para montar una respuesta de linfocitos T citotóxicos específicos de p53 (CTL) contra células tumorales que expresan p53, dando como resultado la lisis de células tumorales. El vector viral de MVA, derivado de la cepa Ankara competente para la replicación, es una cepa muy atenuada, con replicación defectuosa y es incapaz de ensamblaje de virión. El gen p53, un gen supresor de tumores, está mutado en muchos tipos de células cancerosas
DSP-7888 (WT1 protein-derived peptide vaccine DSP-7888) es una vacuna compuesta de péptidos derivados de la proteína del gen 1 del tumor de Wilms (Wilms tumor gene 1; WT1). Tras la administración, la vacuna de péptido derivado de proteína WT1 DSP-7888 puede inducir una respuesta citotóxica específica de linfocitos T (CTL) frente a células tumorales que sobreexpresan WT1. Además, el DSP-7888 induce una respuesta inmune mediada por linfocitos T auxiliares frente a células tumorales que expresan WT1. La proteína WT1 se sobreexpresa en células leucémicas y en muchos tumores sólidos no hematológicos, como el de páncreas.
DCVAC/PCa (prostate tumor antigen-activated autologous dendritic cell vaccine) es una vacuna de células dendríticas alogénicas activadas con un lisado de células tumorales pancreáticas conteniendo antígenos asociados al tumor.
ADXS31-142 (Listeria monocytogenes-LLO-PSA ADXS31-142) contiene cepas vivas atenuadas de Listeria monocytogenes modificadas genéticamente para expresar una proteína de fusión compuesta por el antígeno específico prostático (PSA) humano y un fragmento de listeriolisina O (LLO). Tras la administración de la vacuna, el LLO-PSA expresado es procesado por células presentadoras de antígeno (APC), presentadas al sistema inmune por las moléculas de complejo de histocompatibilidad principal (MHC) I y II, y activa una respuesta inmune innata y adaptativa que implica el reclutamiento y la activación de linfocitos T frente a células tumorales que expresan PSA, así como la inhibición de células T reguladoras de linfocitos T de infiltración de tumores y células supresoras derivadas de mieloides (MDSC ).
pTVG-HP es una vacuna que contienen un plásmido de ADN codificante para la fosfatasa ácida prostática humana (PAP). La vacuna estimula la respuesta inmune frente a las células tumorales pancreáticas, que tienden a sobrexpresar PAP.
La alpha-DC1 es una vacuna de células dendríticas maduradas con un procedimiento diferente al empleado habitualmente (con TNFα, IL-1β, IL-6 y PGE2), utilizando TNFα, IL-1β, poly I: C e interferones alfa y gamma.
La multiepitope TARP-pulsed autologous dendritic cell vaccine está compuesta de células dendríticas autólogas pulsadas con múltiples péptidos antigénicos derivados de la T-cell receptor gamma-chain alternate reading frame protein (TARP), una proteína nuclear intensamente inmunogénica que es expresada por las células tumorales prostáticas y por otros tipos tumorales.
PROSTVAC (rilimogene-galvacirepvec and recombinant fowlpox-prostate specific antigen [PSA] (L155)/ triad of costimulatory molecules [TRICOM] vaccine) es una formulación vacunal consistente en galvacirepvec rilimogene (V-PSA-TRICOM; PROSTVAC-V), un virus vaccinia recombinante, y rilimogene glafolivec (F-PSA-TRICOM; PROSTVAC-F), un virus de la viruela aviar recombinante, B7.1 (CD80), molécula de adhesión intercelular-1 (ICAM-1) y antígeno-3 asociado a la función leucocitaria (lymphocyte function-associated antigen3, LFA-3).
El uso de un régimen de vacunación de sensibilización-refuerzo, con una vacunación primaria de galvacirepvec rilimogene, seguido de múltiples vacunaciones de refuerzo de rilimogene glafolivec, las vacunas PSA-TRICOM infectan las células presentadoras de antígeno (APC), particularmente las células dendríticas. La combinación de PSA y TRICOM mejora en gran medida la activación de células T y destrucción de células tumorales mediada por células T.
CIMAVax® (recombinant human EGF-rP64K/Montanide ISA 51 vaccine) es una vacuna peptídica autorizada inicialmente en Cuba y Perú (2008), que contiene factor de crecimiento epidérmico humano recombinante (rEGF) unido a la proteína portadora inmunogénica recombinante derivada de Neisseria meningitidis P64k (rP64k) y mezclado con el inmunoadyuvante Montanide ISA 51. La vacuna puede desencadenar una respuesta inmune humoral contra el EGF endógeno. La inhibición mediada por anticuerpos de la unión EGF endógena a su receptor, receptor de factor de crecimiento epitelial (EGFR), puede dar como resultado la inhibición de la proliferación de células tumorales.
La viagenpumatucel-L contiene células tumorales alogénicas que expresan una proteína de fusión (gp96-Ig) de la forma secretora recombinante de la proteína de choque térmico gp96 con la fracción constante de una inmunoglobulina quimérica. Tras la administración de viagenpumatucel-L, las células tumorales vivas segregan de forma continua gp96-Ig junto con sus antígenos asociados a tumores (TAA) en la corriente sanguínea. La proteína de fusión Gp96-Ig se construye reemplazando la secuencia de retención de gp96, normalmente un péptido endoplasmático residente en el retículo, con la porción Fc de IgG1 de ratón y humana.
La MUC1 peptide-poly-ICLC adjuvant vaccine contiene mucina 1 (MUC1), una proteína transmembrana expresada en células de pulmón, mama, páncreas, riñón, ovario, colon y otros tejidos; también incluye poly I: C.
TG4010 (MVA-MUC1-IL2 vaccine) es una vacuna bivalente constituida por cepas vacunales modificadas del virus Ankara (MVA) que codifican para mucinae interleucina-2 (IL-2).
La MRC-5 consiste en fibroblastos pulmonares fetales humanos irradiados (Medical Research Council 5; MRC-5) transfectados con ADN procedente de células autólogas de cáncer pulmonar no microcítico. La MRC-5 es una línea celular diploide de fibroblastos pulmonares humanos que es permisiva para un amplio rango de infecciones virales, incluyendo citomegalovirus (CMV) y virus Coxsackie B.
El ID-LV305 es un vector lentiviral dirigido a células dendríticas que contiene ácidos nucleicos que codifican para el antígeno NY-ESO-1 asociado a tumores humanos. Tras la administración intradérmica, el vector lentiviral ID-LV305 se dirige y se une a células dendríticas dérmicas a través del receptor deDC-SIGN (DC-specific intercellular adhesion molecule-3-grabbing non-integrin). Tras la internalización del vector, se expresa la proteína NY-ESO-1, que estimula la maduración de las células dendríticas y activa el sistema inmunológico para montar una respuesta de linfocitos T citotóxicos frente a células que expresan NY-ESO-1. NY-ESO-1 se expresa en testículos normales y en las superficies de varias células tumorales; dentro de estas últimas desempeña un papel clave en la proliferación y supervivencia.
La PGV001 (personalized genomic vaccine 001) es una vacina vacuna terapéutica sintética consistente en múltiples péptidos de origen tumoral específicos de cada paciente, combinados con poly I: C para potenciar la respuesta inmunológica frente a los neoantígenos a través de la liberación de citocinas.
Belagenpumatucel (Lucanix®) utiliza células tumorales enteras alogénicas para estimular el propio sistema inmunológico del paciente que han sido modificadas para bloquear la producción del factor de crecimiento transformante beta (TGF-B), que es uno de los principales métodos que los cánceres usan para esconderse del sistema inmune. En pacientes con carcinoma pulmonar de células escamosas se alcanzó una supervivencia media de 20,7 meses vs. 12,3 en el brazo de control; esta diferencia fue incluso mayor entre los pacientes que había sido sometidos a radioterapia previamente (40,1 vs. 10,3 meses).
