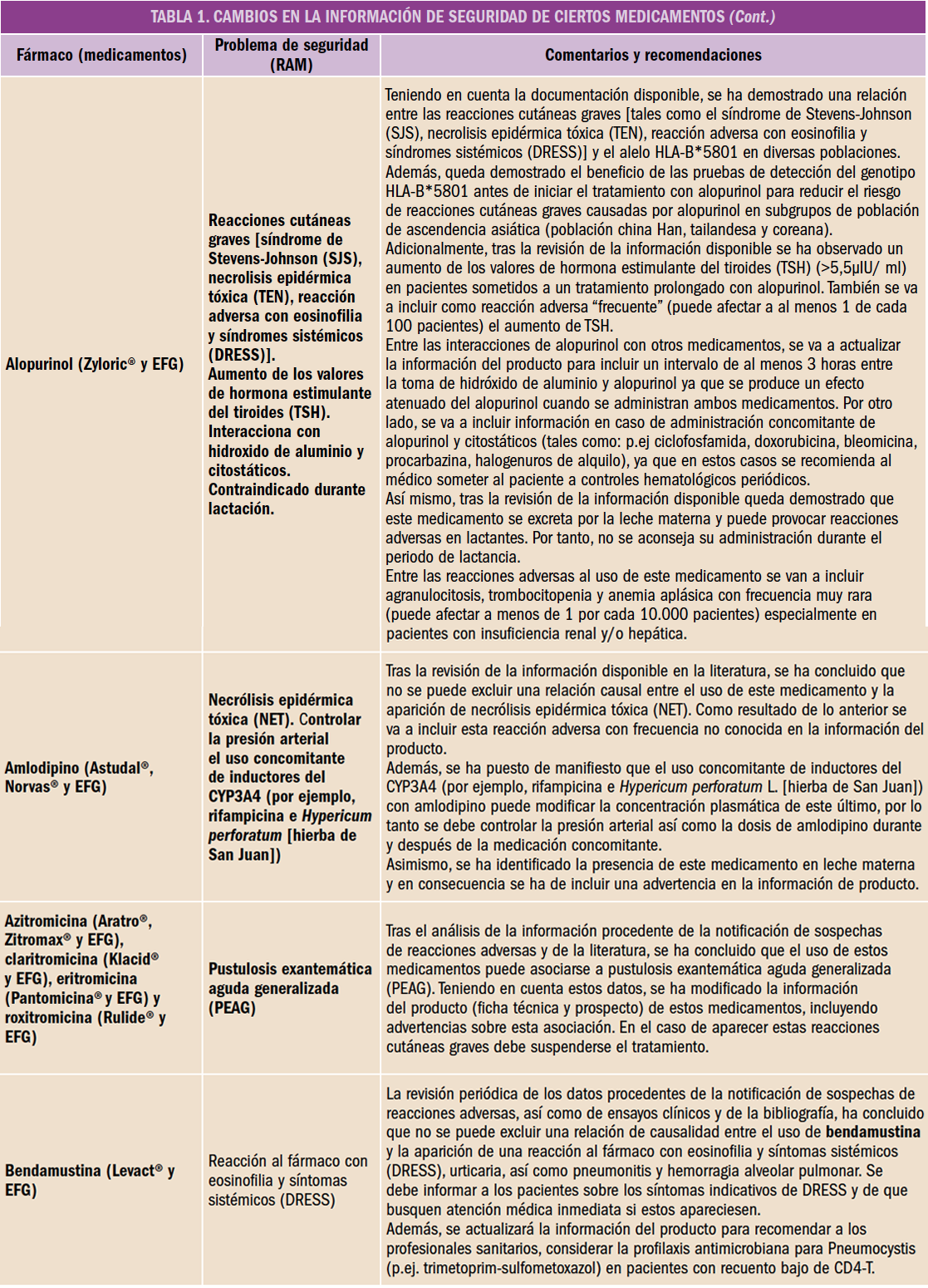
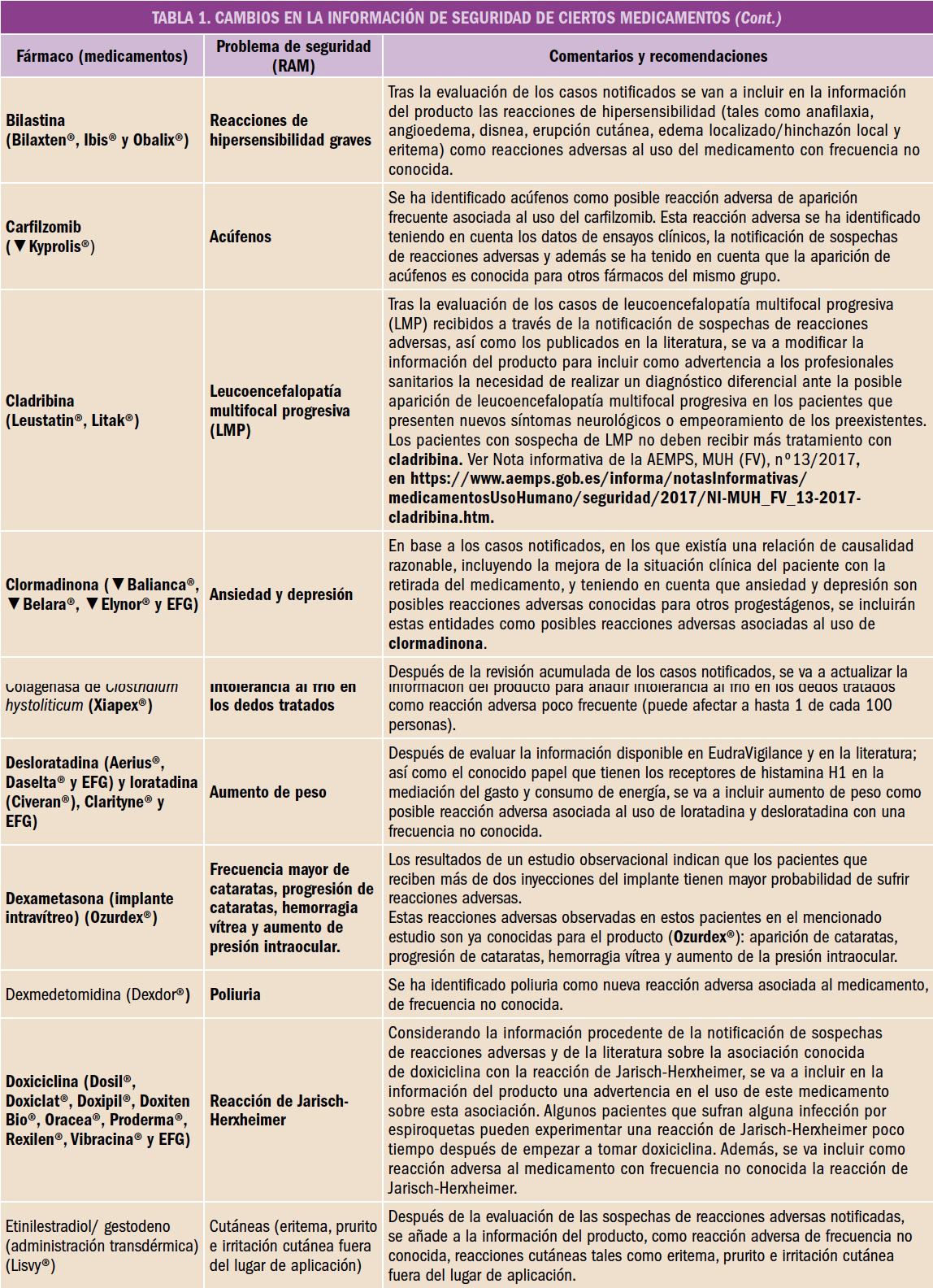
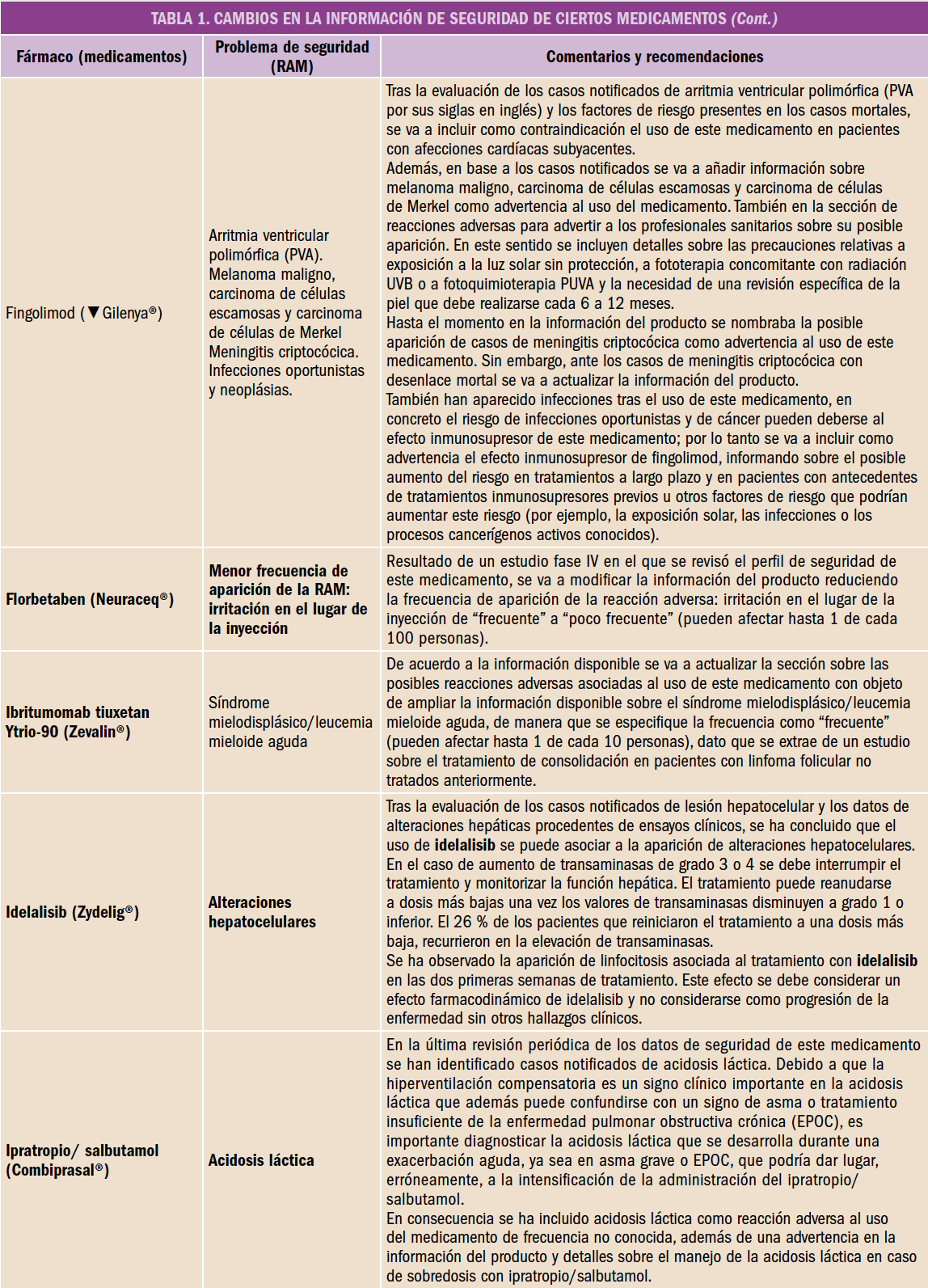
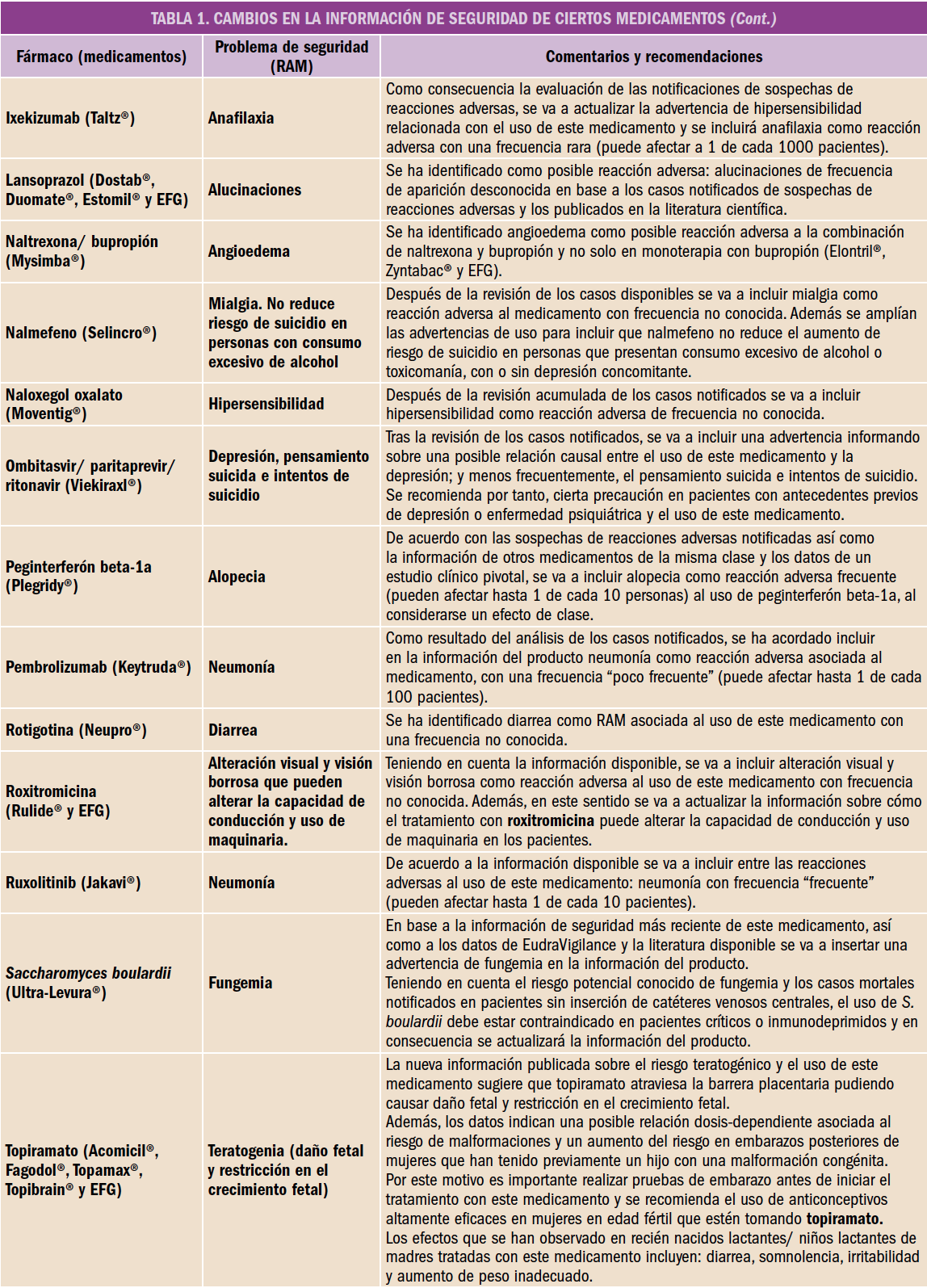
Desde el Consejo General de Colegios Oficiales de Farmacéuticos y los Colegios se está impulsando el avance hacia una Farmacia de Servicios Asistenciales, lo que lleva a plantear, un año más, la puesta en marcha de una nueva Acción, la 4ª de HazFarma, en colaboración con Laboratorios Cinfa.
HazFarma1 es una ambiciosa iniciativa que desde 2014 proporciona al Farmacéutico Comunitario los medios y las herramientas necesarias para el desarrollo de Servicios Profesionales Farmacéuticos Asistenciales (SPFA), reforzando la colaboración con otros profesionales de la salud, e impulsando la valoración de la actuación del farmacéutico.
