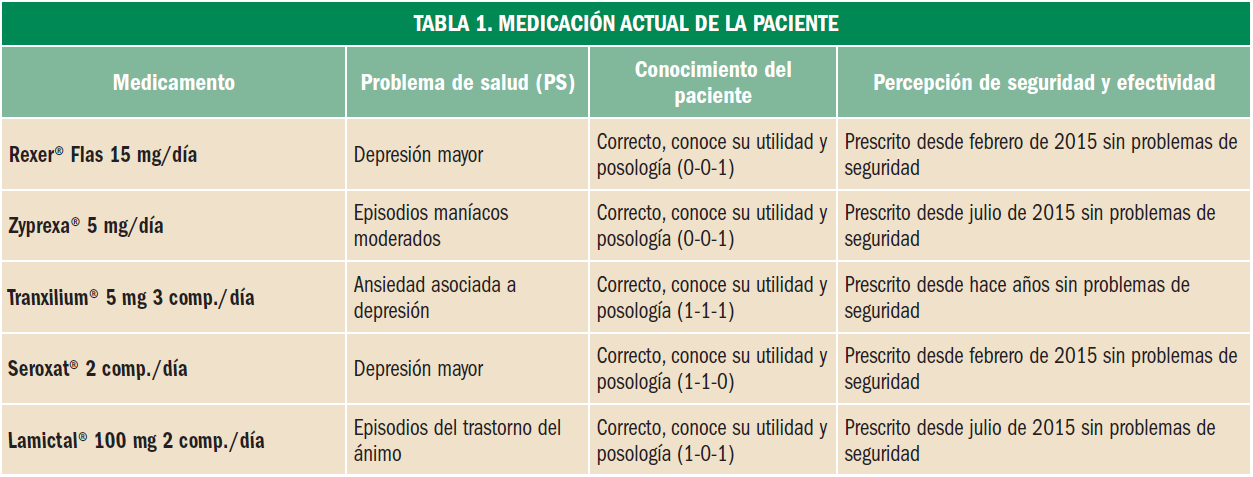
Depresión
La utilidad del tratamiento depresivo en pacientes con VIH
Una reciente revisión Cochrane demuestra la eficacia global de la terapia antidepresiva para el tratamiento de la depresión en las personas infectados por VIH; sin embargo, la baja calidad de los estudios y la ausencia de datos en pacientes de países en desarrollo, limitan la generalización de las conclusiones. Además, los autores recomiendan que los nuevos estudios que se lleven a cabo sobre la materia incorporen una evaluación de los modelos de atención escalonada y las intervenciones del sistema de salud en el diseño del estudio, así como tener presentes los resultados relacionados con la atención del VIH y la terapia antirretroviral.
Las tasas de depresión mayor entre las personas infectadas por el VIH son sustancialmente más altas que las observadas en la población general y esto puede afectar negativamente los resultados del tratamiento antirretroviral. Existen varios factores clínicos y psicosociales que pueden contribuir al desarrollo y la persistencia de la depresión en estas personas, y ello cuestiona la eficacia de la terapia antidepresiva, al menos en comparación con la población general.
Se ha llevado a cabo una revisión Cochrane a partir de ensayos clínicos aleatorizados y controlados con medicamentos antidepresivos en comparación con placebo u otra clase de medicamentos antidepresivos. Los participantes elegibles para la inclusión tenían que ser mayores de 18 años, y tener VIH y depresión. Se seleccionaron para la evaluación diez estudios que incluían 709 participantes, de los que ocho se realizaron en países desarrollados (Estados Unidos e Italia), siete se realizaron antes de 2000 y siete consideraron predominantemente poblaciones de varones; siete estudios evaluaron antidepresivos vs. placebo, dos compararon diferentes clases de antidepresivos y uno tuvo tres brazos que compararon dos clases de antidepresivos con placebo.
Los resultados globales mostraron que el tratamiento antidepresivo resultó en una mayor mejoría en la depresión en comparación con el placebo. Hubo una mejoría moderada en la depresión cuando se evaluó con la puntuación de la escala de depresión de Hamilton (HAM-D) como resultado continuo (variación media 0,59; IC95% 0,21 a 0,96). Sin embargo, no hubo evidencia de mejoría cuando se evaluó con la puntuación HAM-D como resultado dicotomizado (riesgo relativo, RR=1,10; IC95% 0,89 a 1,35) o con la puntuación de Impresión Clínica Global de Mejora (Clinical Global Impression of Improvement, CGI-I) (RR=1,28; IC95% 0,93 a 1,77).
Los métodos de notificación de eventos adversos variaron sustancialmente entre los estudios, lo que resultó en pruebas de muy baja calidad que pudieran contribuir a una estimación combinada adecuada. Por ello, no se pudo determinar si había una diferencia en la proporción de participantes que experimentaron eventos adversos en los grupos con antidepresivos en relación al placebo; sin embargo, la disfunción sexual se informó comúnmente en personas que reciben inhibidores selectivos de la recaptación de serotonina (ISRS), mientras que las que recibieron antidepresivos tricíclicos con frecuencia informaron de efectos adversos típicamente anticolinérgicos, como sequedad de boca y estreñimiento. No se informaron eventos adversos graves (grados 3 o 4) en ningún grupo de estudio; tampoco hubo evidencia de diferencias en el recuento de linfocitos CD4 de seguimiento al finalizar el estudio (variación media de -6,31 células/mm3; IC95%: -72,76 a 60,14). Solo un estudio evaluó el puntaje de calidad de vida (variación media de puntuación +3,60; IC95%: -0,38 a 7,58), pero debido a la baja calidad de los datos no se pudieron sacar conclusiones para este resultado. Las diferencias entre antidepresivos ISRS y ATC no fueron estadísticamente significativas (variación media de -3,20; IC95%: -10,87 a 4,47).
Algunos datos sugieren que la mirtazapina produjo globalmente una mejoría mayor en la depresión que los antidepresivos ISRS (variación media de 9,00; IC95%: 3,61 a 14,39); sin embargo, este resultado no fue consistente para todas las medidas de mejoría en la depresión. Ningún estudio aportó datos sobre la supresión virológica ni ningún otro resultado específico del VIH. Además, como indican los autores de la revisión, los estudios incluidos tenían un riesgo sustancial de sesgo debido a la información insuficiente de los métodos de estudio, elevado riesgo de deserción de pacientes, entre otros tipos de sesgo. Por otro lado, la heterogeneidad entre los estudios y el número limitado de participantes y de eventos degradaron la calidad de la evidencia para diversas variables consideradas.




{kind=link}