Un metanálisis publicado en 2015 hizo una evaluación sistemática de los estudios controlados publicados sobre eficacia terapéutica y seguridad de las vacunas tumorales para el tratamiento del cáncer pulmonar no microcítico avanzado. En total, se realizaron 11 ensayos clínicos controlados, incluyendo a un total de 3.986 pacientes para metanálisis. Los resultados mostraron que el brazo de la vacuna prolongó significativamente la supervivencia global media con respecto al grupo control. La supervivencia a 1, 2 y 3 años también obtuvo beneficios significativos con las vacunas antitumorales en comparación con su correspondiente grupo de control. Además, se observó una mejora significativa en el tiempo medio de progresión, la mediana de supervivencia libre de progresión y una tendencia de mejora en la tasa de respuesta objetiva después del tratamiento con vacuna antitumoral. Se produjeron menos efectos secundarios en el grupo de vacuna en comparación con el grupo de control.
Aproximadamente, del 25% al 30% de los casos de cáncer de células renales son metastásicos en el momento del diagnóstico y un 20-30% de los pacientes que se someten a tratamiento quirúrgico de formas locales acaban recayendo. Este tipo de cáncer es diferente de otros tumores epiteliales en que es intrínsecamente resistente a la quimioterapia citotóxica y durante mucho tiempo no se ha dispuesto de ninguna terapéutica sistémica mínimamente eficaz.
El cáncer de células renales es inmunológicamente sensible, lo que ha permitido la incorporación de los tratamientos con interferón alfa (INF-α) y con dosis altas de aldesleukina (IL-2) para las formas metastásicas. La IL-2 es un potente estimulador de la proliferación y diferenciación de células T, mientras que INF-α tiene efectos antiangiogénicos, promoviendo la presentación de antígenos y maduración de células dendríticas. La tasa de respuesta al IFN-α está en torno al 15%, con un aumento de la supervivencia global de 3 a 7 meses; sin embargo, la mayoría de estas respuestas son de duración limitada y sólo un pequeño número de pacientes experimentan respuestas completas. Por su parte, el empleo de dosis altas de aldesleukina (IL-2) se asocian a tasas de respuesta objetiva entre 10 y 20%, muchas duraderas; la tasa de respuestas parciales es el 13% y la de respuestas completas del 7%; la tasa de supervivencia a los 5 años es de apenas un 10%. Además, el uso a largo plazo presenta importantes limitaciones de toxicidad multiorgánica, especialmente elevada sobre el corazón, los pulmones, los riñones y el sistema nervioso central.
AGS-003 es una vacuna elaborada a partir de células dendríticas sanguíneas autólogas y generada mediante electroporación de ARN derivado de tumores y ligandos CD40 en células inmunitarias del huésped, que se ha estudiado en combinación con sunitinib. El tratamiento se administra mediante inyección intradérmica y las células dendríticas maduras cargadas con ARN del tumor presentan antígenos tumorales únicos específicos del paciente a través del complejo de histocompatibilidad principal de clase I a células T en los ganglios linfáticos que drenan los tumores. Además, la ligación con CD40 promueve el reclutamiento de linfocitos T CD8+ a través de la producción regional de IL-12. En un estudio de fase 2 sobre 21 pacientes con una categoría de riesgo intermedio o de mal pronóstico, se les trató con un ciclo de sunitinib (4 semanas, 2 semanas de descanso), seguido de inmunoterapia concomitante con AGS-003 cada 3 semanas (5 dosis) y luego cada 12 semanas hasta la progresión del tumor o al final del estudio. Nueve de los 21 pacientes (43%) tuvieron una respuesta parcial y otros (19%) cuatro mostraron enfermedad estable. La mediana de supervivencia libre de progresión y supervivencia global fueron de 11,2 meses y 30,2 meses, respectivamente, y 5 pacientes (24%) vivieron más de 5 años y 2 pacientes tuvieron respuestas duraderas durante más de 5 años. El tratamiento con AGS-003 fue bien tolerado, con reacciones en el sitio de inyección como el principal evento adverso. Sobre la base de estos resultados prometedores, se está realizando un ensayo aleatorizado multicéntrico de fase 3 (ADAPT).
La AdGMCAIX es una vacuna consistente en células dendríticas autólogas transducidas con un vector recombinante adenoviral de replicación defectuosa que expresa GM-CSF y la anhidrasa IX (CA-IX o CA9) (GMCA-9). El CA9 es un antígeno asociado al carcinoma de células renales y un miembro de la familia de la anhidrasa carbónica que contiene un epítopo presente la mayoría de los carcinomas de células renales pero que está ausente en la mayoría de los tejidos normales.
Oncophage® es una vacuna autorizada por la Federación Rusa en 2008 para el tratamiento del carcinoma de células renales, pero cuya autorización fue desestimada por la Unión Europea y por Estados Unidos. El principio activo de Oncophage, el vitespén, está formado por un complejo de proteínas y péptidos del choque térmico 96 (gp HSP96; heat shock protein 96) extraídas de las células cancerosas del enfermo. Existe un estudio realizado sobre 818 adultos con carcinoma de células renales localizado que se había extirpado quirúrgicamente y que presentaban un riesgo elevado de reaparición, comparando a los pacientes tratados con Oncophage® con otros que no lo recibieron. Sin embargo, la EMA emitió en 2009 un dictamen desfavorable, por considerar que dicho estudio no había demostrado que Oncophage® fuera eficaz para prolongar la vida de los pacientes antes de la reaparición del cáncer; igualmente, señaló que no había información adecuada sobre el contenido del medicamento y el proceso de fabricación, ni datos suficientes para esclarecer su mecanismo de acción ni para determinar la dosis adecuada.
Hybricell® es una vacuna muy discutida y únicamente autorizada en Brasil (2005), para el tratamiento del cáncer de células renales y el melanoma. No se han realizado estudios clínicos de fase 3 (confirmatorios de eficacia y seguridad). Para producir la vacuna, se extrae sangre del paciente y los monocitos específicos del paciente se separan de la sangre mediante aféresis. Las células dendríticas se derivan entonces añadiendo citocinas a los monocitos extraídos. Las células dendríticas se fusionan luego con células tumorales mediante un procedimiento de electrofusión, creando supuestas “células híbridas” que son incorporadas de nuevo al paciente, donde son reconocidas y destruidas, también supuestamente, por el sistema inmune.
La vacuna peptídica frente a HER-2/neu es una vacuna formada a partir de péptidos derivados del domino extracelular del antígeno asociado a tumores HER-2/neu. También se está estudiando en pacientes con cáncer de ovario. La adenovirus HER2-transduced autologous dendritic cell vaccine está compuesta por células dendríticas autólogas transducidas con un vector de adenovirus de replicación deficiente que codifica HER-2 con potencial actividad antineoplásica. Como es sabido, HER-2 es un receptor de tirosina quinasa para el factor de crecimiento epidérmico (EGF) (también conocido como neu y ErbB2), que es sobreexpresado por algunos cánceres, particularmente de mama, ovario, colon y gástrico.
La vacuna que está más avanzada en esta indicación es posiblemente NeuVax®, que es una vacuna del complejo principal de histocompatibilidad (MHC) de clase I que consiste en el péptido E75 derivado de HER2 (nelipepimut-S) combinado con el factor estimulante de colonias de granulocitos macrófagos (GM -CSF) como inmunoadyuvante. E75 es un péptido de 9 aminoácidos derivado del dominio extracelular de la proteína HER2 que se une de forma estable a HLA-A2 y puede estimular las células T in para lisar las células cancerosas que expresan HER2. Varios experimentos in vivo han confirmado que los linfocitos T citotóxicos pulsados con E75 son capaces de lisar el carcinoma de colon con expresión de HER2 y células de carcinoma de células renales en modelos murinos.
Sin embargo, los datos clínicos más relevantes corresponden a su uso en pacientes con cáncer de ovario y, sobre todo, de mama HER2+. Los datos procedentes de estudios clínicos de fases 1 y 2 son alentadores; en este sentido, la utilización de la vacuna en asociación a GM-CSF en un grupo de 187 pacientes con cáncer de mama se tradujo en que la supervivencia libre de progresión tumoral a los cinco años fue del 89,7% en el grupo vacunado frente al 80,2% en el grupo control (GM-CSF solo). Debido al diseño del ensayo, el 65% de los pacientes no recibieron la dosis óptima de la vacuna y, de hecho, entre aquellos que sí la recibieron la tasa de supervivencia libre de enfermedad a cinco años fue del 94,6 vs. 87,1% (P = 0,05) con la dosis subóptima.
