|
Principio Activo |
Nombre |
Laboratorio |
Indicación |
|
ABATACEP |
Orencia |
BRISTOL MYERS SQUIBB |
Solo o en combinación con metotrexato (MTX): artritis psoriásica activa en pacientes adultos cuando la respuesta a los fármacos antirreumáticos modificadores de la enfermedad (FAME), incluido el metotrexato, ha sido inadecuada y para aquellos que no requieran tratamiento sistémico adicional para las lesiones cutáneas psoriásicas. |
|
ABIRATERONA ACETATO |
Zytiga |
JANSSEN-CILAG |
Indicado con prednisona o prednisolona para el tratamiento de cáncer de próstata hormono-sensible recién diagnosticado, con alto riesgo de metástasis, en combinación con tratamiento de deprivación de andrógenos. |
|
ÁCIDO 5-AMINOLEVULÍNICO |
Ameluz |
BIOFRONTERA PHARMA GMBH |
Carcinoma basocelular superficial y/o nodular cuando la intervención quirúrgica esté contraindicada por riesgo de morbilidad relacionada con el tratamiento o un mal re-sultado cosmético en adultos |
|
ADALIMUMAB |
Humira |
ABBVIE |
Hidradenitis supurativa (HS) activa (acné inverso) de moderada a grave en adultos y adolescentes mayores de 12 años con una respuesta inadecuada a la terapia convencional sistémica. Uveítis anterior no infecciosa crónica en pacientes a partir de 2 años de edad que han presentado una respuesta inadecuada o son intolerantes al tratamiento convencional o para los que el tratamiento convencional no es apropiado |
|
BEVACIZUMAB |
Avastin |
Roche Farma |
Cáncer de ovario: en combinación con carboplatino y gemcitabina o en combinación con carboplatino y paclitaxel, para el tratamiento de pacientes adultos con cáncer de ovario epitelial sensible a platino tras primera recaída. Carcinoma de la trompa de Falopio, o carcinoma peritoneal primario que no hayan recibido tratamiento previo con bevacizumab, otros inhibidores VEGF o agentes dirigidos frente a receptores VEGF. |
|
CANAKINUMAB |
Ilaris |
NOVARTIS FARMACEUTICA |
Síndromes febriles en adultos, adolescentes y niños mayores de 2 años: – Síndromes Periódicos Asociados a la Criopirina, incluidos: Síndrome de Muckle-Wells (MWS). Enfermedad Neonatal Multisistémica Inflamatoria (NOMID) / Síndrome Infantil Neu-rológico Cutáneo y Articular Crónico (CINCA). Manifestaciones graves del Síndrome Autoinflamatorio Familiar inducido por el frío (FCAS)/ Urticaria Familiar Fría (FCU) que presente signos y síntomas más allá de la erupción de tipo urticaria inducido por el frío. Síndrome Periódico asociado al Factor de Necrosis Tumoral (TRAPS). Síndrome de hiperinmunoglobulina E (HIDS)/ deficiencia de mevalonato quinasa (MKD). Fiebre mediterránea familiar (FMF). – Enfermedad de Still activa incluyendo la Enfermedad de Still del Adulto (ESA) y la artritis idiopática juvenil sistémica (AIJS) en pacientes de 2 años de edad o mayores que no hayan respondido adecuadamente al tratamiento previo con antiinflamatorios no esteroideos (AINEs) y corticosteroides sistémicos, en monoterapia o en combinación con metotrexato. – Gota artrítica, en adultos con ataques frecuentes (al menos 3 ataques en los 12 meses previos), que no responden a otros tratamientos |
|
CINACALCET |
Mimpara |
AMGEN |
Hiperparatiroidismo secundario: – en adultos con insuficiencia renal crónica en diálisis. – en niños mayores de 3 años con enfermedad renal terminal (ERT) en tratamiento de mantenimiento con diálisis en la que el hiperparatiroidismo secundario no se controla adecuadamente con el tratamiento Carcinoma de paratiroides e hiperparatiroidismo primario en adultos Reducción de la hipercalcemia en pacientes adultos con: – Carcinoma de paratiroides. – Hiperparatiroidismo primario para los que, según sus niveles de calcio sérico, estaría indicada la paratiroidectomía (según las principales guías de tratamiento) pero que, no obstante, ésta no es clínicamente adecuada o está contraindicada. |
|
DABRAFENIB |
Tafinlar |
NOVARTIS |
Cáncer de pulmón no microcítico (CPNM), en combinación con trametinib para el tratamiento de pacientes que presentan la mutación BRAF V600. |
|
DAPTOMICINA |
Cubicin |
MSD |
Pacientes adultos y pediátricos (de 1 a 17 años de edad) con infecciones complicadas de piel y partes blandas (IPPBc). Pacientes adultos con endocarditis infecciosa del lado derecho (EID) debida a Staphylococcus aureus. Pacientes adultos y pediátricos (de 1 a 17 años de edad) con bacteriemia por Staphylococcus aureus. En pacientes adultos, la bacteriemia debe estar asociada con EID o IPPBc, y en pacientes pediátricos, la bacteriemia debe estar asociada con IPPBc. La daptomicina es activa contra las bacterias gram-positivas solamente. En el caso de infecciones mixtas en que se sospecha la presencia de bacterias gram-negativas y/o ciertos tipos de bacterias anaeróbicas, debe ser administrado simultáneamente con agentes antibacterianos apropiados. |
|
DARATUMUMAB |
Darzalex |
JANSSEN-CILAG |
Mieloma Múltiple: En combinación con lenalidomida y dexametasona, o con bortezomib y dexametasona, para pacientes que han recibido al menos un tratamiento previo. |
|
DOLUTEGRAVIR |
Tivicay |
GLAXO SMITHKLINE |
Virus de Inmu-nodeficiencia Humana (VIH), en combinación con otros antirretrovirales, en adultos, adolescentes y niños mayores de 6 años. |
|
ECULIZUMAB |
Soliris |
ALEXION PHARMA |
Miastenia gravis generalizada (MGg) refractaria en pacientes con anticuerpos anti-receptor de acetilcolina (AChR) positivos. |
|
ELVITEGRAVIR/ COBICISTAT/ EMTRICITABINE/ TENOFOVIR DISOPROXIL |
Stribild |
GILEAD SCIENCES |
VIH-1 en adultos de 18 años de edad o mayores que nunca han recibido tratamiento antirretroviral o que están infectados por un VIH-1 sin mutaciones conocidas asociadas con resistencia a ninguno de los tres fármacos antirretrovirales de Stribild, así como en adolescentes de entre los 12 y los 18 años con peso ≥ 35 kg que están infectados por un VIH-1 sin mutaciones conocidas asociadas con resistencia a ninguno de los tres fármacos antirretrovirales de Stribild y que han experimentado toxicidad que impide el uso de otros regímenes que no contengan tenofovir disoproxil fumarato. |
|
(EMTRICITABINA / TENOFOVIR DISOPROXIL |
Truvada |
GILEAD |
Tratamiento de la infección por VIH-1, de adolescentes de 12 a 18 años infectados por VIH-1 con resistencia a ITIAN o toxicidad que excluya el uso de agentes de primera línea. |
|
ESLICARBAZEPINA |
Zebinix |
Bial |
– Convulsiones de inicio parcial, con o sin generalización secundaria, en adultos con epilepsia recién diagnosticada, en monoterapia. |
|
EVEROLIMUS |
Votubia |
NOVARTIS FARMACEUTICA |
Crisis refractaria asociada con el complejo esclerosis tuberosa (TSC), adyuvante en mayores de 2 años con crisis de inicio parciales refractarias, con o sin generalización secundaria, asociadas con el complejo esclerosis tuberosa (TSC). Angiomiolipoma renal asociado con el complejo esclerosis tuberosa (TSC) que presentan riesgo de pero que no requieren cirugía inmediata Astrocitoma subependimario de células gigantes (SEGA) asociado con el complejo esclerosis tuberosa (TSC) que requieren intervención terapéutica pero no son susceptibles a cirugía. |
|
EXENATIDA |
Bydureon |
ASTRAZENECA |
Tratamiento de la diabetes mellitus tipo 2 en combinación con otros fármacos antidiabéticos, incluyendo insulina, en adultos que no hayan alcanzado un control glucémico adecuado con las dosis máximas toleradas de estos tratamientos, junto con dieta y ejercicio. |
|
FULVESTRANT |
Faslodex |
ASTRAZENECA |
En monoterapia para el tratamiento de mujeres posmenopáusicas con cáncer de mama localmente avanzado o metastásico y con receptor de estrógeno positivo: – no tratadas previamente con terapia endocrina, o – cuya enfermedad ha recidivado durante o después del tratamiento adyuvante antiestrogénico, o bien cuya enfermedad ha progresado durante tratamiento antiestrogénico. En combinación con palbociclib, está indicado para el tratamiento de mujeres con cáncer de mama localmente avanzado o metastásico y con receptor de estrógeno positivo y receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo, que han recibido previamente terapia endocrina (ver sección 5.1 de la ficha técnica). En mujeres pre o perimenopáusicas, el tratamiento en combinación con palbociclib debe ser combinado con un agonista de la hormona liberadora de hormona luteinizante (LHRH). |
|
HEXAFLUORURO AZUFRE |
Sonovue |
ROVI |
Uso intravesical: Ultrasonografía del tracto urinario excretor, en recién nacidos hasta los 18 años para detectar reflujo vesicoureteral. Para la limitación en la interpretación de una urosonografía negativa. |
|
ICATIBANT |
Firazyr |
SHIRE PHARMACEUTICALS |
Crisis agudas de angioedema hereditario (AEH) en adultos, adolescentes y niños mayores de 2 años (con deficiencia del inhibidor de la esterasa C1). |
|
INHIBIDOR DE LA C1 ESTERASA |
Cinryze |
SHIRE PHARMACEUTICALS |
Tratamiento y prevención preoperatoria de las crisis de angioedema en adultos, adolescentes y niños mayores de 2 años de edad con angioedema hereditario (AEH). Prevención rutinaria de las crisis de angioedema en adultos, adolescentes y niños (mayores de 6 años) con crisis recurrentes y severas de angioedema hereditario (AEH), que no responden a otro tratamiento. |
|
LACOSAMIDA |
Vimpat |
UCB PHARMA |
Crisis de inicio parcial con o sin generalización secundaria en pacientes adultos y adolescentes (16-18 años) con epilepsia, en monoterapia o combinada. Crisis de inicio parcial con o sin generalización secundaria en pacientes adultos y, adolescentes (16-18 años) y niños mayores de 4 años con epilepsia, en monoterapia y como terapia concomitante. |
|
LARAGLUTIDA |
Victoza |
NOVO NORDISK |
Adultos con diabetes mellitus tipo 2: – En monoterapia: Cuando la metformina se considera inadecuado debido a intolerancia o contraindicaciones. – Junto a otros medicamentos para el tratamiento de la diabetes. |
|
LEDIPASVIR/ SOFOSBUVIR |
Harvoni |
GILEAD SCIENCES |
Hepatitis C crónica (HCC) en adultos y en adolescentes con edades comprendidas entre 12 y < 18 años. |
|
LENALIDOMIDA |
Revlimid |
CELGENE |
Mieloma múltiple, en monoterapia para el tratamiento de mantenimiento de pacientes de diagnóstico reciente y que han sido sometidos a trasplante autólogo de células madre. |
|
LINAGLIPTINA |
Trajenta |
BOEHRINGER INGELHEIM |
Diabetes mellitus tipo en pacientes adultos En monoterapia, cuando metformina no es adecuada. En combinación, incluyendo insulina. |
|
LINAGLIPTINA/METFORMINA |
Jentadueto |
BOEHRINGER INGELHEIM |
Adultos con diabetes mellitus tipo 2 en: – Pacientes que no estén adecuadamente controlados con su dosis máxima tolerada de metformina sola. – En combinación con otros medicamentos, incluyendo insulina, en pacientes inadecuadamente controlados con metformina y estos medicamentos. |
