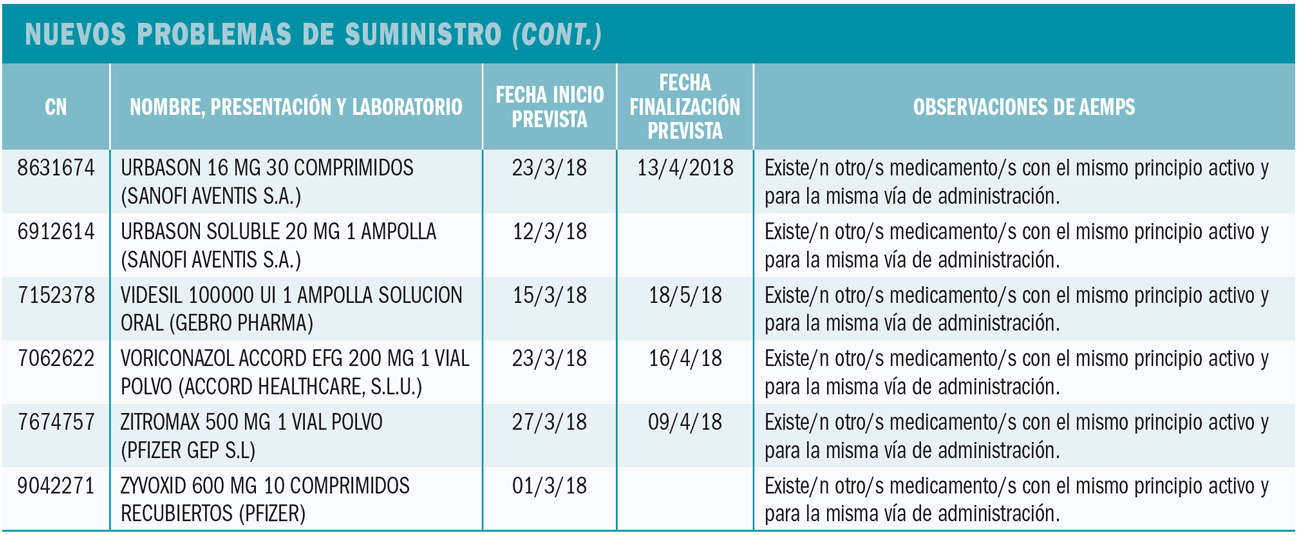
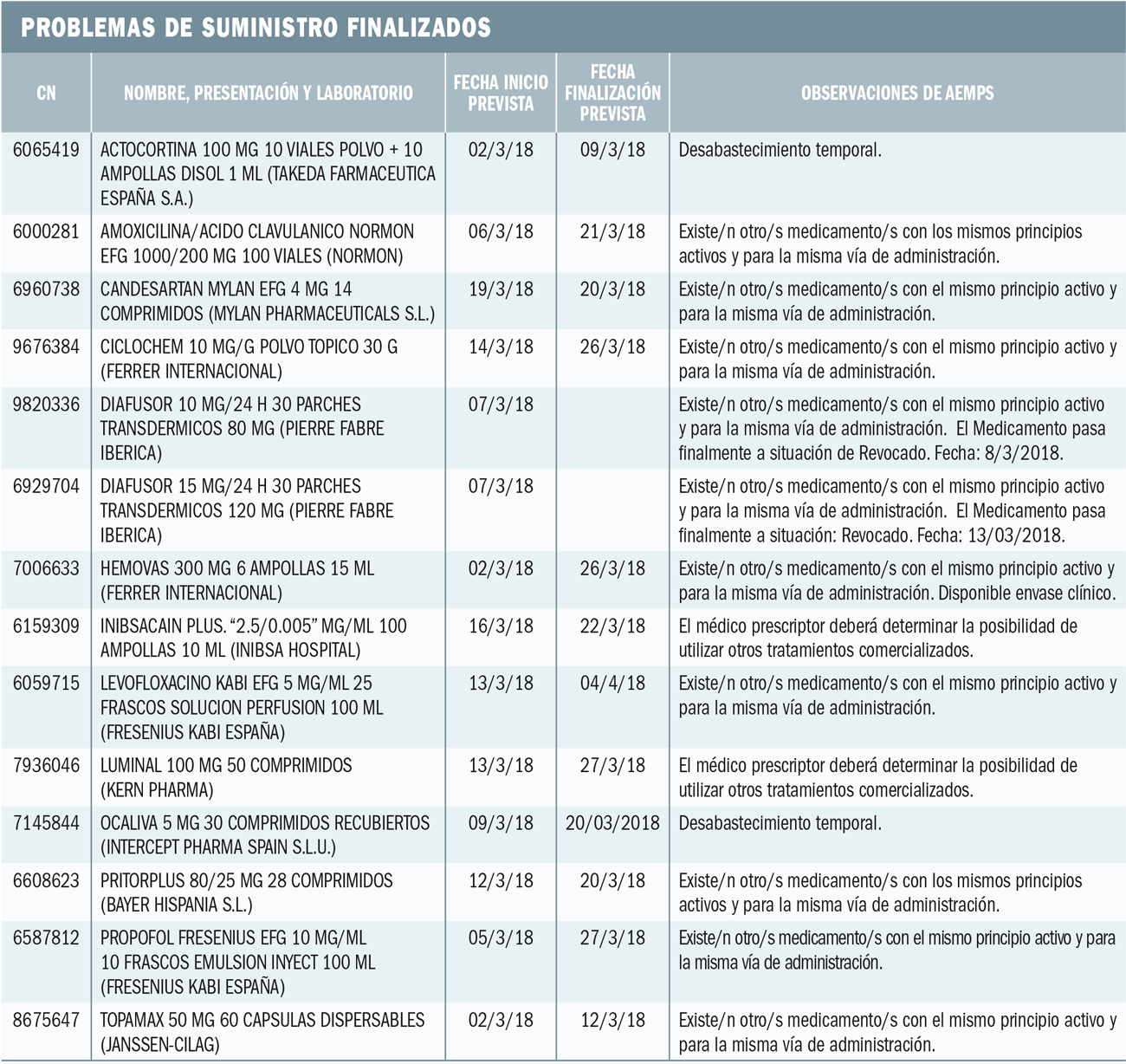
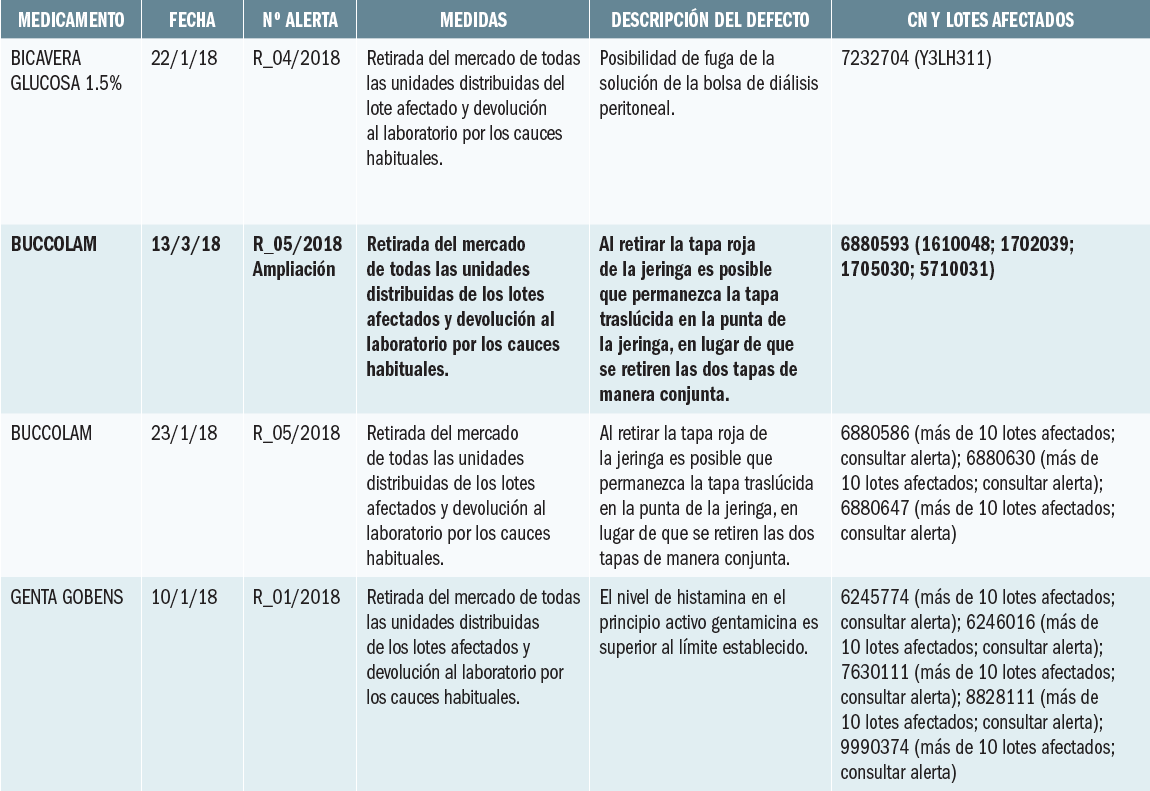
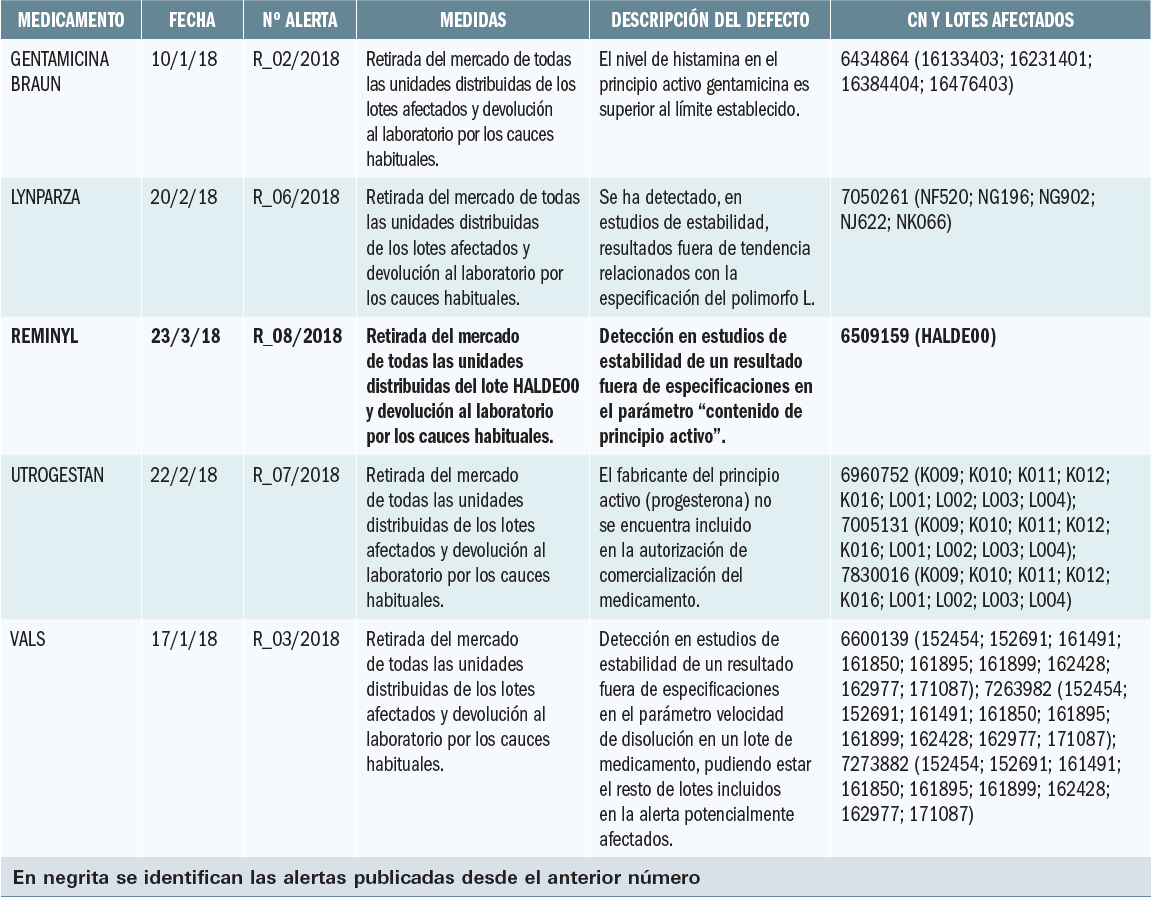
La Agencia Francesa de Seguridad de Medicamentos ha informado de los resultados de un estudio clínico de Dinamarca, en el que se constata que ibuprofeno a dosis altas y periodos prolongados de tratamiento puede afectar a la fisiología de los testículos.
Recientemente la Agencia Francesa de Seguridad de Medicamentos y productos para la Salud (ANSM) ha informado1 de los resultados de un estudio llevado a cabo en Dinamarca con ibuprofeno. El ibuprofeno es un medicamento antiinflamatorio no esteroideo (AINE) utilizado principalmente para el dolor y la fiebre.