Por su parte, la vacuna peptídica frente al receptor alfa de folato (FRα) se fundamenta en la observación de que el FRα tiene una limitada expresión en los tejidos sanos y una elevada expresión en los cánceres de mama, especialmente en los triplemente negativos (células tumorales con ausencia de receptores de estrógeno, ER, de receptores de progesterona, PR, y de HER2; es decir, ER-, PR-, HER2-; triple negative breast cancer, TNBC) en los que el FRα es expresado en cerca del 90% de los casos; además, hay estudios que han mostrado que es un marcador biológicamente importante que se asocia con malos resultados clínicos y se mantiene en las lesiones metastásicas. Los cánceres de mama triplemente negativos (TNBC) ocurren en aproximadamente el 20-25% de todas las pacientes con cáncer de mama, las cuales raramente obtienen algún beneficio con las terapias actualmente disponibles.
La vacuna FRα incluye un grupo de 5 péptidos que son epítopos inmunogénicos y genera respuestas inmunitarias de células T CD4+. Se están realizando actualmente estudios clínicos de fase 2 con esta vacuna en combinación con GM-CSF como adyuvante, con o sin cebamiento inmune con ciclofosfamida, como terapia de consolidación después de tratamiento neoadyuvante o adyuvante de pacientes con cáncer de mama triple negativo en estadio IIb-III.
También está utilizando una vacuna multipeptídica con transcriptasa inversa humana de la telomerasa humana (hTERT) /survivin/citomegalovirus (CMV), emulsificada con montanidaISA 51 VG (un emulgente con actividad inmunoadtuvante). Como ya se ha indicado, la telomerasa (y su unidad catalítica de transcriptasa inversa) se expresa en la mayoría de las células cancerosas humanas, pero su expresión es baja o inexistente en las células normales.
La STEMVAC (CD105/Yb-1/SOX2 CDH3/MDM2-polyepitope plasmid DNA vaccine) es una vacuna formada por plásmidos de ADN que codifica para múltiples antígenos expresados en células tumorales mamarias, especialmente procedentes de tumoraes resistente a la quimioterapia. Está comenzando su investigación clínica en pacientes cáncer de mama avanzado HER2-negativo.
La mammaglobin-ADN plasmid vaccine contiene un plásmido de ADN que codifica el gen de la mamaglobina A, una glicoproteína que se expresa en más del 80% de los cánceres de mama humanos.
WOKVAC (pUMVC3-IGFBP2-HER2-IGF1R) es una vacuna conteniendo un plásmido de ADN codificante para tres antígenos tumorales característicos del cáncer de mama: la proteína 2 de unión al factor de crecimiento de tipo insulínico (IGFBP2), HER2 y el receptor del factor 1 de crecimiento de tipo insulínico (IGF-1R).
Se ha mostrado que la vacunación con células dendríticas alogénicas pulsadas con lisado de células tumorales genera respuesta de células T CD8 + específica en pacientes con leucemia linfocítica crónica de células B (CLL-B). Un grupo de 12 pacientes en estadio clínico 0-2 fueron vacunados intradermicamente hasta ocho veces con una media de 7,4 x 106 células dendríticas pulsadas con lisado de células B-CLL, observándose una disminución de los leucocitos de sangre periférica y las células leucémicas CD19 + / CD5 + en cinco pacientes, tres pacientes mostraron una enfermedad estable, aunque cuatro pacientes progresaron a pesar de la vacunación. En los pacientes con una respuesta clínica, se observó un aumento de los niveles séricos de interleucina 12 (IL-12) y una disminución de la frecuencia de células reguladoras CD4+ CD25 + FOXP3 + linfocitos T reguladores. Actualmente, está siendo objeto de un estudio de fase 1/2 en combinación con lenalidomida.
Se ha desarrollado una vacuna autóloga a partir de células leucémicas enteras, fusionando ex vivo formas blásticas leucémicas con células dendríticas extraídas de pacientes, con el objetivo de provocar una respuesta inmunitaria policlonal antileucémica. Se ha realizado un ensayo clínico en el que 16 pacientes (60 años de media) con leucemia mieloide aguda que no eran candidatos para el trasplante alogénico, que recibieron al menos dos vacunaciones mensuales (5 x 106 células fusionadas), alcanzándose una remisión con ayuda de quimioterapia, manteniendo la misma un 75% de ellos (12/16) durante al menos 45 meses.
Oncoquest® es una vacuna frente a la leucemia linfocítica crónica consistente en proteínas de membrana específicas extraídas directamente de células tumorales autólogas de pacientes e incorporadas en liposomas junto con Interleucina 2 (IL-2) para producir proteoliposomas. Está siendo objeto de estudios clínicos de fase 1.
La vacuna celular GM-K562 está compuesta de células K562 transfectadas con el gen del factor estimulante de colonias de macrófagos de granulocitos (GM-CSF). Las células K562 se derivan de la línea celular de eritroleucemia humana K562.
M-Vax® es una vacuna consistente en células tumorales autólogas conjugada con dinitrofenilo, un hapteno intensamente inmunogénico. La vacuna recibió el estatus de medicamento huérfano por la FDA para el tratamiento del melanoma en estadio III en enero de 1999. En 2001, la FDA suspendió dos ensayos pivotales de fase 3 debido a violaciones de fabricación en la planta estadounidense de la compañía (AVAX Technologies), aunque presentó con éxito una nueva aplicación de Investigación de Medicamentos Nuevos (IND) en 2002. En un estudio de fase 2 en el que participaron 214 pacientes con melanoma en estadio III, los pacientes que recibieron terapia adyuvante con M-Vax® después de una linfadenectomía estándar tuvieron una tasa de supervivencia global a cinco años del 44% (p <0,035). Fue aprobada en Suiza en octubre de 2005 para el tratamiento del melanoma de las etapas III (avanzado) y IV (metastásico).
La vacuna hsp110-gp100 está compuesta de un complejo entre la proteína de choque térmico (heat shock protein; HSP) hsp110 y el antígeno gp100, sobreexpresado por las células del melanoma humano y otros tipos tumorales. Hsp110 actúa como un inmunoadyuvante.
Las vacunas anti-idiotipo GD2 vacuna son una clase de vacunas que consisten en anticuerpos monoclonales anti-idiotipo contra el antígeno asociado al tumor disialogangliósido GD2. La vacunación con este tipo de vacunas produce una respuesta de inmunoglobulina contra GD2 con posterior destrucción de las células tumorales que expresan GD2, a través de anticuerpos dependientes de citotoxicidad celular. GD2 está sobreexpresado en melanoma, neuroblastoma, sarcomas de tejido blando y carcinoma de células pequeñas del pulmón.
La GRN–1201 es una vacuna de péptidos cancerígenos compuesta de cuatro péptidos restringidos por antígenos de leucocitos humanos (HLA) -A2 (HLA-A * 02) derivados de cuatro antígenos asociados a tumores (TAA) específicos y separados, expresados por células de melanoma.
La allogeneic GM-CSF-transfected myeloma cell vaccine es una vacuna a base de células tumorales alogénicas conteniendo células tumorales de mieloma transfectadas con GM-CSF.
La vacuna peptídica SurVAx M (SVN53-67/M57-KLH) contiene un péptido modificado a partir de la survivina, una proteína antiapoptótica, y conjugado con hemocianina de lapa californiana (KLH). La survivina es un miembro de la familia de los inhibidores de la apoptosis (IAP), expresada durante el desarrollo embrionario pero está ausente en la mayoría de las células adultas normales; está regulada positivamente en una variedad de cánceres humanos (mieloma múltiple, glioblastoma) y se asocia con un fenotipo tumoral más agresivo, disminución de la supervivencia y aumento de la resistencia a la quimioterapia. La hemocianina KLH es un inmunoadtuvante que puede mejorar el reconocimiento inmune y puede promover una respuesta mejorada, ya que SVN53-67 tiene un débil poder inmunogénico en seres humanos.