|
LOPINAVIR/RITONAVIR |
Kaletra |
ABBVIE |
En combinación con otros medicamentos antirretrovirales, para el tratamiento de adultos, adolescentes y niños de 14 días y mayores infectados por el virus de la inmunodeficiencia humana (VIH-1). |
|
MARAVIROC |
Celsentri |
VIIV HEALTHCARE |
VIH: en combinación con otros medicamentos antirretrovirales, para el tratamiento de adultos pretratados, adolescentes y niños de 2 años de edad o mayores cuyo peso sea al menos de 10 kg infectados solo por el VIH-1 con tropismo CCR5 detectable. |
|
MENINGOCOCO GRUPOS A, C, W135, e Y (VACUNA CONJUGADA |
Nimenrix |
GLAXO SMITHKLINE |
Inmunización en personas a partir de 6 semanas de edad frente a enfermedad meningocócica invasiva causada por Neisseria meningitidis de los grupos A, C, W-135, e Y. |
|
NILOTINIB |
Tasigna |
NOVARTIS FARMACEUTICA |
Adultos y niños con leucemia mieloide crónica (LMC) cromosoma Filadelfia positivo, de nuevo diagnóstico en fase crónica. Adultos con LMC cromosoma Filadelfia positivo en fase acelerada con resistencia o intolerancia a tratamientos previos, incluyendo el imatinib. Pacientes pediátricos con LMC cromosoma Filadelfia positivo con resistencia o intolerancia a tratamientos previos, incluyendo imatinib. |
|
NIVOLUMAB |
Opdivo |
BRISTOL MYERS SQUIBB |
– Carcinoma de células escamosas de cabeza y cuello en adultos que hayan progresado durante o después de un tratamiento basado en platino. – Carcinoma urotelial localmente avanzado irresecable o metastásico, que han progresado durante o después de un tratamiento basado en platino. |
|
OBINUTUZUMAB |
Gazyvaro |
ROCHE |
En combinación con quimioterapia, seguido de tratamiento de mantenimiento con Obinutuzumab en los pacientes que hayan alcanzado respuesta, en pacientes con linfoma folicular avanzado no tratado previamente. |
|
OFATUMUMAB |
Arzerra |
GLAXO SMITHKLINE |
Leucemia linfocítica crónica (LLC) en recaída, en combinación con fludarabina |
|
PASIREOTIDA |
Signifor |
NOVARTIS |
– Adultos con acromegalia para los que la cirugía no es una opción terapéutica o ha fracasado y que no han sido controlados adecuadamente con tratamiento con otros análogos de la somatostatina |
|
PEGINTERFERON ALFA-2A |
Pegasys |
Roche |
Hepatitis B crónica en: – Pacientes adultos: • Tratamiento de la hepatitis B crónica (HBC) con antígeno de superficie de hepatitis B (AgHBe) positivo o AgHBe negativo en pacientes adultos con enfermedad hepática compensada y evidencia de replicación viral, ALT aumentada e inflamación del hígado comprobada histológicamente y/o fibrosis. – Pacientes pediátricos a partir de 3 años de edad: • Tratamiento de la hepatitis B crónica (HBC) con antígeno de superficie de hepatitis B (AgHBe) positivo o AgHBe negativo en niños y adolescentes a partir de 3 años de edad con evidencia de replicación viral y ALT. |
|
PEMBROLIZUMAB |
Keytruda |
MSD |
– Melanoma avanzado (irresecable o metastásico). – Cáncer de pulmón no microcítico (CPNM) en tumores que expresan PD-L1 con un índice de proporción en el tumor ≥ 50% y sin mutaciones positivas de EGFR o ALK. – Cáncer de pulmón no microcítico (CPNM) localmente avanzado o metastásico que expresen PD-L1 con un índice de proporción de tumor ≥1% y hayan recibido al menos un tratamiento de quimioterapia previo. – En monoterapia, Linfoma de Hodgkin clásico (LHc) recidivante o refractario después de haber fracasado a un trasplante autólogo de progenitores hematopoyéticos (TAPH) y de tratamiento con brentuximab vedotina (BV), o que no son candidatos al trasplante y han fallado al tratamiento con BV. – Carcinoma urotelial localmente avanzado o metastásico, en pacientes que ya han recibido tratamiento previo de quimioterapia basada en platino. |
|
REGORAFENIB |
Stivarga |
BAYER |
En monoterapia para el tratamiento de carcinoma hepatocelular que haya sido previamente tratado con sorafenib. |
|
SAXAGLIPTINA |
Onglyza |
BRISTOL MYERS SQUIBB |
Adultos de 18 años o mayores con diabetes mellitus tipo 2, junto con la dieta y el ejercicio, para mejorar el control glucémico: – En monoterapia cuando metformina es inapropiado. – En combinación con otros antidiabéticos, incluyendo insulina, cuando estos no proporcionan un control glucémico adecuado. |
|
SAXAGLIPTINA / METFORMINA |
Komboglyce |
BRISTOL MYERS SQUIBB |
Adultos de 18 años o mayores con diabetes mellitus tipo 2, junto con la dieta y el ejercicio, – En no controlados con la dosis máxima de metformina sola. – En combinación con otros antidiabéticos incluyendo insulina, en no controlados con metformina y estos medicamentos. – En pacientes que ya han sido tratados con la combinación de saxagliptina y metformina por separado. |
|
SEVELÁMERO, Carbonato. |
Renvela |
SANOFI-AVENTIS |
Hiperfosfatemia en pacientes pediátricos (>6 años de edad y con un área de superficie corporal >0,75 m2) con enfermedad renal crónica. |
|
SOFOSBUVIR |
Sovaldi |
GILEAD |
Hepatitis C crónica (HCC) en adultos y en adolescentes de entre 12 y <18 años, en combinación. |
|
TOCILIZUMAB |
RoActemra |
ROCHE |
Arteritis de células gigantes (GCA) en pacientes adultos. |
|
TRAMETINIB |
Mekinist |
NOVARTIS |
Cáncer de pulmón no microcítico (CPNM), en combinación con dabrafenib para el tratamiento de pacientes adultos que presentan la mutación BRAF V600. |
|
VANDETANIB |
Caprelsa |
SANOFI AVENTIS |
Cáncer medular de tiroides (CMT) agresivo y sintomático en pacientes con enfermedad no resecable localmente avanzada o metastásica, en adultos, niños y adolescentes mayores de 5 años, en los que la mutación del oncogén Reorganizado durante la Transfección (RET) no se conoce o es negativa. |
Archive
Revista PAM: 409
Número 409, Diciembre 2017
Medicamentos con nuevos principios activos o biosimilares comercializados en españa en los últimos doce meses
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS o biosimilares COMERCIALIZADOS EN ESPAÑA EN LOS ÚLTIMOS DOCE MESES
|
Principio |
Medicamento® |
Laboratorio |
GT |
Huér |
Biosi |
Indicación principal |
Autorización |
Comerciali |
Valora |
PAM |
|
SEBELIPASA ALFA |
KANUMA |
Alexion |
A16AB |
* |
|
Deficiencia de lipasa ácida lisosomal |
28/8/2015 |
1/12/2017 |
|
|
|
LONOCTOCG ALFA |
AFSTYLA |
CSL Behring |
B02BD |
|
|
Hemofilia A |
4/1/2017 |
1/12/2017 |
|
|
|
DERMATOPHAGOIDES SPP |
ACARIZAX 12 SQ-HDM |
Alk Abelló |
V01AA |
|
|
Rinitis/Asma alérgica polvo doméstico |
19/9/2016 |
14/11/2017 |
|
|
|
OLARATUMAB |
LARTRUVO |
Lilly |
L01XC |
* |
|
Sarcoma de tejidos blandos |
5/12/2016 |
1/11/2017 |
*** |
409 |
|
RIBOCICLIB |
KISQALI |
Novartis |
L01XE |
|
|
Cáncer de mama |
4/9/2017 |
6/11/2017 |
** |
409 |
|
PALBOCICLIB |
IBRANCE |
Pfizer |
L01XE |
|
|
Cáncer de mama |
23/12/2016 |
1/11/2017 |
** |
409 |
|
SOFOSBUVIR/ |
VOSEVI |
Gilead |
J05AP |
|
|
Hepatitis C |
13/8/2017 |
15/11/2017 |
** |
409 |
|
GLECAPREVIR/ |
MAVIRET |
AbbVie |
J05AX |
|
|
Hepatitis C |
3/8/2017 |
1/11/2017 |
** |
409 |
|
CEFTAZIDIMA/ |
ZAVICEFTA |
Pfizer |
J01DD |
|
|
Infecciones abdominales y renales complicadas. Neumonía hospitalaria |
20/7/2016 |
1/11/2017 |
* |
409 |
|
ISAVUCONAZOL |
CRESEMBA |
Pfizer |
J02AC |
* |
|
Aspergilosis y mucormicosis |
15/10/2015 |
18/10/2017 |
** |
408 |
|
ETELCALCETIDA |
PARSAVIB |
Amgen |
H05BX |
|
|
Hiperparatiroidismo secundario |
11/11/2016 |
1/10/2017 |
** |
408 |
|
BARICITINIB |
OLUMIANT |
Lilly |
L01XE |
|
|
Artritis reumatoide |
6/3/2017 |
1/9/2017 |
*** |
407 |
|
PONATINIB |
ICLUSIG |
Incyte |
L01XE |
|
|
Leucemia linfoide aguda, leucemia mieloide crónica |
13/9/2016 |
1/9/2017 |
** |
407 |
|
RESLIZUMAB |
CINQAERO |
Teva |
R03DX |
|
|
Asma |
16/8/2016 |
1/9/2017 |
* |
406 |
|
TOFACITINIB |
XELJANZ |
Pfizer |
L01XE |
|
|
Artritis reumatoide |
22/3/2017 |
1/9/2017 |
*** |
407 |
|
CABOZANTINIB |
CABOMETYX |
Ipsen |
L01XE |
|
|
Cáncer renal de células claras |
9/9/2016 |
26/7/2017 |
** |
406 |
|
SOFOSBUVIR/ |
EPCLUSA |
Gilead |
J05AX |
|
|
Hepatitis C |
1/4/2017 |
6/7/2017 |
** |
404 |
|
RITUXIMAB |
TRUXIMA |
Kern |
L01XC |
|
* |
Artritis reumatoide, granulomatosis de Wegener, leucemia linfoide crónica, linfoma no. Hodgkin |
7/4/2017 |
1/7/2017 |
* |
– |
|
INSULINA GLARGINA |
ABASALGLAR |
Lilly |
A10AE |
|
* |
Diabetes mellitus |
23/6/2016 |
1/7/2017 |
* |
– |
|
ALBUTREPENONACOG ALFA |
IDELVION |
CSL Behring |
B02BD |
* |
|
Hemofilia B |
11/5/2016 |
19/6/2017 |
** |
405 |
|
PEGASPARGASA |
ONCASPAR |
Baxalta |
L01XX |
|
|
Leucemia linfocítica aguda |
14/1/2016 |
1/6/2017 |
** |
406 |
|
SELEXIPAG |
UPTRAVI |
Actelion |
B01AC |
|
|
Hipertensión pulmonar |
15/5/2016 |
2/5/2017 |
** |
403 |
|
CEFTOBIPROL MEDOCARILO |
ZEVTERA |
Basilea |
J01DI |
|
|
Neumonía |
2/4/2014 |
24/4/2017 |
* |
403 |
|
ÁCIDO DESOXICÓLICO |
BELKYRA |
Allergan |
D11AX |
|
|
Grasa submentoniana (papada) |
18/1/2017 |
10/4/2017 |
** |
403 |
|
BOSUTINIB |
BOSULIF |
Pfizer |
L01XE |
|
|
Leucemia mieloide crónica |
27/3/2013 |
1/4/2017 |
** |
402 |
|
OPICAPONA |
ONGENTYS |
Bial |
N04BX |
|
|
Enfermedad de Parkinson |
24/6/2016 |
1/4/2017 |
* |
403 |
|
ELIGLUSTAT |
CERDELGA |
Sanofi Aventis |
A16AX |
* |
|
Enfermedad de Gaucher |
2/2/2016 |
2/1/2017 |
** |
400 |
|
CONDROITIN SULFATO |
CARTILEX |
Tarbis |
M01AX |
|
* |
Artrosis |
13/11/2015 |
1/1/2017 |
* |
– |
|
TRIFLURIDINA + |
LONSURF |
Servier |
L01BC |
|
|
Cáncer colorrectal |
1/1/2016 |
1/1/2017 |
** |
400 |
|
INFLIXIMAB |
FLIXABI |
Biogen |
L04AB |
|
* |
Artritis reumatoide; artritis psoriásica; psoriasis en placas; espondiloartritis; enfermedad de Corhn; colitis ulcerosa |
7/7/2016 |
15/12/2016 |
* |
– |
|
DARATUMUMAB |
DARZALEX |
Janssen Cilag |
L01XC |
* |
|
Mieloma múltiple |
23/6/2016 |
1/12/2016 |
** |
400 |
|
IXEKIZUMAB |
TALTZ |
Lilly |
L04AC |
|
|
Psoriasis en placas |
15/6/2016 |
1/12/2016 |
* |
400 |
|
LENVATINIB |
LENVIMA |
Eisai |
L01XE |
* |
|
Cáncer de tiroides |
28/05/2015 |
1/12/2016 |
|
|
|
ELBASVIR + |
ZEPATIER |
Merck Sharp Dohme |
J05AX |
|
|
Hepatitis C |
1/8/2016 |
28/11/2016 |
** |
400 |
VALORACIÓN DE LA INNOVACIÓN TERAPÉUTICA EN PANORAMA ACTUAL DEL MEDICAMENTO
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta ese momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
– SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
– INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
– INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
– Evidencia clínica: mediante estudios controlados, específicamente diseñados y desarrollados para demostrar la eficacia y la seguridad del nuevo medicamento, con demostración fehaciente de lo que puede ser un avance o mejora sobre la terapia estándar hasta ese momento, en el caso de que exista.