A través de Acciones anuales, HazFarma da respuesta a los principales objetivos de esta iniciativa como son:
- Comprometerse con la sociedad y el paciente.
- Protocolizar y formar en Servicios.
- Proporcionar una asistencia sanitaria de calidad.
- Promover la práctica colaborativa.
- Valorar el trabajo del farmacéutico para obtener resultados en salud.
- Llegar a todos los profesionales sanitarios, a las administraciones y a la sociedad.
De acuerdo con la definición consensuada en 2016 por Foro de Atención Farmacéutica en Farmacia Comunitaria (Foro AF-FC), los Servicios Profesionales Farmacéuticos Asistenciales2 son aquellas actividades sanitarias prestadas desde la farmacia comunitaria por un farmacéutico que emplea sus competencias profesionales para la prevención de la enfermedad y la mejora tanto de la salud de la población como la de los destinatarios de los medicamentos y productos sanitarios, desempeñando un papel activo en la optimización del proceso de uso y de los resultados de los tratamientos.
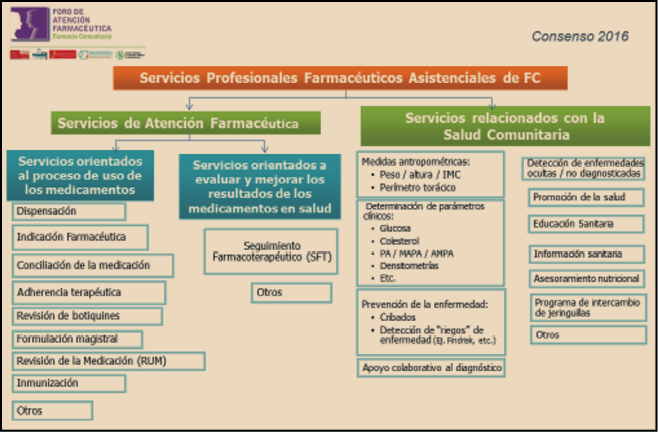
Figura 1. Clasificación de Servicios Profesionales Farmaceuticos Asistenciassles en Farmacia Comunitaria. Consenso Foro AF-FC 2016.
Dichas actividades, alineadas con los objetivos generales del sistema sanitario, tienen entidad propia, con definición, fines, procedimientos y sistemas de documentación, que permiten su evaluación y retribución, garantizando su universalidad, continuidad y sostenibilidad.
El desarrollo de los SPFA clasificados (Figura 1) implica la cooperación con el médico y otros profesionales sanitarios, para conseguir resultados óptimos en la salud de la población, así como su intervención en actividades que promocionen la salud y prevengan las enfermedades.
En este sentido, los farmacéuticos, siendo parte del Sistema Nacional de Salud, compartimos con los pacientes, los médicos, otros profesionales de la salud y las Autoridades Sanitarias, la misión de garantizar el uso seguro, efectivo y eficiente de los medicamentos. En este entorno multidisciplinar, el farmacéutico ha de aportar conocimientos y habilidades específicas para mejorar la calidad de vida de los pacientes en relación con la farmacoterapia y sus objetivos.
Este planteamiento responde a las demandas de una sociedad cada vez más preocupada por alcanzar su bienestar, informada y formada en todos los aspectos relacionados con el concepto de salud.

Este año se plantea una propuesta de trabajo para la aplicación de la práctica asistencial en salud de la mujer, concretamente en las alteraciones hormonales que padecen las mujeres en edad adulta, con el objetivo de actualizar los conocimientos del farmacéutico comunitario en el abordaje asistencial de este perfil.
La Acción finalizará el 31 de julio de 2018.
Justificación
La atención a la mujer adulta, especialmente durante la transición climatérica, es considerada en la actualidad como una prioridad sanitaria por los profesionales de la salud, las autoridades sanitarias y la sociedad en general. Además, supone un reto debido al volumen de población afectada y a la creciente demanda de un abordaje más personalizado.
El climaterio (del griego klimater: escalón) es el periodo que abarca desde la madurez sexual hasta la senectud. En él se producen importantes cambios hormonales, pudiéndose diferenciar diferentes etapas3 (Figura 2).