La vacuna multipeptídica PVX-410 (XBP1-US/XBP1-SP/CD138/CS1) contiene epítopos inmunogénicos específicos de HLA-A2 derivados de la XBP1-US (X-box–binding protein 1-unspliced) XBP1-spliced (SP), sindecano-1 (syndecan-1; CD138), y CS1 (CD2 subset 1, CRACC, SLAMF7, CD319), todos los cuales se sobreexpresan sobre la superficie de células de mieloma múltiple.
El glioblastoma multiforme es un tumor cerebral que tiene un mal pronóstico. En una reciente revisión sistemática y metanálisis se analizaron los resultados de los ensayos clínicos que compararon la inmunoterapia con la terapia convencional para el tratamiento de los gliomas malignos. En los seis ensayos comparativos seleccionados en la revisión sistemática la inmunoterapia se asoció con una supervivencia global a dos años significativamente más larga en comparación con la terapia convencional.
La ICT-107 una vacuna compuesta por células dendríticas autólogas pulsadas con seis péptidos sintéticos de glioblastoma (GBM): absent in melanoma 2 (AIM-2), antígeno asociado a melanoma 1 (melanoma-associated antigen 1; MAGE-1), proteína 2 relacionada con tirosinasa (tyrosinase-related protein 2; TRP-2), glicoproteína 100 (gp100), receptor 2 del factor de crecimiento epidérmico 2 (HER-2) y la subunidad alfa-2 del receptor de interleucina-13 (IL-13Ra2).
Por su parte, la ERC1671 está compuesta por una combinación de células tumorales de glioblastoma autólogas y alogénicas (procedentes de tres diferentes donantes cancerosos), y un lisado de todas estas células.
PEPIDH1M es una vacuna peptídica que consiste en un péptido derivado de la isocitrato deshidrogenasa 1 (IDH1) que contiene la mutación puntual R132H (IDH1R132H), la cual se expresa frecuentemente en gliomas y se asocia con una producción aumentada del oncometabolito R-2-hidroxiglutarato (2HG).
La vacuna DCVax brain para el tratamiento del glioblastoma fue autorizada en Suiza en 2007.
En muchos de los estudios clínicos de vacunas terapéuticas del cáncer que están ahora en marcha, las vacunas se administran conjuntamente con otras formas de terapéutica antineoplásica. Las terapias que se han combinado con vacunas de tratamiento de cáncer incluyen la cirugía, la quimioterapia, la radioterapia, etc. Asimismo, es frecuente la utilización de agentes que tienen como objeto reforzar determinados mecanismos defensivos generales del sistema inmunitario, entre los que cabe citar a citocinas, factores de crecimiento de colonias, etc. En concreto, algunas citocinas aumentan la actividad de las células B y de las células T citotóxicas; entre ellas cabe citar a la interleucina 2 (IL-2, aldesleukina), el interferón alfa (INF–a), y el factor estimulador de colonias de granulocitos y macrófagos (GM–CSF, sargramostim), etc. Con ello, el papel colaborador de todas las terapias se refuerza y permite optimizar el efecto inmune selectivo frente a las células tumorales de cada paciente.
Por ejemplo, estudios preclínicos y estudios clínicos en fase inicial han demostrado que la radioterapia puede intensificar la eficacia de las vacunas terapéuticas anticancerosas y, viceversa, vacunas terapéuticas parecen haber aumentado la efectividad de otras terapias antineoplásicas. Asimismo, la extirpación quirúrgica de grandes tumores puede intensificar la efectividad de las vacunas, ya que en pacientes con enfermedad tumoral extensa, el sistema inmunitario puede sufrir un deterioro importante.
Un aspecto metodológico que los investigadores valoran para realizar el diseño de los ensayos clínicos es si las vacunas terapéuticas funcionan mejor cuando se administran antes, después o al mismo tiempo que otras terapias. Las respuestas a estas cuestiones no solo son fundamentales para optimizar la utilización de una vacuna específica sino también para establecer principios básicos adicionales que guíen la creación futura de combinaciones terapéuticas que incluyan vacunas.
Se han hecho simulaciones matemáticas que muestran que la eficacia de la terapia vacunal depende tanto del tamaño del tumor como de la condición del sistema inmunitario, así como de la respuesta del organismo a la vacunación. En particular, se ha mostrado que la terapia anticancerosa con vacunas se hace más eficaz cuando se usa sin retraso de tiempo desde una fecha de vacunación prescrita después de la cirugía y es ineficaz sin tratamiento preliminar. Un estudio predice que la secuencia más eficaz consiste quimioterapia seguida de inmunoterapia.
Los adyuvantes son sustancias que se añaden a las vacunas o a otras formas de terapéutica antineoplásica para reforzar la capacidad del sistema inmune para enfrentarse a las células tumorales. Su origen es muy diverso, aunque los primeros fármacos fueron desarrollados a partir de fracciones inmunogénicas de microorganismos y posteriormente fueron desarrollados análogos sintéticos de algunos de sus componentes más inmunogénicos. Entre ellos, los más estudiados son el bacilo Calmette-Guérin (BCG), una forma muy poco virulenta del Mycobacterium bovis y ala mifurtida, un análogo sintético de uno de los componentes de las paredes celulares de las especies de Mycobacterium; ambos productos están comercializados desde hace varios años.
Entre 1908 y 1920, Calmette y Guerin consiguieron seleccionar un conjunto de cepas Mycobacterium bovis de muy escasa virulencia patogénica, con el objetivo de desarrollar una vacuna frente a la tuberculosis: el bacilo Calmette Guerin (BCG). En 1929, Raymond Pearl realizó observaciones destacando que, de alguna manera, “la tuberculosis tiene efectos antitumorales”; treinta años más tarde (1959), se observó que ratones vacunados con BCG mostraban una alta resistencia al trasplante experimental de tumores. Finalmente, en 1976, se presentó el primer tratamiento exitoso con BCG para pacientes con cáncer de vejiga, obteniendo tasas de eficacia de hasta un 65% en tumores no invasivos de alto grado e incluso del 75% en carcinomas in situ, pero completamente ineficaces en los tumores invasivos.
El Bacilo Calmette-Guerin (BCG) vivo es capaz de promover, tras su administración intravesical, una reacción inflamatoria aguda y granulomatosa subaguda con infiltración de macrófagos y linfocitos en el urotelio y en la capa propia de la vejiga urinaria. El mecanismo de acción antitumoral no ha sido totalmente esclarecido, pero parece ser dependiente de la activación de linfocitos T. Probablemente, la respuesta celular es llevada a cabo sobre todo por la linfocitos T facilitadores (helper, Th); en este sentido, se ha implicado a diversas interleucinas activadoras y a otras células inmunitarias, requiriéndose que el BCG esté en contacto con la pared vesical, uniéndose a la fibronectina, una glucoproteína presente la membrana basal, urotelial y en la submucosa.
Los granulocitos y monocitos son atraídos hacia la pared vesical, así activando una cascada inmunológica a través de secreción de diversas citocinas, quienes estimulan a las células asesinas naturales (NK) activadas por el BCG, las cuales son capaces de diferenciar células neoplásicas y del epitelio urinario normal. En la pared vesical se encuentra un medio ambiente de citocinas principalmente del tipo Th1 y se forman focos celulares de tipo granulomatoso. Dentro de este escenario los mecanismos efectores más importantes parecen ser la actividad antitumoral directa de los interferones y la actividad citotóxicas de las células citotóxicas naturales (Natural Killers, NK).
Posiblemente, el paso crítico inicial de la inmunoterapia con el BCG parece ser la unión de la micobacteria al urotelio mediante la interacción de una proteína presente en la superficie del bacilo, el antígeno 85, con la fibronectina presente en la pared de la vejiga. Además, las células uroepiteliales, pero no las inmunitarias, responden al BCG a través de la señalización por la vía de los receptores tipo Toll (Toll-like receptors, TLR) 2, 3, 4 y 9, de localización transmembranal. La señalización en las células del urotelio por la vía de los TLR conduce, después de una serie de pasos, a la activación del factor nuclear κB, con producción de citocinas como IL-1, IL-6, IL-8 y el factor de necrosis tumoral (TNF) en respuesta al estímulo por la micobacteria.