– Plausibilidad científica (potencialidad): existencia de aspectos en el medicamento que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostrados mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
Modificaciones de medicamentos diciembre 2017
|
CAMBIO EN LAS CONDICIONES DE DISPENSACIÓN/OFERTA AL SNS (acumulado desde enero 2017) |
|
|
MEDICAMENTO |
ACTUAL |
|
960294 Apiretal niños 250 mg 5 supositorios |
Exclusión SNS. No facturable marzo 2017 |
|
745695 Bactil 10 mg 20 comprimidos |
Exclusión SNS. No facturable febrero 2017 |
|
914879 Bactil Forte 20 mg 20 comprimidos |
Exclusión SNS. No facturable febrero 2017 |
|
973487 Canesmycospor crema 20 g |
Exclusión SNS. No facturable diciembre 2017 |
|
664880 Coversoral 5 mg 30 comprimidos |
Exclusión SNS. No facturable diciembre 2017 |
|
664881 Coversoral 10 mg 30 comprimidos |
Exclusión SNS. No facturable diciembre 2017 |
|
868091 Efferalgan Vitamina C compr eferv |
Exclusión SNS. No facturable diciembre 2017 |
|
870253 Efferalgan Odis 500 mg 16 compr Bucodisp |
Exclusión SNS. No facturable diciembre 2017 |
|
651132 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
651135 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
651136 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
664292 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
664326 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
700627 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
700628 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
700629 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
700630 Extraneal Icodextrina 7.5 % |
Paso a Hospital. No facturable agosto 2017 |
|
672084 Genoxal 200 mg 1 vial |
Paso a Hospital. No facturable febrero 2018 |
|
700551 Genoxal 1000 mg 1 vial |
Paso a Hospital. No facturable febrero 2018 |
|
654435 Genoxal 50 mg 20 comprimidos |
Paso a Diagnóstico Hospitalario (Visado) |
|
672546 Ibuprofeno (arginina) Ratiopharm 20 sobres |
Exclusión SNS. No facturable diciembre 2017 |
|
672547 Ibuprofeno (arginina) Ratiopharm 40 sobres |
Exclusión SNS. No facturable diciembre 2017 |
|
663060 Ibuprofeno (arginina) Teva 600mg 20sobre |
Exclusión SNS. No facturable diciembre 2017 |
|
663061 Ibuprofeno (arginina) Teva 600mg 40sobre |
Exclusión SNS. No facturable diciembre 2017 |
|
708258 Ibuprofeno Mabo Farma 600 mg 40 compr |
Exclusión SNS. No facturable abril 2018 |
|
701705 Junifen 20 mg/ml suspensión 200 ml |
Exclusión SNS. No facturable septiembre 2017 |
|
802140 Junifen 200 mg 24 comprimidos bucodisp |
Exclusión SNS. No facturable marzo 2018 |
|
701703 Junifen 20 mg/ml suspensión 100 ml |
Exclusión SNS. No facturable septiembre 2017 |
|
855429 Junipro 20 mg/ml 150 ml |
Exclusión SNS. No facturable febrero 2017 |
|
779314 Leuco Hubber 5 óvulos vaginales |
Exclusión SNS. No facturable enero 2018 |
|
654768 Lixacol 400 mg 100 comprimidos |
Exclusión SNS. No facturable julio 2017 |
|
688499 Locetar 50 mg/ml barniz uñas |
Exclusión SNS. No facturable septiembre 2017 |
|
677725 Menveo 1 vial |
Inclusión SNS septiembre 2017 |
|
690375 Nimenrix 1 vial |
Inclusión SNS septiembre 2017 |
|
729061 Nolotil 5 ampollas |
Exclusión SNS. No facturable julio 2017 |
|
691352 Nutrineal PD4 1.1% Aminoacidos |
Paso a Hospital. No facturable agosto 2017 |
|
691360 Nutrineal PD4 1.1% Aminoacidos |
Paso a Hospital. No facturable agosto 2017 |
|
691386 Nutrineal PD4 1.1% Aminoacidos |
Paso a Hospital. No facturable agosto 2017 |
|
700632 Nutrineal PD4 1.1% Aminoacidos |
Paso a Hospital. No facturable agosto 2017 |
|
650993 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
650996 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
650997 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
651295 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
691311 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
700631 Nutrineal PD4 1.1% Aminoacidos con S/D |
Paso a Hospital. No facturable agosto 2017 |
|
700639 Oestraclin 0.6 mg gel 80 g |
Exclusión SNS. No facturable octubre 2017 |
|
705051 Omeprazol Abdrug 20 mg 28 cápsulas Fco |
Exclusión SNS. No facturable abril 2017 |
|
705050 Omeprazol Abdrug 20 mg 14 cápsulas Fco |
Exclusión SNS. No facturable abril 2017 |
|
705052 Omeprazol Abdrug 20 mg 14 capsulas Blis |
Exclusión SNS. No facturable abril 2017 |
|
705055 Omeprazol Abdrug 20 mg 28 cápsulas Blis |
Exclusión SNS. No facturable abril 2017 |
|
705056 Omeprazol Abdrug 40 mg 14 cápsulas Fco |
Exclusión SNS. No facturable junio 2018 |
|
705057 Omeprazol Abdrug 40 mg 28 cápsulas Fco |
Exclusión SNS. No facturable junio 2018 |
|
705058 Omeprazol Abdrug 40 mg 14 cápsulas Blis |
Exclusión SNS. No facturable junio 2018 |
|
705059 Omeprazol Abdrug 40 mg 28 cápsulas Blis |
Exclusión SNS. No facturable junio 2018 |
|
684906 Paracetamol Ratio 1 g 20 compr eferv |
Exclusión SNS. No facturable diciembre 2017 |
|
684907 Paracetamol Ratio 1 g 40 compr eferv |
Exclusión SNS. No facturable diciembre 2017 |
|
695673 Paracetamol Teva 1 g 40 compr eferv |
Exclusión SNS. No facturable diciembre 2017 |
|
704006 Paracetamol Teva 1 g 20 compr eferv |
Exclusión SNS. No facturable diciembre 2017 |
|
683193 Pentasa 1000 mg 7 envases rectales |
Exclusión SNS. No facturable abril 2017 |
|
650738 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
650741 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
650742 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
779462 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
858696 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
858811 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
858829 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
858852 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
858878 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
872226 Physioneal 40 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
650732 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
650733 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
650734 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
779579 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858886 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858894 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858902 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858910 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858928 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
858944 Physioneal 40 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
650735 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
650736 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
650737 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
779587 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
858951 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
859249 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
859298 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
859447 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
859470 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
859404 Physioneal 40 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
651137 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
651138 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
651139 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
765230 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
765867 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
766352 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
766360 Physioneal 35 glucosa 1.36% |
Paso a Hospital. No facturable agosto 2017 |
|
651140 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
651143 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
651144 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
766519 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
766576 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
766741 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
766774 Physioneal 35 glucosa 2.27% |
Paso a Hospital. No facturable agosto 2017 |
|
651145 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
651146 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
651147Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
766782 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
766808 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
766972 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
767889 Physioneal 35 glucosa 3.86% |
Paso a Hospital. No facturable agosto 2017 |
|
654443 Prevenar 13 1 jer prec |
Sin cupón precinto. No facturable enero 2017 |
|
654147 Romilar jarabe 200 ml |
Exclusión SNS. No facturable marzo 2018 |
|
819581 Romilar 15 mg 20 comprimidos |
Exclusión SNS. No facturable marzo 2018 |
|
823799 Silarine 80 mg 30 cápsulas |
Exclusión SNS. No facturable julio 2017. |
|
825554 Solu Moderin 40 mg 1 vial |
Exclusión SNS. No facturable marzo 2018 |
|
825562 Solu Moderin 40 mg 3 viales |
Exclusión SNS. No facturable marzo 2018 |
|
759936 Tracleer 125 mg 56 comprimidos |
Exclusión SNS. No financiado enero 2017 |
|
759928 Tracleer 62.55 mg 56 comprimidos |
Exclusión SNS. No financiado enero 2017 |
|
654252 Uromitexan 15 ampollas 2 ml |
Paso a Hospital. No facturable diciembre 2017 |
|
698670 Ursitan 3 mg/ml 20 monodosis |
Exclusión SNS. No facturable septiembre 2017 |
|
918714 Venofusin Bicarbonato sódico 1 M 250 ml |
Paso a Hospitalaria. No facturable enero 2018 |
|
916478 Viaflex glucosa 20 % bolsa 250 ml |
Exclusión SNS. No facturable febrero 2018 |
|
916486 Viaflex glucosa 20 % bolsa 500 ml |
Exclusión SNS. No facturable febrero 2018 |
|
933093 Viaflex glucosa 20 % bolsa 1000 ml |
Exclusión SNS. No facturable febrero 2018 |
|
957209 Viaflex glucosa 50 % bolsa 250 ml |
Exclusión SNS. No facturable febrero 2018 |
|
957217 Viaflex glucosa 50 % bolsa 500 ml |
Exclusión SNS. No facturable febrero 2018 |
|
957225 Viaflex glucosa 50 % bolsa 1000 ml |
Exclusión SNS. No facturable febrero 2018 |
|
962019 Viaflex glucosa 70 % bolsa 250 ml |
Exclusión SNS. No facturable febrero 2018 |
|
962027 Viaflex glucosa 70 % bolsa 500 ml |
Exclusión SNS. No facturable febrero 2018 |
|
962035 Viaflex glucosa 70 % bolsa 1000 ml |
Exclusión SNS. No facturable febrero 2018 |
|
801605 Viaflo Hartman Ringer Lactada 500 ml |
Paso a Hospital. No facturable abril 2017 |
|
728709 Vincristina Pfizer 1 vial 2 ml |
Paso a Hospital. No facturable diciembre 2017 |
|
728279 Vincristina Pfizer 1 vial 1 ml |
Paso a Hospital. No facturable diciembre 2017 |
|
703582 Voltaren 1 mg/ml 10 unidosis |
Exclusión SNS. No facturable noviembre 2017 |
|
699686 Zavedos 5 mg 1 vial polvo |
Paso a Hospital. No facturable octubre 2017 |
|
699694 Zavedos 10 mg 1 vial polvo |
Paso a Hospital. No facturable octubre 2017 |
|
cambio DE NOMBRE (acumulado desde enero 2017) |
|
|
ACTUAL |
ANTERIOR |
|
Ácido Acetilsalicílico Tarbis (Tarbis) |
Ácido Acetilsalicílico Codramol (Farmalider) |
|
Ácido Alendrónico Semanal Bluepharma |
Ácido Alendrónico Semanal Onedose |
|
Äcido Ibandrónico Aurovitas Spain |
äcido Ibandrónico Actavis |
|
Adexyl |
Aripiprazol Mylan |
|
Alprazolam Apotex |
Alprazolam Dermogen |
|
Anastrozol Aristo |
Anastrozol UR |
|
Aristine |
Famylette |
|
Atenolol Placasod |
Atenolol Sandoz |
|
Azitromicina Bluepharma |
Azitromicina Onedose |
|
Capecitabina Aurovitas Spain |
Capecitabina Actavis |
|
Celenib |
Celecoxib Qualigen |
|
Ciprofloxacino Bluepharma |
Ciprofloxacino Onedose |
|
Citalopram Apotex |
Citalopram Lareq |
|
Claritromicina Bluepharma |
Claritromicina Onedose |
|
Clopidogrel Viso Farmacéutica (Aurovitas) |
Clopidogrel Pharmathen (Glenmark) |
|