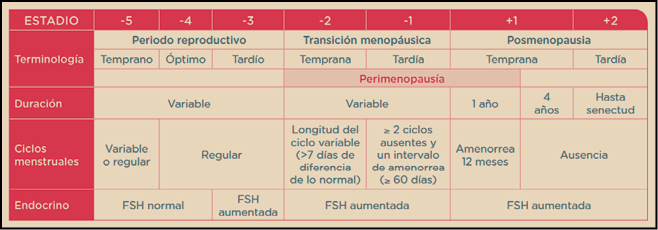
Figura 2. Etapas del Climaterio de la mujer.
La edad adulta en la mujer se presenta como una etapa de profundos cambios en todos los ámbitos. Entre ellos, las transformaciones en el ámbito fisiológico adquieren especial relevancia por su impacto en la vida de la mujer. La mujer experimenta importantes cambios hormonales, que se inician tiempo antes de la menopausia, alrededor de los 45 años y se prolongan durante el tiempo en una etapa en la que la menopausia es la protagonista.
En términos generales, la menopausia se presenta a una edad media de 50 años y aunque la edad de menarquia es cada vez más temprana, la edad de menopausia se mantiene. Debido al aumento progresivo de la esperanza de vida de la población en los países desarrollados, el número de mujeres en climaterio es cada vez mayor (aproximadamente el 50% de la población femenina en España)4. La percepción de la menopausia por parte de la sociedad y de las propias mujeres ha evolucionado a lo largo del tiempo: mientras tiempo atrás se veía como la decadencia tras el periodo fértil de la mujer, en nuestros días se percibe como un proceso fisiológico que forma parte de la vida de la mujer al que hay que prestar especial atención para asegurar el mejor estado de salud posible y una calidad de vida satisfactoria en los años posteriores de vida de la mujer.
Aunque el término “menopausia” (del griego menós: mes y paûsis: cese) hace referencia a la última menstruación de la mujer con el útero intacto, se entiende por menopausia una situación fisiológica determinada que conlleva profundos cambios en la mujer y cuya sintomatología puede afectar de modo importante a su calidad de vida.
El diagnóstico de la menopausia es retrospectivo, de modo que, según la OMS y la Sociedad Española de Ginecología y Obstetricia (SEGO), la menopausia natural se define como “el cese permanente de la menstruación determinado de manera retrospectiva, después de 12 meses consecutivos de amenorrea, sin causas patológicas”. Este proceso viene determinado por la depleción de folículos ováricos con la consiguiente pérdida de secreción de estrógenos ováricos.
La sintomatología de la menopausia es muy variada y presenta grandes diferencias tanto en los síntomas como en su intensidad entre las mujeres. Así, la menopausia está relacionada con la aparición de síntomas del déficit estrogénico (síntomas vasomotores – sofocos y sudoración –, vaginales y urinarios – atrofia urogenital, dispareunia, polaquiuria, infecciones –, alteraciones del estado del ánimo- dolor de cabeza, insomnio, ansiedad, angustia, depresión, pérdida de memoria, falta de concentración-, aumento de peso, alteraciones cutáneas, etc), con alteraciones hormonales (hipo e hipotiroidismo) y con la aparición de enfermedades crónicas (osteoporosis, etc.), por lo que actualmente son muchas las mujeres susceptibles de padecer este tipo de patologías.
En este contexto de epidemiología, es importante identificar qué síntomas están asociados a la pérdida de función ovárica y cuáles son consecuencia de la edad. En muchos casos, la menopausia, cuyo abordaje debe hacerse desde una perspectiva global, abarcando, además del aspecto sanitario, la esfera psicosocial, se ha sobremedicado.
Es por todo ello que la atención a la mujer adulta tiene por objeto, por un lado, mejorar sus síntomas, algunos de los cuales afectan en gran medida a su calidad de vida y, por otro lado, prevenir la aparición de enfermedades crónicas.
El Farmacéutico Comunitario, por su formación y cercanía a esta población, puede ejercer un papel central en las actividades preventivas y de promoción de la salud orientadas a la mejora de la calidad de vida y que contribuyan a la optimización de los resultados de salud en el contexto de continuidad del proceso de seguimiento de la mujer a lo largo de su ciclo vital y la eficiencia global del Sistema Sanitario.