Las células mononucleares de sangre periférica adquieren la capacidad de aniquilar células tumorales de vejiga después de haber sido estimuladas con el BCG; por otro lado, los linfocitos T CD4+ y los monocitos actúan como células accesorias en la citotoxicidad mediada por células inducida por el BCG. La célula efectora de esta citotoxicidad se conoce como célula BAK (BCG activated killer) y comprende a una subpoblación de células asesinas naturales (natural killers) que es capaz de matar a células tumorales a través de la perforina que secreta. La generación de estas células BAK requiere la presencia de IFN- γ, IL-2 e IL-12.
OncoTICE® y Vejicur® son medicamentos constituidos por un liofilizado de bacilos atenuados de Mycobacterium bovis, preparados a partir de cepas especiales de Bacilo Calmette-Guerin (BCG), que se presenta en viales conteniendo entre 2 × 108 y 3 × 109 unidades formadoras de colonias (UFC), que deben ser reconstituidas con 50 mL de suero fisiológico para dar lugar a una suspensión que se emplea por instilación en la vejiga urinaria mediante un catéter apropiado. Estos medicamentos están indicados como tratamiento del carcinoma urotelial superficial in situ de la vejiga y como coadyuvante terapéutico después de la resección transuretral de un carcinoma papilar superficial de vejiga (primario o recurrente) estadio TA (grado 2 o 3) o T1 (grado 1, 2 o 3).
La suspensión instilada debe permanecer en la vejiga durante 2 horas, durante las cuales debe procurarse que contacte con toda la superficie mucosa interna de la vejiga. Por ello, el paciente no debe estar inmovilizado o en caso de que el paciente esté postrado en la cama, debe ser colocado de decúbito supino a prono y viceversa, cada 15 minutos. Transcurridas las 2 horas, el paciente debe eliminar la suspensión instilada en posición de sentado. Deberá añadirse lejía al inodoro antes de limpiar la evacuación de orina durante las 6 horas siguientes al tratamiento, manteniendo en contacto la lejía y la evacuación en el inodoro durante al menos 15 minutos antes de su retirada definitiva.
Es importante tener en cuenta que estos medicamentos son agentes infecciosos y pueden producir una infección sistémica Mycobacterium, por lo que se deben tomar precauciones durante su administración, para no introducir contaminantes en el tracto urinario o no traumatizar indebidamente la mucosa urinaria; en caso de cateterización traumática u otras lesiones en la uretra o la mucosa de la vejiga se recomienda retrasar la administración del medicamento a tales pacientes hasta que la mucosa vesical esté recuperada. Antes de proceder a la primera instilación intravesical del medicamento, debe realizarse la prueba de la tuberculina y en caso de dar un resultado positivo; si se demuestra clínicamente la existencia de una infección de tuberculosis activa, deberá evitarse el tratamiento. En este sentido, no se han identificado predictores que puedan ayudar a identificar a los pacientes en riesgo de desarrollar infección sistémica después de las instilaciones intravesicales y la incertidumbre sobre la seguridad de reiniciar las instilaciones de BCG intravesical conducen con frecuencia al incumplimiento con el régimen terapéutico programado, poniendo en peligro el pronóstico inicial.
Aproximadamente el 90% de los pacientes desarrollan síntomas de irritación local en la vejiga, manifestándose muy frecuentemente polaquiuria y disuria, aunque en la mayoría de los casos los síntomas desaparecen en 2 días tras la instilación y la cistitis no requiere tratamiento; no obstante, durante el tratamiento de mantenimiento los síntomas de cistitis pueden ser más pronunciados y prolongados, pudiendo requerir la administración de isoniazida (300 mg/día) y analgésicos hasta su desaparición completa. Otros efectos adversos que son muy frecuentes (>10% de los pacientes) son los síntomas de tipo gripal, como fiebre, malestar general y fatiga
La mifamurtida es un agente inmunoestimulante, autorizado en niños, adolescentes y adultos jóvenes para el tratamiento del osteosarcoma de alto grado resecable no metastásico después de una resección quirúrgica macroscópicamente completa. Se utiliza en combinación con quimioterapia postoperatoria combinada.
La mifamurtida es un derivado sintético del muramil dipéptido (MDP), uno de los componentes de las paredes celulares de las especies de Mycobacterium. Administrada dentro de liposomas, éstos son captados por macrófagos y monocitos, en cuyo interior la mifamurtida es liberada tras la degradación del liposoma, desarrollando efectos inmunoestimulantes similares al MDP natural al actuar como un ligando específico del NOD2, una proteína que induce la activación del Factor de Transcripción Kappa B (NF-kappa B), implicado en la respuesta inmune y en los procesos inflamatorios celulares, mediante la inducción de diversas citocinas, incluido el factor de necrosis tumoral (TNF-α), interleucina-1 beta (IL-1β), IL-6, IL-8, e IL-12, proteína C reactiva (PCR) y neopterina9 así como moléculas de adhesión, incluido el antígeno 1 asociado a la función de los linfocitos (LFA-1) y la molécula-1 de adhesión intercelular (ICAM-1). La activación de los monocitos y macrófagos en esta línea se relaciona con un incremento de su actividad tumoricida, habiendo demostrado experimentalmente ser susceptibles de destruir diversas células alogénicas y autólogas tumorales, sin ejercer toxicidad aparente sobre las células normales.
La mifamurtida no es otra cosa más que un conjugado de muramil tripéptido (L-alanina-D-isoglutamina-L-alanina) ligado a dipalmitoil fosfatidil etanolamina, (MTP-PE) y, por tanto, un derivado sintético del muramil dipéptido (MDP), un inmunoestimulante natural presente en paredes celulares de diversas bacterias y micobacterias Gram+, incluyendo las especies de Mycobacterium. Las modificaciones estructurales introducidas tienen como misión prolongar la permanencia en el organismo, sin mermar las propiedades inmunoestimulantes, en relación al MDP natural. Por otro lado, el medicamento tiene una formulación liposómica diseñada específicamente para administrarse mediante perfusión intravenosa y alcanzar el interior celular de los macrófagos; gracias la fracción fosfolipídica presente en la molécula de la mifamurtida, ya que ésta tiende a acumularse en la bicapa lipídica de los liposomas.
Desde el punto de vista clínico, la mifamurtida es capaz de prolongar modestamente la supervivencia de los pacientes con osteosarcoma resecable no metastático, sin que reaparezca la enfermedad (68% vs. 61%, tras un seguimiento medio de 7,9 años), reduciéndose significativamente el riesgo de muerte en un 28%. Aunque esta misma tendencia también se observa en los pacientes con osteosarcoma metastático o no resecable, las diferencias al cabo de cinco años en la supervivencia global (53% vs. 40%) o en la supervivencia en ausencia de eventos adversos (42% vs. 26%) no llegaron a ser estadísticamente significativas en el único ensayo clínico de fase 3 presentado para su autorización en la Unión Europea; por este motivo, esta última indicación no fue autorizada por la EMA.
Desde el punto de vista toxicológico, la mifamurtida parece tener un perfil relativamente benigno, con efectos generalmente leves o moderados. Su uso inevitablemente asociado a combinaciones quimioterápicas altamente tóxicas no permite discriminar adecuadamente la responsabilidad de cada fármaco en los eventos adversos registrados ni descartar posibles interacciones. No obstante, los efectos secundarios agudos más comunes atribuibles a la mifamurtida son fiebre y síntomas de tipo gripal (mialgia, artralgia, malestar, escalofríos, etc.), que responden satisfactoriamente a dosis convencionales de ibuprofeno.
Además de los indicados anteriormente, existe una amplia y creciente variedad de agentes inmunoadyuvantes que están siendo progresivamente incorporados no solo en vacunas experimentales anticancerosas sino incluso en vacunas frente a microorganismos actualmente en uso (como ocurre con la vacuna frente al virus del papiloma humano, VPH). Un ejemplo es la emulsión lipídica de monofosforil lípido A procedente de micobacterias o de bacterias como la Salmonella minnesota, el producto sintético poly I: Co la hemocianina de la lapa californiana (KLH).