La ANSM ha tomado nota de los resultados de un estudio2 realizado en Dinamarca, en colaboración con un equipo del francés INSERM (Institut national de la santé et de la recherche médicale), sobre los efectos del ibuprofeno en la fisiología testicular.
Este estudio, que involucró a 31 voluntarios deportivos varones sanos, 14 de los cuales recibieron ibuprofeno en una dosis de 1.200 mg diarios durante 6 semanas, sugiere que el ibuprofeno administrado en estas condiciones puede alterar la fisiología testicular. Sin embargo, los niveles de testosterona observados en estos voluntarios siguen siendo normales. Además, no se encontraron consecuencias clínicas (trastornos de la fertilidad masculina, impotencia, trastornos de la libido) a partir de estas observaciones biológicas.
Este estudio se está analizando a nivel europeo para determinar, en particular, si se necesitan más estudios sobre estos efectos.
En esta etapa, estos resultados no alteran la relación beneficio-riesgo del ibuprofeno cuando se usa de acuerdo con su ficha técnica en las condiciones autorizadas de utilización.
Recomendaciones
La ANSM recuerda a los profesionales sanitarios y a los pacientes la gran importancia de respetar las modalidades de tratamiento (dosis y duración) definidas en el marco de la autorización de comercialización (ver Fichas Técnicas y Prospectos) de ibuprofeno, a saber, un uso con la dosis efectiva más baja, durante el menor tiempo necesario para aliviar los síntomas del paciente.
Referencias
- ANSM. Rappel sur le bon usage de l’ibuprofène après la publication d’une étude qui suggère des perturbations de la physiologie testiculaire – Point d’Information. ANSM, 10 de enero de 2018, Francia. Disponible en: http://ansm.sante.fr/S-informer/Points-d-information-Points-d-information/Rappel-sur-le-bon-usage-de-l-ibuprofene-apres-la-publication-d-une-etude-qui-suggere-des-perturbations-de-la-physiologie-testiculaire-Point-d-Information (consultado 3 abril 2018).
- Kristensen DM, et al. Ibuprofen alters human testicular physiology to produce a state of compensated hypogonadism. PNAS, 2018: Disponible en: www.pnas.org/cgi/doi/10.1073/pnas.1715035115 (consultado 3 abril 2018).