Objetivos de la Acción
- Mejorar la calidad de la atención en un paciente muchas veces “invisible”, como es la mujer adulta.
- Implicar al farmacéutico en Campañas de Educación Sociosanitaria.
- Promover la realización de Servicios Profesionales Farmacéuticos Asistenciales.
- Demostrar el valor añadido del farmacéutico en los Servicios de Dispensación de medicamentos e Indicación Farmacéutica ante los problemas de salud relacionados con las mujeres en la edad adulta.
- Potenciar la imagen del Farmacéutico Comunitario como uno de los pilares básicos del equipo asistencial, tanto para la sociedad como para el resto de profesionales sanitarios y la Administración.
Metodología
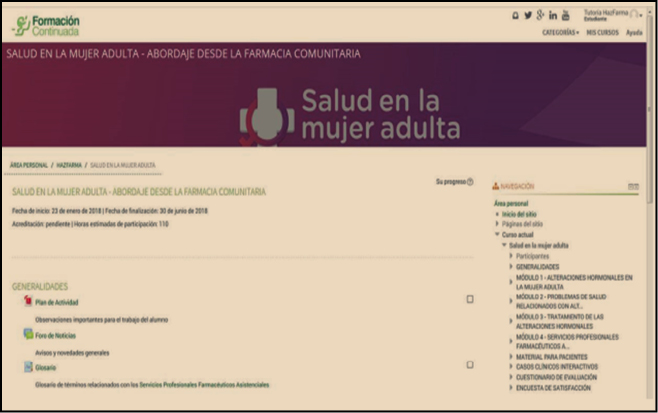
Figura 3. Previsualización del contenido de la Acción en la plataforma on line de formación continuada.
Todos los Colegios disponen de los materiales impresos correspondientes a la Acción, para su distribución entre los farmacéuticos inscritos. Así mismo y de forma individual cada inscrito recibirá por correo electrónico la clave y contraseña para el acceso a la plataforma de formación on line alojada en www.portalfarma.com.
Durante 6 meses, se podrá acceder (Figura 3) al material formativo para poder cumplimentar la parte teórica (lectura y comprensión de los materiales y resolución de los test correspondientes) y la parte práctica del curso (la resolución de casos clínicos interactivos).
Al inicio de la Acción, cada Colegio podrá realizar un taller presencial explicativo. Asimismo, desde el Consejo General se desarrollará un webinar con una mesa redonda en la que se abordarán temas de interés que influyen de manera esencial en la salud de la mujer en la edad adulta por parte de profesionales especializados de la Sociedad Española de Ginecología y Obstetricia (SEGO), en pro de la calidad asistencial.
Adicional actividad voluntaria
Además, y de forma simultánea habrá una actividad voluntaria que implicará el registro de casos de los Servicios de Dispensación e Indicación Farmacéutica en la Farmacia Comunitaria: mediante esta actividad se pretende valorar la actuación del farmacéutico comunitario en relación a la dispensación de la Terapia Hormonal Sustitutiva (THS) y frente a la demanda por parte de una paciente de algún remedio para un problema de salud relacionado con la menopausia.
El registro se realizará en el Módulo de Servicios de Atencion Farmacéutica. El acceso para la realización de dicha actividad estará disponible en la Plataforma de Formación a partir del mes de marzo de 2018.
Es una actividad voluntaria que no es un requisito indispensable para optar a la acreditación. Los farmacéuticos que colaboren recibirán un reconocimiento especial y aparecerán como autores en las publicaciones a las que se remita.
Materiales
Para el farmacéutico
On line
- Guía Práctica dividida en 4 módulos que se irán alojando mensualmente en la plataforma on line:
- Módulo 1: Alteraciones hormonales en la mujer adulta
- Módulo 2: Problemas de salud relacionados con las alteraciones hormonales en la mujer adulta
- Módulo 3: Tratamiento de las alteraciones hormonales en la mujer adulta y abordaje desde la Farmacia Comunitaria
- Módulo 4: Servicios Profesionales Farmacéuticos Asistenciales; Servicios de Atención Farmacéutica y Salud Comunitaria
- Casos clínicos interactivos relacionados con cada Módulo: casos clínicos tipo, de situaciones concretas que pueden darse en la farmacia al atender a estos pacientes.