En concreto, el monofosforil lípido A es un derivado lipopolisacarídico que se comporta como un potente agonista del eje de señalización TLR4-TICAM1 que está presente como inmunoadyuvante en la vacuna Cervarix® frente al VPH. Como ya se ha mencionado anteriormente, los receptores tipo Toll (o Toll-like receptors; TLR) forman una familia de proteínas presentes en la membrana de numerosas células del sistema inmunitario innato. Estos receptores reconocen patrones moleculares expresados por un amplio espectro de agentes infecciosos y otras células anómalas, estimulando respuestas inflamatorias. Además, la señalización mediada por los TLR en las células presentadoras de antígeno (APC) constituye un vínculo importante entre la respuesta inmune innata y la adaptativa. La TICAM1 (toll like receptor adaptor molecule 1) es una proteína intracelular señalizadora que intermedia en las interacciones entre los receptores tipo toll (TLR) y los componentes transductores de la señal bioquímica.
Por su parte, el ácido poliinosínico: policitidílico (poly I: C) es un agente inmunoestimulante sintético ampliamente utilizado en investigación que actúa sobre el receptor 3 de tipo Toll (TLR3) que es expresado en la membrana de los linfocitos B, macrófagos y células dendríticas. Es estructuralmente similar a un ARN de doble cadena.
Montanide ISA 51 VG es una emulsión W/O (agua en aceite) con actividad inmunoadyuvante, que actúa potenciando la actividad de los linfocitos T citotóxicos en respuesta a los antígenos tumorales. Contiene monooleato de dianhidro-D-manitol (mannide) como agente surfactante. En este tipo de emulsiones W/O el antígeno puede suspenderse en la fase acuosa, para ser emulsionada posteriormente. La administración de estas emulsiones adyuvantes da lugar a la formación de un depósito en el sitio de inyección, que permite la liberación lenta del antígeno y la estimulación de las células plasmáticas productoras de anticuerpos. Los efectos secundarios frecuentes de las emulsiones incluyen reacciones inflamatorias, granulomas y úlceras en el sitio de la inyección.
El empleo de un adyuvante compuesto de saponina, derivado de la corteza de Quillaia saponaria, colesterol y fosfolípido en combinación con diversos antígenos, incluyendo los del virus del papiloma humano (HPV), el virus de la hepatitis C (HCV) y el antígeno de cáncer humano NY-ESO-1, ha mostrado potenciar la respuesta de células T facilitadoras (Th; CD4+) y citotóxicas (CD8+) contra los antígeno mencionados. Además, este agente podría reducir la cantidad de antígeno necesaria para inducir una respuesta inmune eficaz en el huésped, facilitando con ello la producción de vacunas basadas en células dendríticas.
Las hemocianinas son glicoproteínas con un grupo prostético que contiene cobre10 (en lugar del hierro, como en la hemoglobina) con capacidad para transportar oxígeno que están presentes en algunos moluscos y artrópodos. El término hemocianina alude al color azul característico. Además de esta propiedad biológica, las hemocianinas procedentes de moluscos son capaces de inducir una potente respuesta inmunológica en mamíferos, lo que posiblemente está relacionado con su gran tamaño molecular (4-8 millones de Da) y la presencia de restos oligosacáridos y de una estructura cuaternaria caracterizada por múltiples epítopos repetidos.
Se ha sugerido que la inmunogenicidad de las hemocianinas podría relacionarse con la existencia de linfocitos B de memoria, generados por una estimulación previa con xenoantígenos similares, de tal manera que los anticuerpos preformados facilitarían su incorporación y procesamiento por células presentadoras de antígeno. Estas propiedades inmunoestimulantes de las hemocianinas las han llevado a ser utilizadas como proteínas transportadoras (carrier) para producir anticuerpos contra haptenos y péptidos, particularmente en la formulación de vacunas experimentales de células dendríticas contra diversos tipos de cáncer. Además, por sí solas han sido estudiadas como inmunoestimulantes no específicos en la terapia del carcinoma superficial de vejiga como alternativa al BCG. La hemocianina de la lapa californiana(Megathura crenulata) o KHL (keyhole limpet)ha venido siendo utilizada durante más de tres décadas para esta finalidad; otra variante de ésta es la hemocianina del molusco loco11.
Bibliografía
La aterosclerosis carotídea consiste en un engrosamiento patológico de la capa íntima de la arteria carótida primitiva o la interna, en zonas focales (placas o ateromas). Aunque los ateromas pueden permanecer estables durante muchos años, la ruptura de la cubierta de las placas inestables puede provocar la formación de trombos locales, con la ulterior embolización a los territorios de la arteria oftálmica, cerebral media o cerebral anterior homolateral. Los efectos resultantes pueden ir desde son ceguera fugaz hasta la ceguera persistente o a un ictus de gravedad variable. En este sentido, la progresión del volumen de la placa carotídea en pacientes con diabetes tipo 2 es habitual.
Algunos estudios observacionales previos mostraron una posible asociación entre el consumo moderado de bebidas alcohólicas y la reducción del riesgo de enfermedad coronaria. Esto dio lugar a que un grupo investigara si consumir vino moderadamente afecta a la progresión de la aterosclerosis carotídea. En el estudio CASCADE (CArdiovaSCulAr Diabetes and Ehanol), un ensayo aleatorizado y controlado de 2 años, se asignó aleatoriamente un grupo de abstemios con diabetes tipo 2 a beber diariamente 150 ml de vino tinto, vino blanco o agua, durante 2 años. Además, los grupos fueron instruidos para mantener una dieta mediterránea. Se hizo un seguimiento de los cambios a lo largo de 2 años en el volumen total de la placa carotídea (TPV carotídeo) y el volumen de la pared de los vasos carotídeos (VWV carotídeo), utilizando técnicas de ultrasonidos.
Los resultados mostraron que después de 2 años, no se observó progresión significativa en el TPV carotídeo (agua: –1,4 mm3; vino blanco, –1,2 y vino tinto, –1,3). Sin embargo, en un análisis post hoc, se separaron a los participantes con una placa carotídea detectable en terciles. Aquellos con la carga de placa basal más grande, a quienes se les asignó beber vino, redujeron su volumen de placa significativamente después de 2 años, en comparación con el valor inicial. Las reducciones de dos años en la proporción de ApoB/ApoA se asociaron independientemente con la regresión en el TPV carotídeo (β = 0,4; p <0,001), así como las disminuciones en la presión arterial sistólica se asociaron independientemente con la regresión en el VWV carotídeo (β = 0,2; p = 0,005). En definitiva, no se observó progresión en el TPV carotídeo, pero aquellos con la mayor carga de placa y que bebieron vino sí experimentaron una pequeña regresión de la carga de placa.