Dorzolamida/Timolol Aurovitas |
Dorzolamida/Timolol Actavis |
|
Doxazosina Neo Aurovitas |
Doxazosina Neo Actavis |
|
Edunix |
Hidromorfona Aristo |
|
Epirubicina Aurovitas |
Apirubicina Actavis |
|
Escitalopram VIR Pharma |
Escilan |
|
Fosinopril Aurovitas Spain |
Fosinopril Actavis |
|
Gemfibrozilo Tarbis |
Gemfibrozilo Ur (Germed) |
|
Gexana |
Buprenorfina Aristo |
|
Ibuprofeno Bluepharma |
Ibuprofeno Onedose |
|
Insucor |
Nebivolol Viso Farmacéutica |
|
Irbesartan/Hidroclorotiazida Aristo |
Irbesartan/Hidroclorotiazida Edigen |
|
Levetiracetam Exeltis (Exeltis) |
Levetiracetam Juste (Juste) |
|
Levodopa/Carbidopa/Entacapona Kern Pharma |
Levodopa/Carbidopa/Entacapona Gadur (Vegal) |
|
Levofloxacino Bluepharma |
Levofloxacino Onedose |
|
Memantina Aurovitas Spain |
Memantina Actavis |
|
Micofenolato de mofetilo Aurovitas |
Micofenolato de mofetilo Actavis |
|
Miflodine Breezhaler |
Miflodine |
|
Montelukast Aurovitas Spain |
Montelukast Actavis |
|
Nabila (Exeltis Helathcare) |
Memantina Juste |
|
Olmesartan Tecnigen |
Olmesartan Premium Pharma |
|
Olmesartan/Hidroclorotiazida Combix |
Olmesartan/Hidroclorotiazida Ric |
|
Omeprazol Bluepharma |
Omeprazol Onedose |
|
Pantoprazol Bluepharma |
Pantoprazol Onedose |
|
Paracetamol Bluepharma |
Paracetamol Onedose |
|
Paracetamol Aristo |
Paracetamol Unither |
|
Risperidona Apotex |
Risperidona Farmalider |
|
Risperidona Flas Mylan |
Rispemylan |
|
Rubicrono |
Meclarat |
|
Rupatadina Kern Pharma (Kern Plarma) |
Rupatadina Binomil (Uriach) |
|
Sumapriptan Bluepharma |
Sumatriptan Onedose |
|
Taioma |
Oxicodona Aristo |
|
Topiramato Apotex |
Topiramato Combix |
|
Tramadol/Paracetamol Aurovitas |
Tramadol/Paracetamol Actavis |
|
Tramadol/Paracetamol Bluepharma |
Tramadol/Paracetamol Onedose |
|
Venlafaxina Bluepharma |
Venlafaxina Onedose |
|
Venlafaxina retard Apotex |
Flaxen (Cantabria) |
|
Virelbina Aurovitas |
Vinorelbina Actavis |
|
Zolpidem Sandoz |
Zolpidem Bexalabs |
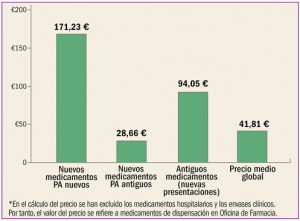
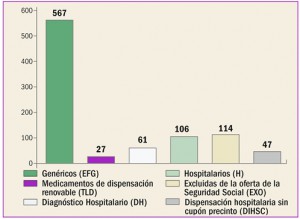
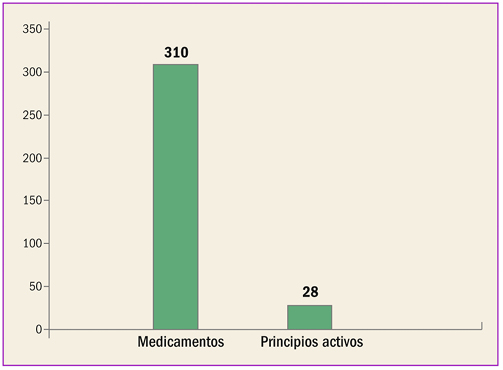
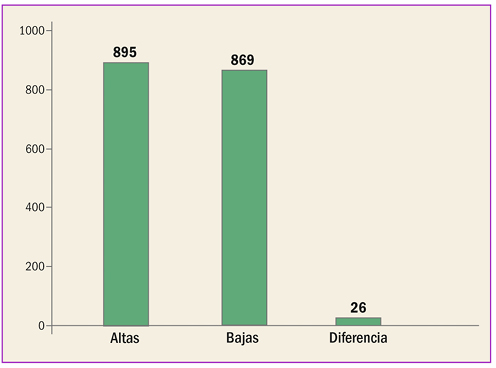
Estadísticas de medicamentos en España (Acumulado anual)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Palbociclib ▼ Ibrance® (Pfizer) y Ribociclib ▼ Kisqali® (Novartis) en cáncer de mama
Resumen
Palbociclib y ribociclin son inhibidores selectivos de las cinasas dependientes de ciclinas CDK4 y 6. A través de la inhibición de CDK4/6, reducen la proliferación celular mediante el bloqueo de la progresión de la célula de la fase G1 a la fase S del ciclo celular. Ambos medicamentos han sido autorizados para el tratamiento del cáncer de mama metastásico o localmente avanzado, positivo para el receptor hormonal (ER) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2), en combinación con un inhibidor de la aromatasa (IA); en el caso del palbociclib también ha sido autorizado para su uso en combinación con fulvestrant en mujeres que hayan recibido hormonoterapia previa. En mujeres pre- o perimenopáusicas la hormonoterapia se debe combinar con un agonista de la hormona liberadora de la hormona luteinizante (LHRH). Los datos clínicos sugieren una eficacia significativa de ambos medicamentos, teniendo en cuenta las características de las pacientes (ER+ y HER-2 negativo), ya que este tipo de cáncer de mama tienen opciones terapéuticas limitadas y un mal pronóstico, de ahí que la disponibilidad de nuevos tratamientos que prolonguen significativamente el tiempo hasta la progresión tumoral debe ser valorada favorablemente, modestas pero relevantes incrementos de la duración de la supervivencia libre de progresión (en relación a los controles), que en el caso del palbociclib fue de una media de 6,6 meses en pacientes que no respondieron a tratamiento hormonal y 10,3 meses en pacientes previamente no tratadas, mientras que con ribociclib fue de una media de 9,3 meses en general y de 11,4 entre aquellos con metástasis hepáticas y/o pulmonares. La toxicidad de ambos medicamentos es importante, aunque clínicamente manejable. Por otro lado, es importante también constatar que palbociclib y ribociclib son los primeros inhibidores selectivos de cinasas dependientes de ciclinas (CDK4/CDK6), lo que supone un nuevo mecanismo antitumoral que ha demostrado ser sinérgico con los tratamiento hormonales habitualmente utilizados.
ASPECTOS FISIOPATOLÓGICOS
El cáncer de mama es el cáncer más frecuente en la mujer española; supone un 29% de todos los cánceres. En 2012 se diagnosticaron en España 25.215 cánceres de mama. Aunque la mortalidad por cáncer de mama ha descendido en los últimos años gracias a los programas de cribado y a la mejora de los tratamientos, el cáncer de mama sigue siendo la primera causa de muerte por cáncer en España en las mujeres. En 2012, 6.075 mujeres fallecieron por esta causa. La edad de máxima incidencia está por encima de los 50 años, pero aproximadamente un 10% se diagnostica en mujeres menores de 40 años. La supervivencia media relativa del cáncer de mama tras 5 años es del 89,2% de forma global. El estadio en el que se ha diagnosticado el cáncer influye en la supervivencia. La supervivencia en el estadio I es de más del 98% y, en cambio, en los estadios III la supervivencia desciende al 24% (Martínez, 2017).
El cáncer de mama es el crecimiento desenfrenado de células malignas en el tejido mamario con capacidad invasora y metastatizante. En el 99% de los casos, el cáncer de mama ocurre en mujeres. Los 2 tipos principales de cáncer de mama son el carcinoma ductal (el más frecuente), que comienza en los conductos que llevan leche desde la mama hasta el pezón, y el carcinoma lobulillar, que comienza en los lobulillos, que producen la leche materna (Figura 1).
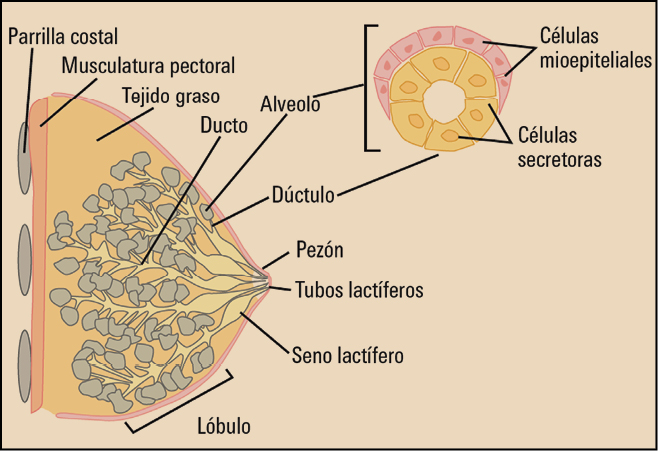{kind=link}
El pronóstico y el tratamiento del cáncer de mama dependen, en gran medida, del estadio (etapa del desarrollo) en que se encuentra el tumor, aunque en los últimos años se está cambiando el concepto en función de la agresividad, según el “perfil genético” que tenga el tumor.
Considerando los dos sexos, el cáncer de mama es el segundo en frecuencia en el mundo después del cáncer de pulmón. El cáncer de mama es el tumor más frecuente en la mujer, representa el 20-30% de todos los casos de cáncer en el sexo femenino y continúa siendo la primera causa de muerte por cáncer. La edad de máxima incidencia está por encima de los 50 años, pero aproximadamente un 6% se diagnostica en mujeres menores de 35 años. En Europa, el pronóstico es relativamente bueno, con una supervivencia a 5 años del 77%.
Su incidencia aumenta con el nivel económico. La incidencia en España es baja, menor que la de los Estados Unidos y Canadá, el Reino Unido, los Países Bajos, Bélgica, Alemania, Francia o Suiza, y similar al resto de los países de Europa Mediterránea, centroeuropeos, Portugal e Irlanda. La tasa ajustada española es de 50,9 casos/100.000 habitantes/año, por lo que en España se diagnostican unos 25.215 casos al año según el último IARC (International Agency for Research on Cancer) 2012 publicado, que representan casi el 30% de todos los tumores del sexo femenino en nuestro país. La mayoría de los casos se diagnostican entre los 35 y los 80 años, con un máximo entre los 45 y los 65.
Tanto el número de casos como las tasas de incidencia aumentan lentamente en España y en el mundo, probablemente debido al envejecimiento de la población y a un diagnóstico cada vez más precoz. El aumento de la incidencia se estima en un 1-2% anual y el riesgo de padecer cáncer de mama es de, aproximadamente, 1 de cada 8 mujeres. En España existe una distribución geográfica de incidencia notablemente variable según las provincias. Así, la tasa de incidencia en Cataluña es de 83,9 casos/100.000 habitantes, mientras que la media nacional se sitúa en 50,9 casos/100.000 habitantes.
Los principales factores de riesgo para el cáncer de mama son:
- Edad: el riesgo se incrementa al aumentar la edad. La mayoría de los casos ocurre en mujeres mayores de 60 años.
- Raza: es más frecuente en mujeres de raza blanca.
- Alcohol: su consumo excesivo aumenta el riesgo.
- Aparición temprana de la primera regla.
- Menopausia tardía.
- Historia familiar: aumenta el riesgo si existe un familiar de primer grado (madre, hermana o hija) que haya padecido la enfermedad. Si la enfermedad apareció en familiares más lejanos, el riesgo es menor. Los genes involucrados en este padecimiento son: BRCA1 en 20%, BRCA2 en 20%, CHEK2 en 5%, TP53 en 1%; sin embargo, en más del 50% de los casos se desconoce en gen asociado. El BRCA1 es un gen localizado en el cromosoma 17q21, supresor de tumor, involucrado en la regulación del ciclo celular, la reparación del ADN dañado, el mantenimiento de la estabilidad genómica y la regulación de la transcripción. Existen indicaciones precisas para la búsqueda intencionada del gen BRCA en pacientes con historia familiar o personal de cáncer de mama y ovario.
- Antecedentes personales: una mujer que ha tenido cáncer de mama tiene más riesgo de padecer otro cáncer en la mama contralateral.
- Enfermedades previas de la mama: algunas enfermedades de la mama como la hiperplasia atípica o el carcinoma lobulillar in situ pueden aumentar el riesgo.
- Primer embarazo tardío.
- Nuliparidad (ausencia de embarazo).
- Terapia hormonal sustitutiva prolongada.
En los estadios que pueden tratarse quirúrgicamente con éxito, los factores pronósticos marcan el riesgo que tiene una paciente de tener una recaída de la enfermedad tras completar el tratamiento. La mayoría depende de las características anatomopatológicas del tumor, pero algunas dependen del propio paciente. Los factores pronósticos “clásicos” más importantes son:
- Dependientes del tumor:
- Estadio clínico: tamaño tumoral y afectación ganglionar axilar. Ambos siguen considerándose el factor más importante, fundamentalmente la afectación axilar. La supervivencia a 5 años es prácticamente del 100% en el estadio I y de aproximadamente el 20% en el estadio IV.
- Grado de diferenciación celular: cuanto menos diferenciado sea el tumor es de peor pronóstico.
- Receptores hormonales: las pacientes con tumores dependientes de hormona se consideran de mejor pronóstico. Además, son sensibles al tratamiento hormonal, lo que amplía el arsenal terapéutico.
- Expresión de Her-2: las pacientes con sobreexpresión de Her-2 tenían peor pronóstico.
- Dependientes del paciente:
- Edad: tener menos de 35 años es un factor de riesgo.
- Perfil genético tumoral: los perfiles de expresión génica buscan definir patrones que permitan predecir la evolución clínica que tendrán grupos de pacientes muy bien definidos (actualmente desarrollado sólo para tumores luminales: receptores hormonales positivos Her2 negativo).
El tratamiento del cáncer de la mama es multidisciplinar y precisa la combinación de diversas modalidades terapéuticas para conseguir un control eficaz de la enfermedad. Estas modalidades son la cirugía, la radioterapia (RT), la quimioterapia (QT), la hormonoterapia (HT) y la terapia biomolecular. Las dos primeras actúan a nivel local, es decir, sobre la enfermedad en la mama y los ganglios linfáticos, y constituyen el tratamiento de elección en la enfermedad localizada no metastásica. Las restantes actúan tanto a nivel local como general de todo el organismo, en lo que se denomina tratamiento sistémico, y se utilizan de forma complementaria al tratamiento local con cirugía y/o RT o como tratamiento de primera elección en la enfermedad metastásica o diseminada.
