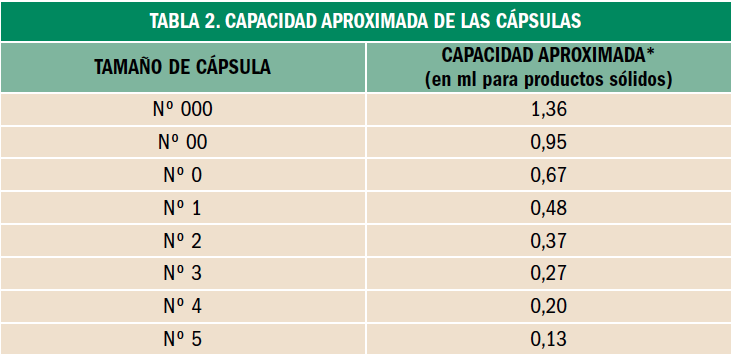
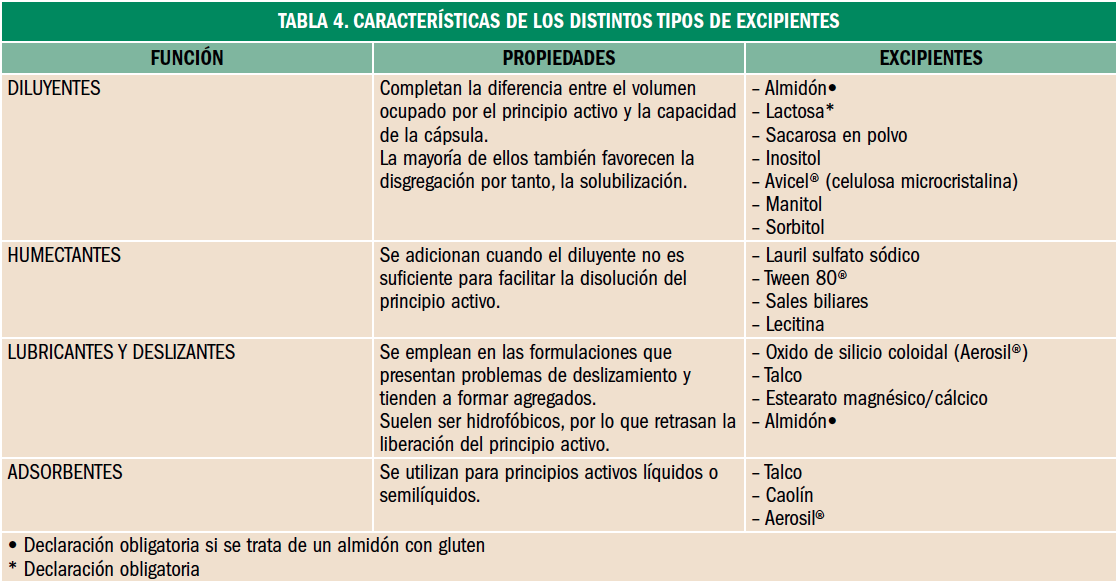
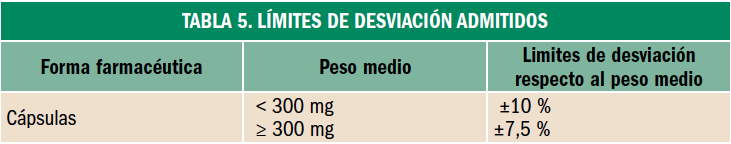
Osteoporosis
Los suplementos de calcio y vitamina D no reducen el riesgo de fractura ósea
Un reciente metanálisis de ensayos clínicos aleatorizados ha mostrado que el uso de suplementos de calcio, vitamina D o ambos, en comparación con placebo o ningún tratamiento, no se asoció con un menor riesgo de fracturas entre los adultos mayores que habitan en la comunidad. Estos resultados parecen no respaldan el uso rutinario de estos suplementos en las personas mayores que viven en la comunidad.
El aumento de la relevancia social y la carga económica de las fracturas relacionadas con la osteoporosis en todo el mundo hace que la prevención de dichas lesiones se haya convertido un importante objetivo de salud pública. Estudios previos han llegado a conclusiones contradictorias con respecto a la asociación entre el calcio, la vitamina D o los suplementos combinados de calcio y vitamina D y la incidencia de fracturas en adultos mayores.
Con el fin de investigar si la utilización de dichos suplementos podrían estar asociados o no con una menor incidencia de fracturas en los adultos mayores que viven en la comunidad, se ha realizado una revisión sistemática de las bases de datos de PubMed, Cochrane Library y EMBASE desde las fechas de inicio hasta el 24 de diciembre de 2016, con el fin de identificar revisiones sistemáticas o metanálisis. Asimismo, se identificaron los ensayos clínicos aleatorios primarios incluidos en las revisiones sistemáticas o los metanálisis, y se realizó una búsqueda adicional de los ensayos aleatorios publicados recientemente desde el 16 de julio de 2012 hasta el 16 de julio de 2017. Se realizó un metanálisis para calcular las tasas de riesgos relativo (RR) y las diferencias de riesgo absoluto (DRA). La fractura de cadera se definió como la variable primaria, mientras que la fractura no vertebral, la vertebral y fractura total fueron variables secundarias.
Un total de 33 ensayos aleatorios con 51.145 participantes cumplieron los criterios de inclusión de la revisión, no encontrándose ninguna asociación significativa entre el consumo de calcio o vitamina D con respecto del riesgo de fractura de cadera, en comparación con placebo o ningún tratamiento:
- Calcio: RR=1,53; IC95% 0,97 a 2,42; DRA= 0,01; IC95% 0,00 a 0,01.
- Vitamina D: RR=1,21; IC95% 0,99 a 1,47; DRA= 0,00; IC95% 0,00 a 0,01.
- Combinación de calcio y vitamina D: RR=1,09; IC95% 0,85 a 1,39
El análisis de subgrupos mostró que estos resultados fueron generalmente consistentes independientemente de la dosis de calcio o vitamina D, el sexo, el historial de fracturas, la ingesta de calcio en la dieta y la concentración sérica basal de 25-hidroxivitamina D.


{kind=link}