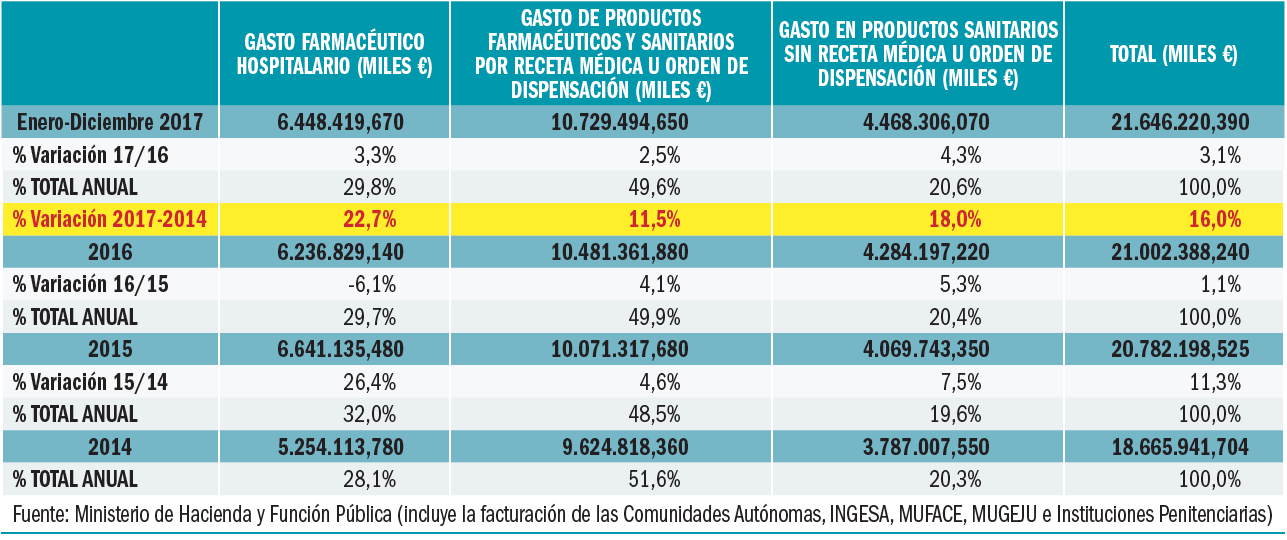
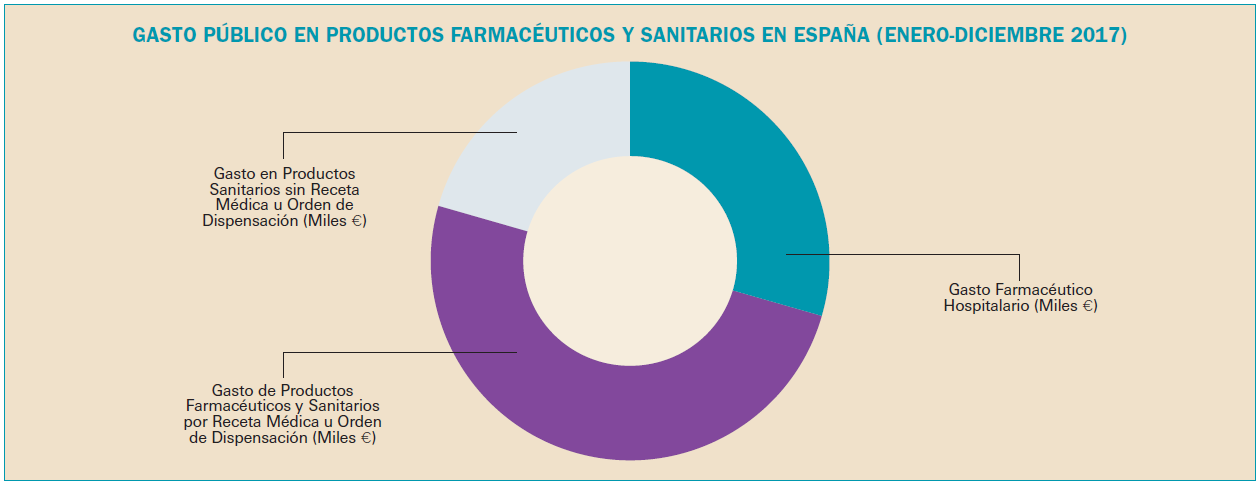
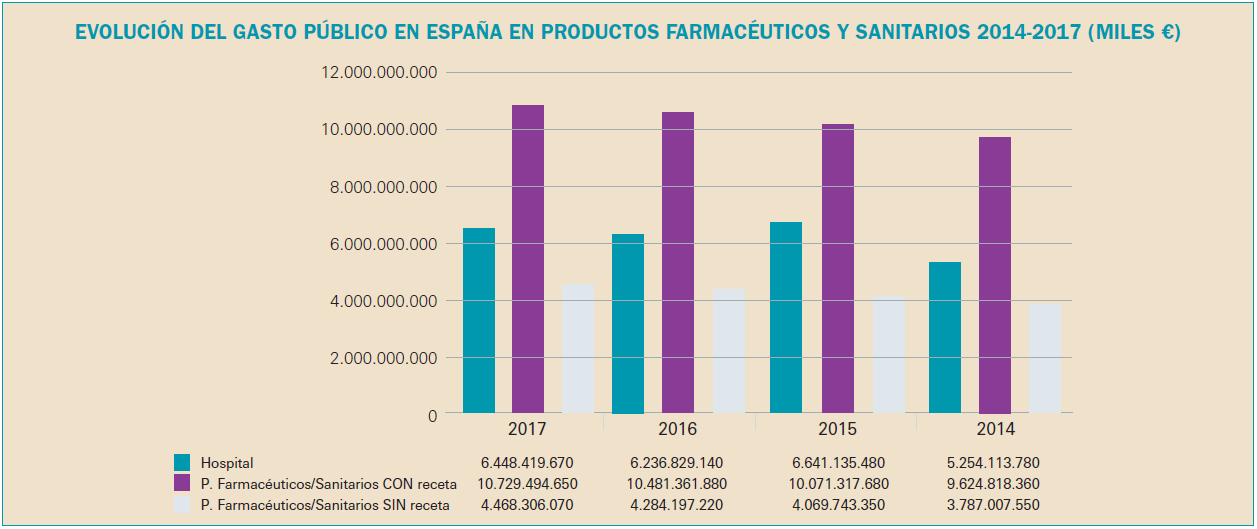
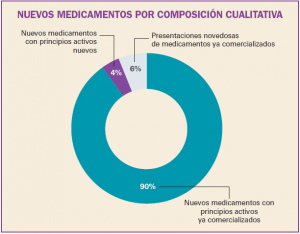
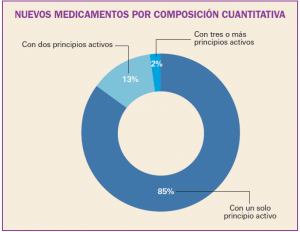
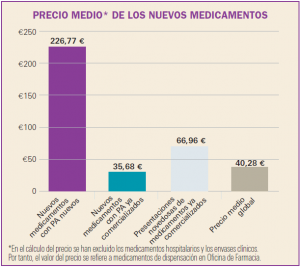
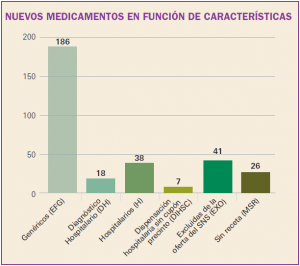
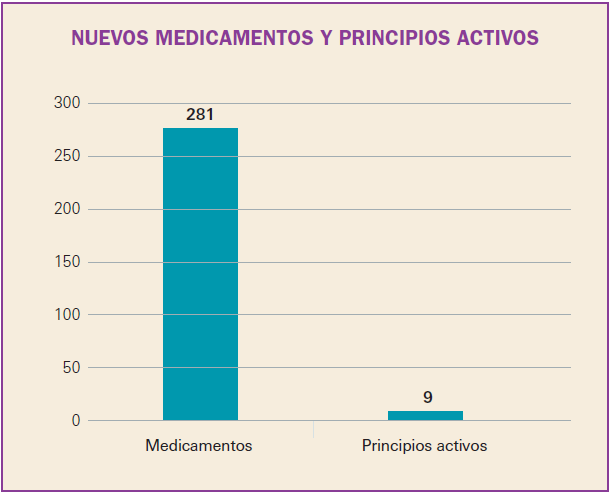{kind=link}
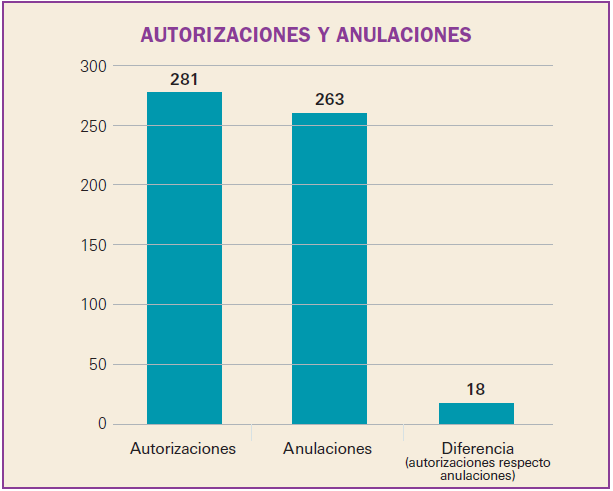{kind=link}