Las modalidades de aplicación de la QT en los pacientes con cáncer de mama se pueden clasificar en neoadyuvante, adyuvante y enfermedad metastásica o paliativa. Se considera quimioterapia neoadyuvante a la que se administra antes de la cirugía. Está indicada en los cánceres de mama localmente avanzados y en aquellos que midan más de 2 cm o que tengan adenopatías axilares. Los fármacos más utilizados son fundamentalmente las antraciclinas y los taxanos, en combinación con otros agentes (ciclofosfamida, 5-fluorouracilo, carboplatino…). La intención de la neoadyuvancia es principalmente la disminución del tamaño tumoral para practicar una cirugía conservadora con márgenes libres de enfermedad, en los tumores localmente avanzados e inflamatorios, además de la valoración de la respuesta a la QT para posteriores tratamientos. Por todo ello, la QT neoadyuvante se ha convertido en una estrategia cada vez más utilizada en el tratamiento multidisciplinario del cáncer de mama y constituye, además, un marcador pronóstico de respuesta y un criterio de selección en el tratamiento posterior locorregional y/o sistémico del cáncer de mama localmente avanzado.
Por su parte, la quimioterapia adyuvante es la que se administra después de la cirugía. Su indicación depende de los factores pronósticos clásicos, que son la edad, el tamaño tumoral, la afectación ganglionar axilar, el grado de diferenciación celular (grado histológico) y los receptores hormonales presentes. La QT utilizada depende del riesgo de recaída, aunque en la actualidad en un 80% se utilizan taxanos y antraciclinas, obviando los taxanos en aquellos casos de tumores menores de 2 cm: grado I, sin afectación ganglionar y posmenopáusicas, ya que su utilización no ha demostrado mejoría en la supervivencia libre de progresión (SLP). En las pacientes con tumores que sobreexpresan la proteína HER-2, se debe asociar al tratamiento el anticuerpo monoclonal específico frente el receptor HER-2 (trastuzumab, etc.). El HER-2 es un receptor transmembrana, con actividad tirosina cinasa intrínseca, que está sobreexpresado en un 20-30% de los carcinomas de mama. Esta sobreexpresión está asociada a un alto riesgo de recurrencia y muerte.
El cáncer de mama metastásico de inicio se observa en un 10% de los casos. En el seguimiento de las pacientes diagnosticadas previamente de enfermedad local, se observan metástasis en el 50%. Los objetivos del tratamiento paliativo del cáncer metastásico son lograr un alivio sintomático y una atención emocional, social y espiritual para el enfermo y la familia, utilizando un tratamiento oncoespecífico (QT, HT, RT) que haya demostrado en un análisis de riesgo-costo-beneficio previo la conveniencia de su empleo.
La QT antitumoral ha mostrado beneficio en el incremento de la supervivencia y de la calidad de vida en las pacientes con cáncer de mama, pero no siempre está indicada. Por ello, inicialmente suele plantearse podríamos el tratamiento hormonal (HT), en los tumores hormonosensibles, Her-2 negativos de bajo riesgo, con afectación ósea o de partes blandas, intervalo libre de progresión desde que ha finalizado el tratamiento prolongado y en pacientes asintomáticas. La duración media del tratamiento está establecida en 1 año, con tasas de respuestas entre el 30 y el 50%.
Se suele pautar QT de primera línea ante tumores con receptores hormonales negativos, enfermedad visceral, intervalo de recaída corto desde la cirugía o sintomatología relacionada con la recaída o la metástasis, o en pacientes Her-2 positivas. La tasa de respuesta varía entre el 25 y el 60% según las series, alcanzado las mejores tasas cuando se utiliza la poli-QT; aunque ésta sólo se utiliza cuando queremos un rápido control de síntomas o una disminución rápida de la carga tumoral, ya que se ha demostrado que la supervivencia global (SG) no se modifica si se utiliza mono- o poli-QT, aumentando en esta última modalidad los efectos secundarios.
Las pacientes con sobreexpresión HER-2 positiva son candidatas a recibir tratamiento anti-HER-2, asociado a la QT. En la actualidad hay disponibles 4 fármacos: el trastuzumab, un anticuerpo monoclonal frente a este receptor; pertuzumab, anticuerpo monoclonal que inhibe la dimerización HER-2; trastuzumab emtansina, un conjugado del anticuerpo y el agente citotóxico antimicrotúbulos DM1, unidos mediante un enlace estable; y el lapatinib, que inhibe su actividad tirosina cinasa. Actualmente, la investigación va dirigida a otras dianas, con la idea de pautar un tratamiento que actúe no sólo en las distintas fases de la replicación celular, sino en un aspecto crucial para los tumores como es la angiogénesis. Se han estudiado inhibidores de la tirosina cinasa del receptor del factor de crecimiento endotelial vascular (VEGF) y el anticuerpo monoclonal contra el VEGF (bevacizumab). En el cáncer de mama estadio IV, se han ensayado en estudios fase 3 sunitinib, sorafenib y bevacizumab, aunque solo este último ha demostrado que, en combinación con QT (paclitaxel, docetaxel y capecitabina), mejora las tasas de respuesta y aumenta el tiempo hasta la progresión tumoral, por lo que ha sido autorizado como primea línea de tratamiento de la enfermedad avanzada.
ACCIÓN Y MECANISMO
Palbociclib y ribociclib son inhibidores selectivos de las cinasas dependientes de ciclinas CDK4 y 6. A través de la inhibición de CDK4/6, reducen la proliferación celular mediante el bloqueo de la progresión de la célula de la fase G1 a la fase S del ciclo celular. Ambos medicamentos han sido autorizados para el tratamiento del cáncer de mama metastásico o localmente avanzado, positivo para el receptor hormonal (ER) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2), en combinación con un inhibidor de la aromatasa (IA); en el caso del palbociclib también ha sido autorizado para su uso en combinación con fulvestrant en mujeres que hayan recibido hormonoterapia previa. En mujeres pre- o perimenopáusicas la hormonoterapia se debe combinar con un agonista de la hormona liberadora de la hormona luteinizante (LHRH).
Las ciclinas son proteínas sintetizadas durante la interfase y destruidas al final de la mitosis de cada ciclo celular. Actúan como reguladoras de la actividad enzimática de las cinasas dependientes de ciclinas (CDK) y su concentración varía a lo largo de las diferentes fases del ciclo celular. Las ciclinas C, D y E funcionan durante la fase G1 y en la transición de G1-S; por su parte, las A y B son ciclinas mitóticas que permanecen estables durante la interfase, pero son rápidamente proteolizadas durante la mitosis.
Las cinasas dependientes de ciclinas (CDK) son enzimas que regulan el desarrollo correcto del ciclo celular. Se trata de heterodímeros constituidos por una subunidad cinasa y una subunidad ciclina; su función es fosforilar residuos de serina y treonina de proteínas reguladoras específicas. Estos procesos se llevan a cabo cuando las CDK están activadas por las correspondientes ciclinas, las cuales dotan de especificidad a la enzima y permiten la regulación de su actividad, fundamentalmente mediante proteolisis dependiente de ubiquitinación y posterior traslado al proteasoma. Por el momento, se han descrito 12 CDK (CDK1 a CDK12).
Destacan entre ellas por ser potenciales dianas farmacológicas la CDK4 y la CDK6, las cuales forman complejos con las ciclinas D y funcionan durante la transición G0/G1, fosforilando a la proteína del retinoblastoma (Rb) y activando así la expresión de genes necesarios para la entrada en la fase S. Mutaciones en el gen cdk4, así como en los de sus proteínas asociadas, incluyendo las ciclinas D, p16 (TINTA4un) y pRb, han sido asociadas con la carcinogénesis en una amplia variedad de órganos, incluyendo el de mama y el de esófago.
Los datos experimentales indican que el palbociclib es capaz de desarrollar una elevada actividad frente a los cánceres de mama luminales, en particular los cánceres de mama con receptores estrogénicos (ER+). La pérdida de la proteína del Rb se asocia con una pérdida de actividad de palbociclib; por otro lado, la combinación de palbociclib con fármacos antiestrógenos (como el letrozol) aumenta la reactivación de la Rb mediante la inhibición de la fosforilación de Rb, dando lugar a la reducción de la vía de señalización de E2F y la interrupción del crecimiento (Cuéllar, 2017).
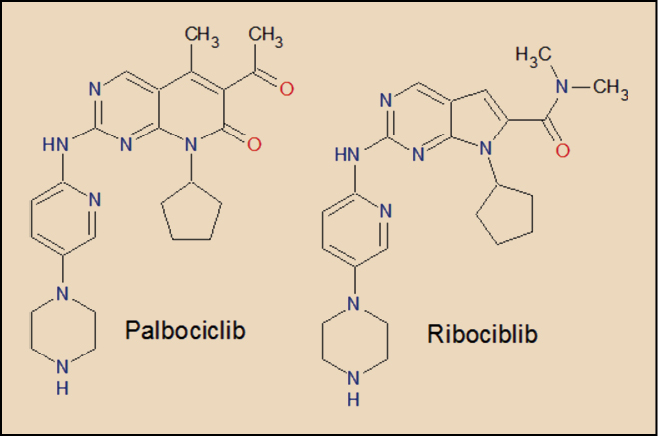
ASPECTOS MOLECULARES
El palbociclib y el ribociclib contienen núcleos análogos al de la purina. Tal estructura emula la del ATP y, como tal, actúa como inhibidor selectivo y reversible de las cinasas dependientes de ciclinas CDK4 y 6, al competir con el ATP en el proceso de fosforilación, bloqueándolo.
EFICACIA Y SEGURIDAD CLÍNICAS
{kind=link}
La eficacia y la seguridad clínicas del palbociclib y del ribociclib han sido adecuadamente contrastadas en las indicaciones autorizadas mediante ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados, multicéntricos, internacionales, doblemente ciegos y controlados con placebo.
Palbociclib
El primero de estos estudios (PALOMA-3; Cristofanilli, 2016) incluyó a 521 mujeres adultas (media de 57 años: 29-88) con cáncer de mama metastásico HER2 negativo y receptores hormonales positivo (HR+) que habían progresado tras una terapia endocrina anterior, procedentes de 144 centros en 17 países. Las pacientes podían estar en cualquier estado menopáusico, enfermedad mensurable o enfermedad ósea solamente, y recaída de la enfermedad o progresión después del tratamiento endocrino previo para la enfermedad avanzada durante el tratamiento o dentro de los 12 meses de la finalización de la terapia adyuvante, Las pacientes recibieron 125 mg al día de palbociclib oral durante las primeras tres semanas de un ciclo de cuatro, seguido de una semana de inactividad, más 500 mg de fulvestrant (IM) los días 1 y 15 del ciclo 1, luego el día 1 de los siguientes ciclos de 28 días, o placebo más fulvestrant. La variable clínica primaria fue la supervivencia libre de progresión evaluada por el investigador. Como variables clínicas secundarias se determinaron la supervivencia global, la tasa de respuesta objetiva (completa y parcial), la duración de la respuesta y la tasa de beneficio clínico.
La mediana de supervivencia libre de progresión fue de 11,2 meses (IC95% 9,5 a 12,9) en el grupo de fulvestrant más palbociclib y de 4,6 meses (IC95% 3,5 a 5,6) en el de fulvestrant más placebo (HR=0,46; IC95% 0,36 a 0,59; p<0,0001); la supervivencia global fue de 21,0 vs 8,6 (OR=2,78; IC95% 1,56 a 5,60; p=0,0001). Los eventos adversos de grado 3 o 4 ocurrieron en el 73% de las pacientes en el grupo de fulvestrant más palbociclib y en el 22% de las fulvestrant más placebo. Los eventos adversos de grado 3 o 4 más comunes fueron neutropenia (65% en el grupo fulvestrant más palbociclib vs 1% en el de fulvestrant más placebo), anemia (3 vs 2%) y leucopenia (28 vs 1%). Se presentaron eventos adversos graves en el 13% en el grupo de fulvestrant más palbociclib vs 17% en el grupo de fulvestrant más placebo. Ni el estado mutacional PIK3CA (con mutación en el 33% de los pacientes) ni el nivel de expresión del receptor hormonal afectaron significativamente la respuesta al tratamiento.
El segundo estudio (PALOMA-2; Finn, 2016) incluyó a 666 mujeres posmenopáusicas con cáncer de mama ER-positivo, HER2-negativo (media de 61 años: 28-89), que no habían tenido tratamiento previo para la enfermedad avanzada, para recibir por vía oral palbociclib (125 mg/día durante los primeros 21 días de cada ciclo de 28) más letrozol (2,5 mg/día, durante todo el ciclo) o placebo más letrozol. La variable primaria de eficacia fue la misma que en el estudio anterior.
Los resultados mostraron una mediana de supervivencia libre de progresión de 24,8 meses (IC95% 22,1 a no estimable) en el grupo de palbociclib-letrozol vs 14,5 meses (IC95% 12,9 a 17,1) en el grupo placebo-letrozol (HR= 0,58; IC95% 0,46 a 0,72; p<0,001); la supervivencia global fue de 42,1 vs 34,7 meses (OR=1,40; IC95% 0,98 a 2,01; p=0,0310). Los eventos adversos de grado 3 o 4 más frecuentes fueron neutropenia (66,4% con palbociclib-letrozol vs 1,4% con placebo-letrozol), leucopenia (24,8 vs 0%), anemia (5,4 vs 1,8%) y fatiga (1.8 vs 0,5%). Se registró neutropenia febril en el 1,8% con palbociclib-letrozol vs 0% con placebo-letrozol. La interrupción permanente de cualquier tratamiento del estudio como resultado de eventos adversos ocurrió en el 9,7 vs 5,9%.