Impreso
- Fichas prácticas (Figura 4)

Figura 4. Carpeta contenedora y fichas de información para el farmacéutico y para la mujer adulta.
- Para el farmacéutico, a modo de resumen de las cuestiones más relevantes tratadas en cada Módulo: anatomía y fisiología de la mujer, las alteraciones hormonales, educación sanitaria, las opciones terapéuticas, el tratamiento no farmacológico y otros aspectos importantes sobre los problemas de salud relacionados.
- Para la mujer adulta, Recomendaciones para las mujeres con problemas derivados de la menopausia
- Recomendaciones para un uso correcto del tratamiento (IPM)
- Recomendaciones sobre problemas de salud
- Recomendaciones para la prevención de infecciones
Acreditación
- Solicitada acreditación a la Comisión de Formación Continuada de los profesionales sanitarios.
Normas para obtener la acreditación
Para optar al diploma acreditativo SON REQUISITOS OBLIGATORIOS:
- Superar el cuestionario de evaluación (70% de las respuestas correctas).
- Completar los 4 casos prácticos interactivos disponibles en la Plataforma de Formación on line del Consejo General.
Sobre el Cuestionario de evaluación:
- El test consta de 15 preguntas, SÓLO UNA CONTESTACIÓN es la correcta. Cada respuesta correcta tiene el valor de 1 punto (máximo 15). Las respuestas erróneas no computan en negativo. La superación del cuestionario requiere la obtención de, al menos, 11 puntos.
- Al cuestionario se accede a través de la Plataforma. Sólo hay una única oportunidad para enviar el cuestionario pulsando sobre el botón “enviar todo y terminar”.
No obstante, se podrá acceder al cuestionario las veces que desee y las respuestas se irán guardando automáticamente.
- El resultado del cuestionario se obtiene de forma inmediata y se tendrá la posibilidad de revisar las preguntas falladas con su correspondiente respuesta razonada.
- La fecha límite para el envío del cuestionario de evaluación será el 31 de junio de 2018.
Información de interés
- El periodo de reclamación sobre correcciones del cuestionario y/o casos prácticos finalizará a los 6 meses del cierre efectivo del curso. Pasado este periodo, sólo se admitirán reclamaciones relativas a extravíos de certificados y diplomas de farmacéuticos aprobados.
- El certificado de acreditación se hará disponible de forma electrónica a través de la Plataforma de Formación a todos los que superen los requisitos de acreditación señalados.
- Solicitud de duplicados de certificados y diplomas:
- Vía fax: escrito en el que conste si solicita copia de Certificado o de Diploma, motivo por el cual se solicita (extravío, destrucción total, destrucción parcial o modificación del original) nombre de la actividad para la cual lo solicita, teléfono de contacto, fotocopia del DNI y firma, al número 91 432 81 00.
- A través de la sección de Formación Continuada de Portalfarma (www.portalfarma.com): Solicitud de Duplicados de Certificados/Diplomas para actividades de Formación.
En caso de duda:
- COF: los responsables de cada COF ofrecerán información a los alumnos sobre las fechas de realización de talleres; los plazos de desarrollo de la acción; los requisitos para conseguir la acreditación; los resultados de los cuestionarios de evaluación, la visualización y finalización de los casos prácticos interactivos.
- CCGOF: para resolver dudas de tipo técnico, sobre los materiales formativos o sobre el manejo de la Plataforma de Formación se puede contactar con atfarmaceutica@redfarma.org o en el 91 431 25 60.
Para dudas de tipo administrativo o acerca de inscripciones, diplomas o duplicados el alumno puede contactar con secretariatecnicacgcof@redfarma.org o en el 91 432 41 00.
Si estás inscrito, solicita más información al Comité HazFARMA de tu Colegio.




{kind=link}
{kind=link}