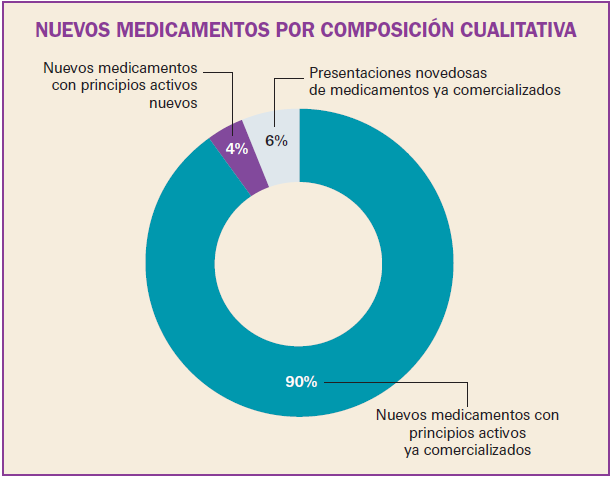{kind=link}
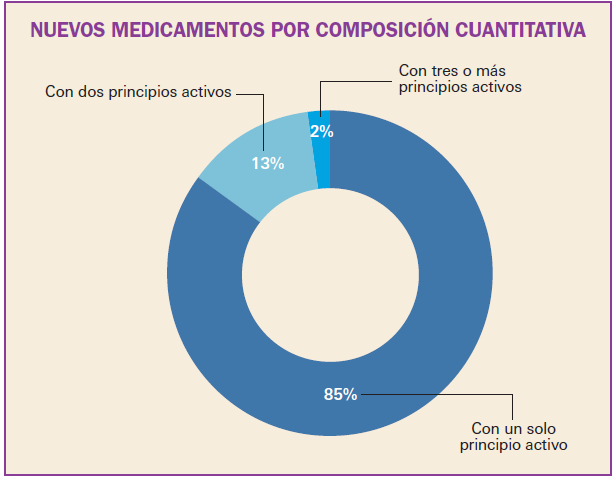{kind=link}
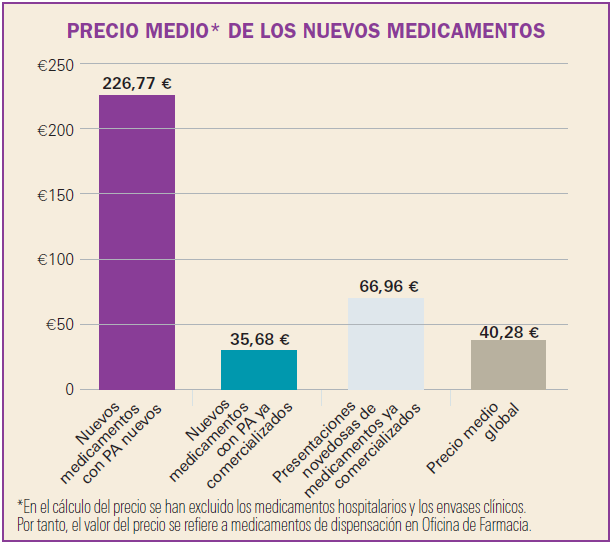{kind=link}
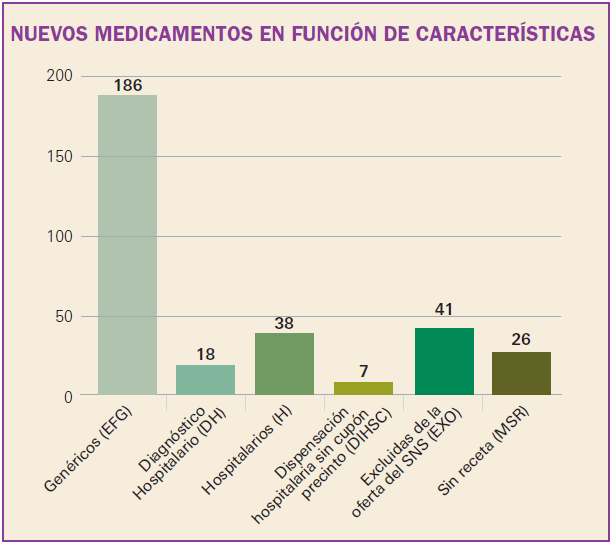{kind=link}
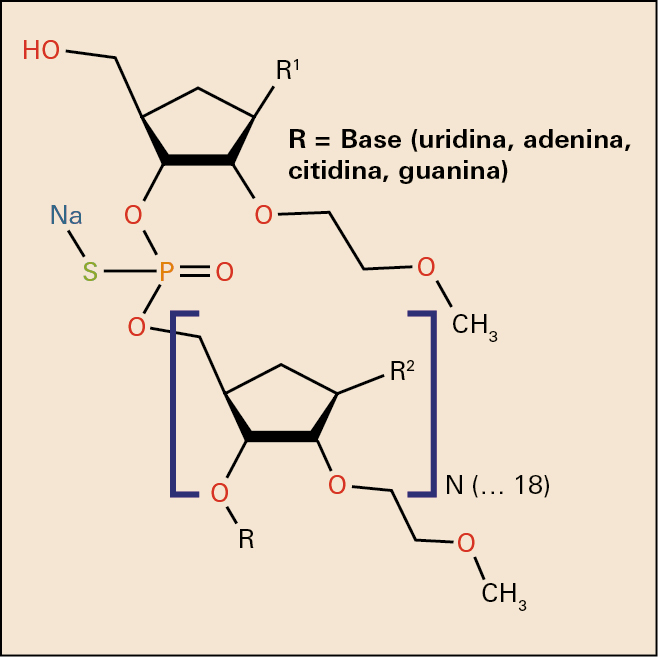{kind=link}
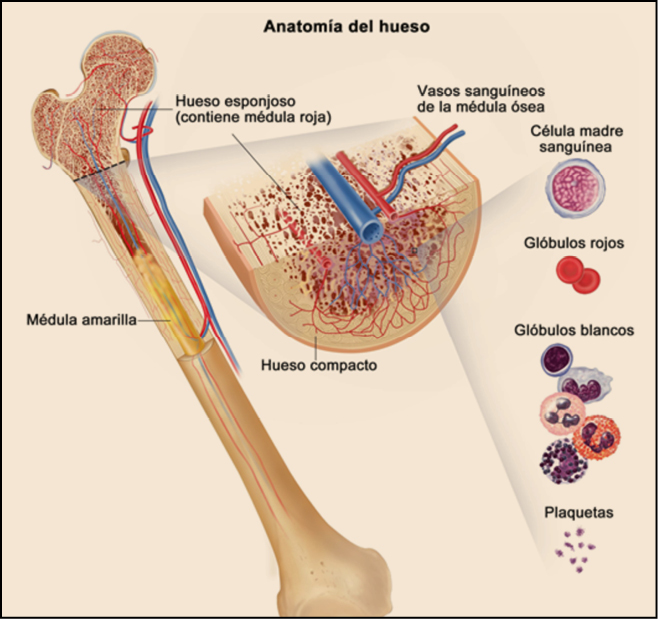{kind=link}
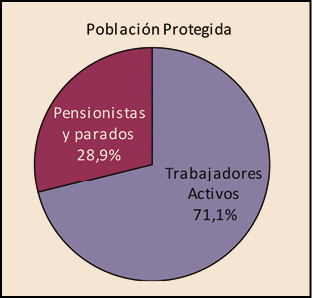{kind=link}
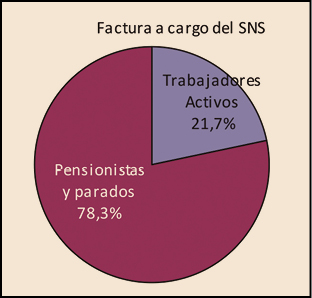{kind=link}
{kind=link}