Los efectos adversos más frecuentes del palbiciclib (≥20%) fueron neutropenia, infecciones, leucopenia, cansancio, náuseas, estomatitis, anemia, alopecia y diarrea. Los efectos adversos graves más frecuentes (≥2%) fueron neutropenia, leucopenia, anemia, cansancio e infecciones.
Ribociclib
El estudio MONALEESA-2 (Sonke, 2017) fue desarrollado con el fin de determinar la eficacia y la seguridad del ribociclib más letrozol de primera línea en un grupo de 668 pacientes con cáncer de mama avanzado HR+ y HER2-negativos, sin tratamiento sistémico previo, con una mediana de edad de 62 años (23 a 91); 295 pacientes (44%) tenían edades ≥65 años. Los pacientes fueron asignados al azar a ribociclib (600 mg/día durante 3 semanas, seguido de una semana de descanso) o placebo, más letrozol (2,5 mg/día) hasta progresión de la enfermedad, toxicidad inaceptable, muerte o interrupción del tratamiento. Un 44% presentaban metástasis en hígado y/o pulmón; un 44% habían recibido quimioterapia adyuvante o neoadyuvante y el 52% había recibido tratamiento antihormonal, mientras que el 34% de las pacientes no habían recibido ningún tratamiento previo (naïve).
La variable clínica principal fue la supervivencia libre de progresión tumoral (SLP), que se evaluó en pacientes de edad avanzada (≥ 65 años) y menores (<65 años). Los resultados mostraron que la combinación de ribociclib más letrozol mejoró significativamente la SLP frente a placebo más letrozol en términos globales (HR=0,608; IC95% 0,394 a 0,937; p<0,00001), siendo la mediana de SLP de 25,3 (IC95% 23,0 a 30,3) vs 16,0 meses (IC95% 13,4 a 18,2); a los 24 meses la tasa de pacientes libres de enfermedad era del 54,7 vs 35,9%. Considerando los efectos de forma estratificada por la edad, la mejora de la SLP fue consistente en todos los grupos: en pacientes ≥65 años (HR=0,608; IC95% 0,394 a 0,937) y <65 años (HR=0,523; IC95% 0,378 a 0,723), así como en pacientes con metástasis hepáticas y/o pulmonares (mediana de SLP de 24,8 vs 13,4 meses; HR=0,561; IC95% 0,424 a 0,743) o sin ellas (mediana de SLP de 27,6 vs 18,2 meses; HR=0,597; IC95% 0,426 a 0,837).
La combinación de ribociclib con letrozol es sustancialmente más tóxica que el letrozol solo. En este sentido, la tasa de suspensión del tratamiento por eventos adversos fue del 15,0 vs 3,0%, y la de pacientes que requirieron ajustes posológicos o interrupciones parciales fue del 73 vs 16%. La incidencia de eventos adversos graves fue del 21 vs 12%. Los eventos adversos más comunes fueron neutropenia (75 vs 5,2%), náusea (52 vs 29%), diarrea (31 vs 22%), vómitos (21 vs 16%), erupciones exantemáticas (17 vs 7,9%), anemia (18 vs 4,5%), elevaciones de los valores de enzimas hepáticas (15 vs 3,7%) y trombocitopenia (6,0 vs 0,6%).
ASPECTOS INNOVADORES
Palbociclib y ribociclib son inhibidores selectivos de las cinasas dependientes de ciclinas CDK4 y 6. A través de la inhibición de CDK4/6, reducen la proliferación celular mediante el bloqueo de la progresión de la célula de la fase G1 a la fase S del ciclo celular. Ambos medicamentos han sido autorizados para el tratamiento del cáncer de mama metastásico o localmente avanzado, positivo para el receptor hormonal (HR+) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2-negativo), en combinación con un inhibidor de la aromatasa (IA); en el caso del palbociclib también ha sido autorizado para su uso en combinación con fulvestrant en mujeres que hayan recibido hormonoterapia previa. En mujeres pre- o perimenopáusicas la hormonoterapia se debe combinar con un agonista de la hormona liberadora de la hormona luteinizante (LHRH).
En el caso del palbociclib, la evidencia clínica contrastada procede de dos estudios clínicos de fase 3, multicéntricos, multinacionales, aleatorizados, doblemente ciegos y controlados con placebo, realizados en mujeres con cáncer de mama con receptores hormonales positivo (ER-positivo) y HER2-negativo. En ambos estudios, el criterio principal de valoración de la eficacia fue el tiempo de vida de las pacientes sin que la enfermedad empeorase (supervivencia sin progresión, valorada por los investigadores).
En el primer estudio (PALOMA-3; Cristofanilli, 2016) participaron 521 mujeres con cáncer de mama metastásico que había empeorado después del tratamiento con un medicamento hormonal. Recibieron palbocilcib o placebo, más fulvestrant. El tratamiento con palbociclib/fulvestrant se asoció con una mediana de supervivencia de 11,2 meses vs 4,6 con placebo/fulvestrant, con una diferencia media estadísticamente significativa de 6,6 meses, mientras que la supervivencia global fue de 21,0 vs 8,6 meses, lo que supone una diferencia de 12,4 meses, también estadísticamente significativa. En el segundo estudio participaron 666 mujeres que habían pasado la menopausia y cuyo cáncer de mama había empezado a extenderse, pero que todavía no habían recibido tratamiento antitumoral, comparando la combinación de palbociclib/letrozol vs placebo/letrozol. En este caso, la mediana de supervivencia libre de progresión fue 24,8 meses (palbociclib/letrozol) vs 14,5 meses (placebo/letrozol), siendo la diferencia media de 10,3 meses, estadísticamente significativa; por su parte, la supervivencia global fue de 42,1 vs 34,7 meses (OR=1,40; IC95% 0,98 a 2,01; p=0,0310), una diferencia estadísticamente significativa de 7,4 meses. Los efectos adversos más frecuentes del palbiciclib (≥20%) fueron neutropenia, infecciones, leucopenia, cansancio, náuseas, estomatitis, anemia, alopecia y diarrea. Los efectos adversos graves más frecuentes (≥2%) fueron neutropenia, leucopenia, anemia, cansancio e infecciones.
Por su parte, la combinación de ribociclib con letrozol mejoró significativamente la supervivencia libre de enfermedad en 9,3 meses (25,3 vs 16,0), con un porcentaje de pacientes libres de progresión a los dos años de 55 vs 36%, manteniendo esta superioridad con independencia de la edad o del estatus metastásico de las pacientes. La combinación de ribociclib con letrozol es sustancialmente más tóxica que el letrozol solo, con una incidencia de eventos adversos graves del 21 vs 12% y una la tasa de suspensión del tratamiento por eventos adversos sea también diferente (15,0 vs 3,0%). Los eventos adversos más comunes (>20%) fueron neutropenia, náusea, diarrea y vómitos.
Los datos sugieren una eficacia significativa de ambos medicamentos, dadas las características de las pacientes (ER+ y HER-2 negativo). Es importante tener en cuenta que este tipo de cáncer de mama tienen opciones terapéuticas limitadas y un mal pronóstico, de ahí que la disponibilidad de nuevos tratamientos que prolonguen significativamente el tiempo hasta la progresión tumoral debe ser valorada favorablemente, aunque las mejoras conseguidas no son, obviamente, espectaculares:
- Palbociclib: media de 6,6 meses en pacientes que no respondieron a tratamiento hormonal y 10,3 meses en pacientes previamente no tratadas.
- Ribociclib: media de 9,3 meses en general y de 11,4 entre aquellos con metástasis hepáticas y/o pulmonares
La toxicidad de ambos medicamentos es importante, aunque clínicamente manejable.
Por otro lado, es importante también constatar que palbociclib y ribociclib son los primeros inhibidores selectivos de cinasas dependientes de ciclinas (CDK4/CDK6), lo que supone un nuevo mecanismo antitumoral que ha demostrado ser sinérgico con los tratamiento hormonales habitualmente utilizados.
|
VALORACIÓN |
|
PALBOCICLIB
 IBRANCE® (Pfizer) RIBOCICLIB
KISQALI® (Novartis) |
|
Grupo Terapéutico (ATC): L01XE. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Inhibidores directos de proteína cinasas |
|
Indicaciones autorizadas: Tratamiento del cáncer de mama metastásico o localmente avanzado, positivo para el receptor hormonal (HR) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2), en combinación con un inhibidor de la aromatasa; o en combinación con fulvestrant en mujeres que hayan recibido hormonoterapia previa (palbociclib). En mujeres pre o perimenopáusicas la hormonoterapia se debe combinar con un agonista de la hormona liberadora de la hormona luteinizante (LHRH). |
|
INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |
|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Palbociclib |
Ibrance |
Pfizer |
2017 |
|
Ribociclib |
Kisqali |
Novartis |
2017 |
BIBLIOGRAFÍA
Glecaprevir/pibrentasvir ▼ Maviret® (Abbvie) y Voxilaprevir/velpatasvir/sofosbuvir ▼ Vosevi® (Gilead) en hepatitis C
Resumen
Las asociaciones de pibrentasvir y de glecaprevir (Maviret®), y de velpatasvir, sofosbuvir y voxilaprevir (Vosevi®) son sendas combinaciones de antivirales de acción directa activos sobre los virus de la hepatitis C (VHC) que actúan sobre varios pasos consecutivos del proceso final de replicación del ARN viral. Dichas combinaciones presentan una elevada actividad frente todos los genotipos de VHC y han sido autorizadas para el tratamiento de la hepatitis C en adultos. El pibrentasvir y el velpatasvir actúan inhibiendo específicamente la proteína NS5A, alterando su localización subcelular, sus procesos de hiperfosforilación e inhibiendo la síntesis de ARN viral; por su parte, el glecaprevir y el voxilaprevir actúan inhibiendo específicamente el complejo de serina proteasa NS3/4A, lo que impide la replicación viral. Finalmente, el sofosbuvir actúa inhibiendo selectivamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral.
Los datos clínicos para glecaprevir/pibrentasvir muestran tasas de respuesta viral sostenida a las 12 semanas (RVS12) – una variable subrogada robusta de la eficacia clínica, considerada como estándar en hepatitis C – que oscilan entre el 90% y el 100% para todos los genotipos virales (salvo el 5, para el que no hay datos) en tratamientos de ocho semanas de duración, y del 95,3% al 100% para todos los genotipos en tratamientos de 12 semanas. Por lo que respecta a los datos clínicos correspondientes sofosbuvir/velpatasvir/voxilaprevir, también resultan altamente positivos, con tasas de RSV12 que oscilan entre el 91% y el 100% para todos los serotipos de VHC, tanto en pacientes naïve (no tratados previamente con antivirales de acción directa, AAD) con tratamientos de 8 semanas de duración, como en aquellos con historial de tratamiento previo en tratamiento de 12 semanas.
Actualmente, las tasas de respuesta viral sostenida registradas en España son muy elevadas y, por tanto, el margen de mejora por parte de los nuevos medicamentos es, afortunadamente, pequeño; no obstante, la combinación pibrentasvir/glecaprevir – farmacológicamente similar a las de otros inhibidores selectivos de NS5A/NS3/4a como ombitasvir/paritaprevir y elbasvir/grazoprevir – presenta algún aspecto innovador desde el punto de vista de las resistencias virales – relevante especialmente para el genotipo viral 3 – y de su utilidad en pacientes con insuficiencia renal grave, sin olvidar la alta tasa de respuesta incluso con formas abreviadas (8 semanas) de tratamiento en los pacientes no cirróticos. Asimismo, es resaltable la elevada eficacia y la actividad sobre todos los tipos virales de VHC de la combinación sofosbuvir/velpatasvir/voxilaprevir.
ASPECTOS FISIOPATOLÓGICOS
Se estima que la infección por el virus de la hepatitis C (VHC) afecta a 170-180 millones de personas en todo el mundo (2%), con una incidencia anual de 4 millones de nuevos casos y alrededor de 500.000 muertes. En España, la prevalencia de la infección por VHC C se sitúa en torno al 1,7% de la población general, lo que supone alrededor de 700.000 personas infectadas, todavía muchas de ellas sin diagnosticar.
Generalmente, la infección aguda por VHC es asintomática y muy raramente se asocia a una enfermedad mortal. Sin embargo, si no se trata, 15% de los pacientes con hepatitis C crónica acabará por desarrollar cirrosis hepática al cabo de 20 años, porcentaje que aumenta por encima del 40% a los 30 años; muchos de ellos acabarán por padecer un cáncer de hígado (hepatocarcinoma), a un ritmo de un 2% anual. Más del 50% de los pacientes que han necesitado un trasplante de hígado en España son pacientes con hepatitis C que han evolucionado a una enfermedad hepática terminal.
El virus de la hepatitis C (VHC) pertenece al género Hepacivirus, de la familia Flaviviridae. Se trata de un virus con cubierta de un diámetro de unos 50 nm, que tiene como material genético una única cadena de ARN en sentido positivo (+ssRNA). Existen 6 genotipos (1-6) y numerosos subgenotipos (a, b, c…) del VHC; en Europa y en Estados Unidos el más frecuente es el 1 (70% de los casos, mayoritariamente el 1b, que en España supone el cerca del 45% de todas las infecciones por VHC, mientras que el 1a representa el 25%), seguido por el 3 (3a; 20%), el 4 (4%) y el 2 (2a, 2b y 2c; 3%).
La expresión del ARN del VHC utiliza la maquinaria genética de los hepatocitos humanos; para ello, induce la síntesis de una única proteína o poliproteína(C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B), formada por 3.011 aminoácidos, lo que implica la necesidad de que varias proteasas celulares y virales (NS2 y NS3, junto con su cofactor NS4A, que forman parte de la propia poliproteína) actúen para liberar las formas activas de las 3 proteínas estructurales (E) y de las 7 no estructurales (NS) que la constituyen (Figura 1).
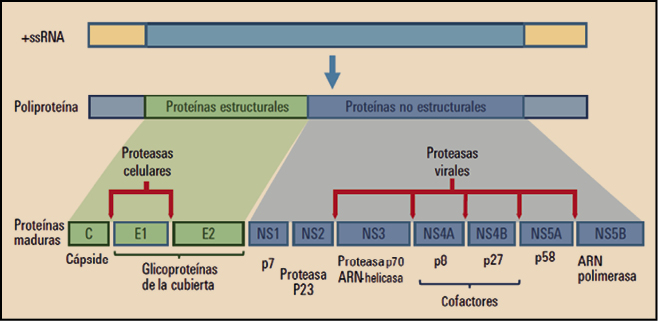
Figura 1. Expresión del ARN del virus de la hepatitis C (VHC) y resultado del procesamiento de la poliproteína.
El procesamiento de la poliproteína se inicia por la señal peptidasa del retículo endoplásmico; liberando la proteína C que forma la cápside viral, las E1 y E2 de la envoltura de la partícula y la p7, forma los canales iónicos en la envoltura viral, necesarios para la liberación del genoma. Por su parte, la región de las proteínas no estructurales (NS2 a NS5B), es procesada por NS2 y NS3 que producen un corte autocatalítico de las regiones NS2-NS3 y NS3-NS4A, respectivamente. Esta última es un complejo enzimático que inicia el procesamiento del resto de la poliproteína viral; está constituido por la NS3, una enzima multifuncional con actividades de serina-proteasa y de ARN helicasa DexH/D (revierte el enrollamiento de cadenas de ARN formadas durante la replicación viral); por su parte, la NS4A es responsable de reorganizar la estructura y optimizar la actividad de NS3 y facilitar la localización del complejo NS3/4A sobre la membrana del retículo endoplásmico, donde es procesada la poliproteína viral.
La NS5A es una proteína intensamente fosforilada y forma parte del complejo de replicación; se une a la fracción 3’-terminal del ARN viral recién sintetizado e interacciona con la ARN polimerasa dependiente del ARN viral. Esta última es la proteína NS5B, obviamente esencial en la replicación del ARN viral, que utiliza la cadena de éste como molde para nuevas cadenas y cataliza el proceso de polimerización de los ribonucleótido-trifosfato (rNTP) durante este proceso de replicación viral. En definitiva, produce la hebra de ARN complementaria – ARN(–) – que servirá de molde para fabricar las auténticas hebras de ARN viral – ARN(+) –, que a su vez podrán ser de nuevo replicadas y traducidas, o bien empaquetadas en las proteínas estructurales para formar nuevas partículas virales de VHC, que son liberadas mediante un proceso de exocitosis (Cuéllar, 2016).
El VHC posee un elevada variabilidad genética (1.400-1.900 sustituciones/nucleótido/año), que es debida principalmente a la sustitución nucleotídica que ocurre durante el proceso replicativo y por la falta de actividad de exonucleasa de la polimerasa viral, que impide la corrección de errores en la secuencia replicada. Sin embargo, esta variabilidad no es uniforme, ya que existen regiones altamente conservadas, como la secuencia que codifica para la proteína de la cápside viral (C), la región para la serina proteasa NS3 y los extremos 5’ y 3’ no traducibles de la cadena de ARN; otras regiones, como las que codifican las proteínas NS4B, NS5A y las regiones hipervariables 1 y 2 (RHV-1 y RHV-2) en la proteína de envoltura E2, pueden diferir hasta en más del 50%, dando lugar a las múltiples variantes genotípicas del VHC mencionadas anteriormente.
El tratamiento farmacológico de la hepatitis C crónica ha ido evolucionando de forma muy rápida en los últimos años. Hasta hace algunos años, se basaba en la administración de interferón alfa (generalmente, como peginterferón alfa) subcutáneo y ribavirina oral durante 24-72 semanas. Este tratamiento se asocia con una respuesta viral sostenida no demasiado satisfactoria (30-50%) y un perfil notable de efectos adversos.
En el año 2011 se produjo un cambio notable en el panorama terapéutico de la hepatitis C, al comercializarse el boceprevir y el telaprevir – la primera generación de inhibidores selectivos y reversibles de la proteasa NS3 – para el tratamiento de pacientes infectados por VHC de tipo 1, tanto no tratados previamente (naïve) como tratados, en combinación con peginterferón alfa y ribavirina (terapia triple). En estas circunstancias, las tasas de respuesta tanto en pacientes naïve como en pretratados llegaban a alcanzar hasta un 70% en algunas subpoblaciones de pacientes, permitiendo acortar la duración del tratamiento en algunos casos de 48 a 24 semanas. Sin embargo, boceprevir y telaprevir presentan un perfil toxicológico importante que obliga a suspender el tratamiento en un porcentaje de pacientes netamente superior a los tratados solo con peginterferón alfa y ribavirina, amén de un amplio abanico de interacciones farmacológicas, lo cual, asociado con la propia complejidad del tratamiento, dejaba un amplio margen para la mejora.
Tras esta primera generación de inhibidores de la proteasa del VHC, en 2014 llegó otra oleada de nuevos agentes con propiedades farmacodinámicas, farmacocinéticas y toxicológicas más satisfactorias que los anteriores, formada por el simeprevir, inhibidor dual de NS3 y NS4A, el daclatasvir, inhibidor de la NS5A, y el sofosbuvir, inhibidor de la NS5B, todos ellos con tasas de respuesta viral sostenida por encima del 80% para la mayoría de los genotipos y condiciones clínicas.
La forma de uso de todos ellos consiste en combinar dos o más para optimizar los resultados y, especialmente, reducir la duración de los tratamientos, excluir o reducir el uso de la terapia triple (peginterferón y ribavirina, más boceprevir o telaprevir), y reducir el riesgo de emergencia de cepas virales resistentes. Por este motivo, muchos de los nuevos medicamentos que están llegando actualmente consisten en combinaciones a dosis fijas de inhibidores de NS3/NS4A, NS5A y NS5B, con el objetivo adicional de optimizar la adherencia al tratamiento. Entre ellas, cabe citar las de:
- NS5A/NS3/4A: Ombitasvir/Paritaprevir (+ritonavir1), Pibrentasvir/Glecaprevir y Elbasvir/Grazoprevir
- NS5A/NS5B: Ledipasvir/Sofosbuvir y Velpatasvir/Sofosbuvir.
- NS5A/NS5B/NS3/4A: Velpatasvir/Sofosbuvir/Voxilaprevir
El criterio fundamental en la estrategia terapéutica es considerar el nivel de fibrosis hepática que presenta el paciente en el momento del diagnóstico, que determina la potencial utilidad de los tratamientos disponibles. Se suele cuantificar el nivel de fibrosis hepática mediante Fibroscan, considerándose como leve para niveles 0 y 1 (F0 y F1), moderada (F2), avanzada (F3) y grave (F4), típicamente cirrosis, que puede estar compensada o descompensada. La estrategia terapéutica es actualizada por el Plan Estratégico para el abordaje de la hepatitis C en el Sistema Nacional de Salud.
Según el genotipo, se han propuesto diferentes combinaciones de tratamientos considerando separadamente la condición de pacientes previamente no tratados (naïve) o con recaída tras un éxito inicial con terapia triple, con la de pacientes con fracaso previo a terapia triple. Entre las diversas opciones terapéuticas propuestas en la Estrategia ninguna tiene un carácter preferencial dado que no hay datos comparativos directos entre ellas. Actualmente (MSSSI, 2017), las tasas de respuesta viral sostenida (RVS) registradas en España con los medicamentos comercializados oscilan, según el grado de fibrosis entre un mínimo del 94,3% (F4) y un máximo del 96,7% (F1), siendo la media del 95,5%; asimismo, según el genotipo viral, van desde un 93,3% (genotipo 3), hasta el 96,2% (1b).
ACCIÓN Y MECANISMO
Las asociaciones de pibrentasvir y de glecaprevir (Maviret®), y de velpatasvir, sofosbuvir y voxilaprevir (Vosevi®) son sendas combinaciones de antivirales de acción directa activos sobre los virus de la hepatitis C (VHC) que actúan sobre varios pasos consecutivos del proceso final de replicación del ARN viral. Dichas combinaciones han sido autorizadas para el tratamiento de la hepatitis C en adultos.
El pibrentasvir y el velpatasvir actúan inhibiendo específicamente la proteína NS5A, alterando su localización subcelular, sus procesos de hiperfosforilación e inhibiendo la síntesis de ARN viral. La proteína NS5A es una proteína intensamente fosforilada que, una vez escindida de la poliproteína viral, localiza a las membranas donde se une a la fracción 3’-terminal del ARN viral recién sintetizado y participa en la replicación del genoma viral, en parte a través de interacciones con la ARN polimerasa dependiente del ARN viral (NS5B). Por su parte, el glecaprevir y el voxilaprevir actúan inhibiendo específicamente el complejo de serina proteasa NS3/4A, lo que impide la replicación viral. La proteína NS3 del VHC es una enzima multifuncional ya que presenta en el primer tercio de su estructura, una actividad de proteasa de serina y en el resto función de ARN helicasa DexH/D que revierte el enrollamiento de cadenas dobles de ARN formadas durante la replicación viral. El extremo NS4A es una proteína que se asocia con las cadenas de NS3 y provoca una reorganización en su estructura, optimizando así la actividad de proteasa de NS3; además NS4A promueve la localización del complejo NS3/4A a la membrana del retículo endoplásmico donde es procesada la poliproteína del VHC. Finalmente, el sofosbuvir actúa inhibiendo selectivamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral. Dicha NS5B es responsable de producir la hebra de ARN complementaria – ARN(-) – que servirá de molde para fabricar las auténticas hebras de ARN viral – ARN(+) –, que a su vez podrán ser de nuevo replicados y traducidas, o bien empaquetados en las proteínas estructurales para formar nuevas partículas virales de VHC, que son liberadas mediante un proceso de exocitosis.
El pibrentasvir presente una elevada actividad frente al VHC, con valores de EC502 que oscilan entre 0,001 a 0,004 nM para los genotipos para todos diferentes genotipos virales; los correspondientes valores de EC50 para el glecaprevir frente a dichos genotipos oscilan entre 0,85 a 4,6 nM (no se dispone de datos para el genotipo 5a). Por lo que respecta al desarrollo de resistencia viral frente a glecaprevir en cultivos celulares con replicones del VHC, las sustituciones individuales en la posición 168 de la proteasa del VHC redujeron la sensibilidad en más de 50 veces en los genotipos 1, 3 y 4, así como en más de 100 veces para el genotipo 6. Asimismo, algunas sustituciones del aminoácido 156 redujeron la sensibilidad en más de 100 veces en los genotipos del 1 al 4. Por su parte, no se han observado pérdidas significativas de la sensibilidad al pibrentasvir en los replicones mutados estudiados.
El velpatasvir ha mostrado una elevada actividad frente a las formas estables de los genotipos de VHC 1a, 2a, 2b, 3a, 4a, 5a y 6a, con valores de EC503 que van desde 0,014 a 0,11 µM. Los valores medios de EC50 encontrados para el velpatasvir frente a replicones quiméricos que codifican sequencias de NS5A procedentes de aislados clínicos son de 0,029 µM para el genotipo 1; 0,027 para el 2; 14,8 para el 3; 0,005 para el 4; 0,007 para el 5 y 0,11 para el 6. La combinación de sofosbuvir/velpatasvir ha mostrado in vitro que es aditiva. El replicón del mutante NS5B S282T, que confiere un bajo nivel de sensibilidad al sofosbuvir fue susceptible, sin embargo al velpatasvir (EC50 0,004 µM). De igual manera, los mutantes con baja sensibilidad al velpatasvir fueron completamente inhibidor por el sofosbuvir. Incluso los mutantes dobles (NS5A y NS5B) mostraron una baja capacidad de replicación con la combinación sofosbuvir/velpatasvir.
El voxilaprevir presenta una potente actividad frente a todos los genotipos (1-6) de VHC, con valores de EC50 que van de 0,33 a 6,6 nM. Se ha observado la aparición de cepas virales resistentes al voxilaprevir como consecuencia de mutaciones en las posiciones 156 y 168 de la proteína NS3 en todos los genotipos (1 a 6). Los genotipos 1a, 1b, 2a, 3a y 4a seleccionaron las sustituciones A156L, A156V o A156T, todos los cuales mostraron una notable reducción (>256 veces) de la susceptibilidad al voxilaprevir. Asimismo, algunas variantes en la posición 168 se seleccionaron en replicones de genotipo 2a, 5a y 6ª, confiriendo niveles moderadosde resistencia (1,5 a 17,8 veces). En los genotipos 5 y 6, las variantes dobles Q41R + D168H/Y y Q41R + D168H fueron las seleccionadas predominantemente, respectivamente, y confirieron mayores niveles de resistencia (35 a 39 veces) en comparación con las variantes únicas (2,7 a 13,5 veces).
ASPECTOS MOLECULARES
Pibrentasvir y velpatasvir están estructural y farmacológicamente relacionados con elbasvir, daclatasvir y ledipasvir, en tanto que todos ellos son inhibidores de la proteína NS5A del VHC. Presentan una característica estructura prácticamente simétrica, como elbasvir, daclatasvir y ledipasvir. Son destacables dos aspectos fundamentales; en primer lugar, el núcleo central de la molécula que constituye un amplio sistema resonante, de alta densidad electrónica, el cual facilita la unión a la proteína NS5A, estabilizando su estructura y bloqueando sus funciones. En segundo lugar, se aprecian sendas estructuras en los extremos de las moléculas que emulan secuencias peptídicas (son carbamatos, en realidad), que facilitan la inserción del fármaco en los huecos (bolsillos) de la proteína.
{kind=link}
Por su parte, el glecaprevir y el voxilaprevir están farmacológica y estructuralmente relacionados con el grazoprevir y el simeprevir, inhibidores específicos del complejo de serina proteasa NS3/4A. Estructuralmente destaca la presencia de varios grupos amida que confieren a estas moléculas un carácter de falso péptido, lo que resulta necesario para incorporarse – y bloquear – a determinadas regiones de la proteasa NS3/NS4A. También es peculiar la presencia de una estructura química relativamente compleja caracterizada por la presencia de un anillo con un elevado número de eslabones (macrociclo), de 18 eslabones, como en el grazoprevir, que es de 14 en el simeprevir y de 15 en el paritaprevir. Ambas estructuras también presentan estructuras anulares (ciclopropano, ciclopentano o pirrolidina) que quedan integradas en el macrociclo.
Finalmente, el sofosbuvir es un análogo estructural del uridilato (trifosfato de uridina, UTP), una de las cuatro bases nucleicas que constituyen el ARN; además, incluye un resto de L-alanina. Tras un proceso de fosforilación intracelular, el sofosbuvir es transformado en un derivado trifosfatado, que es la forma biológicamente activa del fármaco, actuando como un falso trifosfato de uridina (UTP) que es capaz de bloquear a la ARN polimerasa dependiente del ARN viral, provocando la finalización prematura del proceso de replicación y, con ello, deteniendo la reproducción del VHC.
EFICACIA Y SEGURIDAD CLÍNICAS
Pibrentasvir/glecaprevir
La eficacia y la seguridad clínicas de la combinación pibrentasvir/glecaprevir han sido adecuadamente contrastadas en las indicaciones autorizadas mediante seis ensayos clínicos de fase 3 (confirmatorios de eficacia y seguridad), con características diversas: doblemente diegos y controlados con placebo (1) o con comparadores activos (1), o abiertos (6), pacientes con o sin cirrosis hepática, refractarios a terapias previas (incluyendo interferón alfa, ribavirina y/o sofosbuvir) o no tratados previamente (naïve), co-infectados o no con VIH, con o sin insuficiencia renal grave, etc. Las características demográficas globales de estos seis estudios son: 56% varones, 80% blancos, 41% procedentes de Norte América y 38% de Europa, y 54 años de mediana de edad (86% ≤65 años).
La variable clínica más utilizada en los ensayos clínicos fue la respuesta viral sostenida durante 12 semanas (RVS12), definida como la persistencia de una concentración plasmática de ARN viral (VHC) inferior a 25 UI/ml durante las 12 semanas posteriores a la finalización del tratamiento. La dosis del medicamento empleada en todos los casos fue de 300 mg de glecaprevir y 120 mg de pibrentasvir, administrados por vía oral, una vez al día; la duración del tratamiento osciló en los diversos estudios entre 8 y 12 semanas. Las principales características de los estudios están reflejadas en la Tabla 1, resumiéndose los resultados de forma agregada en la tabla 2.
|
Tabla 1. Características generales de los ensayos clínicos de fase 3 con Glecaprevir/Pibrentasvir en hepatitis C |
||||||
|
Estudio (referencia) |
Nº pac. |
Genotipo viral (GT) |
Tratamiento previo (TP) o no (naïve) |
Cirrosis |
Tratamiento y duración |
Tipo de estudio |
|
ENDURANCE-1 |
703* |
1 |
Naïve y TR |
NO |
8 y 12 semanas |
Abierto |
|
ENDURANCE-2 |
302 |
2 |
Naïve y TR |
NO |
12 semanas |
Doble ciego, controlado con placebo |
|
ENDURANCE-3 |
505 |
3 |
Naïve |
NO |
8 Y 12 semanas |
Doble ciego, controlado con sofosbuvir/daclatasvir |
|
ENDURANCE-4 |
121 |
4-6 |
Naïve y TR |
NO |
12 semanas |
Abierto |
|
EXPEDITION-1 (Forns, 2017) |
146 |
1,2,4,5,6 |
Naïve y TR |
SI** |
12 semanas |
Abierto |
|
EXPEDITION-4 (Gane, 2017) |
104*** |
1-6 |
Naïve y TR |
SI (19%) |
12 semanas |
Abierto |
* Algunos pacientes estaban coinfectados por VIH.
** Con cirrosis compensada.
*** Con insuficiencia renal.
|
Tabla 2. Resultados agregados por genotipo, duración del tratamiento y estatus cirrótico (modificado de EMA, 2017)* |
|||
|
Genotipo |
Cirrosis |
Duración |
RVS12 (%) |
|
1 |
NO |
8 |
99,0 |
|
NO |
12 |
99,8 |
|
|
SI |
12 |
97,0 |
|
|
2 |
NO |
8 |
98,0 |
|
NO |
12 |
99,1 |
|
|
SI |
12 |
100 |
|
|
3 |
NO |
8 |
94,9 |
|
NO |
12 |
95,3 |
|
|
4 |
NO |
8 |
93,5 |
|
NO |
12 |
99,1 |
|
|
SI |
12 |
100 |
|
|
5 |
NO |
12 |
100 |
|
6 |
NO |
8 |
90,0 |
|
NO |
12 |
100 |
|
* No figuran los datos correspondientes a grupos de menos de 10 pacientes.
|
Tabla 3. Resultados agregados por genotipo, duración del tratamiento y tratamiento previo (modificado de EMA, 2017) |
|||
|
Genotipo |
Duración |
RVS12 (%) Naïve |
RVS12 (%) Pretratados |
|
1 |
8 |
98,8 |
99,3 |
|
12 |
99,0 |
99,5 |
|
|
2 |
8 |
98,9 |
91,3 |
|
12 |
99,0 |
100 |
|
|
3 |
8 |
95,2 |
– |
|
12 |
95,6 |
89,8 |
|
|
16 |
– |
95,5 |
|
|
4 |
8 |
92,3 |
– |
|
12 |
100 |
98,0 |
|
|
5 |
12 |
100 |
– |
|
6 |
12 |
100 |
– |
Desde el punto de vista de la seguridad, la combinación glecaprevir/pibrentasvir presenta un perfil toxicológico benigno, equiparable al del placebo. La incidencia global de eventos adversos relacionados con el tratamiento fue de 40%, siendo los más comunes fatiga (11%), cefalea (13%), náusea (7,6%), diarrea (3,8%), prurito (3,3%), insomnio (2,4%) y dolor abdominal (1,3%). La frecuencia de eventos adversos intensos (severos) emergentes durante el tratamiento fue inferior al 0,1% y el porcentaje de pacientes que suspendieron el tratamiento por este motivo fueron del 0,4%.
Sofosbuvir/Velpatasvir/voxilaprevir
La eficacia y la seguridad clínicas de la combinación han sido adecuadamente contrastadas en las indicaciones autorizadas mediante cuatro ensayos clínicos multinacionales y aleatorizados de fase 3 (confirmatorios de eficacia y seguridad), con características diversas: doblemente diegos y controlados con placebo o con comparadores activos (sofosbuvir/velpatasvir), o abiertos, en pacientes con o sin cirrosis hepática, refractarios a terapias previas con antivirales de acción directa (AAD) o no tratados previamente con estos (naïve). La variable clínica utilizada fue la respuesta viral sostenida durante 12 semanas (RVS12). La dosis del medicamento empleada en todos los casos fue de 400 mg de sofosbuvir, 100 de velpatasvir y 100 mg de voxilaprevir, administrados por vía oral, una vez al día; la duración del tratamiento osciló en los diversos estudios entre 8 y 12 semanas. Las principales características de los estudios están reflejadas en la Tabla 4, resumiéndose los resultados en la tabla 5.
|
Tabla 4. Características de los ensayos clínicos de fase 3 con Sofosbuvir/Velpatasvir/Voxilaprevir en hepatitis C |
|||||||
|
Estudio |
Nº pac. |
Genotipo viral (GT) |
Tratamiento previo (AAD) |
Cirrosis |
Comparador |
Duración (semanas) |
Tipo de estudio |
|
POLARIS-1 (Bourliére, 2017) |
415 |
1, 2, 3, 4, 5 y 6* |
AAD |
SI (41%) |
Placebo |
12 |
Doble ciego |
|
POLARIS-4 (Bourliére, 2017) |
333 |
1, 2, 3 y 4 |
AAD |
Si (46%) |
Sofosbuvir/ Velpatasvir |
12 |
Abierto |
|
POLARIS-2 (Jacobson, 2017) |
941 |
1, 2, 3, 4, 5 y 6 |
Naïve |
SI (19%) |
Sofosbuvir/ Velpatasvir |
8 (SVV) 12 (SV) |
Abierto |
|
POLARIS-3 (Jacobson, 2017) |
219 |
3 |
Naïve |
SI (100%) |
Sofosbuvir/ Velpatasvir |
8 (SVV) 12 (SV) |
Abierto |
* El número de pacientes fue inferior a 10 para los serotipos 2, 5 y 6.
|
Tabla 5. Resultados (% RSV12) de los ensayos clínicos de fase 3 con Sofosbuvir/Velpatasvir/Voxilaprevir (SVV) vs Sofosbuvir/Velpatasvir (SV) en hepatitis C, por serotipos* |
||||||||
|
Estudio |
Tratamiento |
Global |
1 |
2 |
3 |
4 |
5 |
6 |
|
POLARIS-1 |
SVV |
96 |
97 |
– |
95 |
91 |
– |
– |
|
POLARIS-2 |
SV |
98 |
98 |
100 |
97 |
98 |
– |
– |
|
SVV |
95 |
93 |
97 |
99 |
94 |
94 |
100 |
|
|
POLARIS-3 |
SV |
– |
– |
– |
96 |
– |
– |
– |
|
SVV |
– |
– |
– |
96 |
– |
– |
– |
|
|
POLARIS-4 |
SV |
90 |
91 |
97 |
85 |
– |
– |
– |
|
SVV |
98 |
97 |
100 |
96 |
100 |
– |
– |
|
* No figuran los datos correspondientes a grupos de menos de 10 pacientes.
Desde el punto de vista de la seguridad, la combinación sofosbuvir/velpatasvir/voxilaprevir presenta un perfil toxicológico benigno, equiparable al del placebo. La incidencia global de eventos adversos relacionados con el tratamiento fue de 51% (tratamiento de 8 semanas) y 56% (12 semanas), frente a 44% con 12 semanas de sofasbuvir/velpatasvir y 41% con placebo, siendo los más comunes cefalea (19/22/19/14%), fatiga (17/18/15/15%), diarrea (13/14/3,3/9,2%) y náusea (13/12/6,3/6,6%). La frecuencia de eventos adversos de grado 3-4 emergentes durante el tratamiento fue del 2,3/1,6/1,7/2,6% y el porcentaje de pacientes que suspendieron el tratamiento por este motivo fue inferior al 0,1%.
ASPECTOS INNOVADORES
Las asociaciones de pibrentasvir y de glecaprevir (Maviret®), y de velpatasvir, sofosbuvir y voxilaprevir (Vosevi®) son sendas combinaciones de antivirales de acción directa activos sobre los virus de la hepatitis C (VHC) que actúan sobre varios pasos consecutivos del proceso final de replicación del ARN viral. Dichas combinaciones presentan una elevada actividad frente todos los genotipos de VHC y han sido autorizadas para el tratamiento de la hepatitis C en adultos.
El pibrentasvir y el velpatasvir actúan inhibiendo específicamente la proteína NS5A, alterando su localización subcelular, sus procesos de hiperfosforilación e inhibiendo la síntesis de ARN viral; por su parte, el glecaprevir y el voxilaprevir actúan inhibiendo específicamente el complejo de serina proteasa NS3/4A, lo que impide la replicación viral. Finalmente, el sofosbuvir actúa inhibiendo selectivamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral.
Los datos procedentes de los estudios clínicos de fase 3 considerados pivotales para glecaprevir/pibrentasvir muestran tasas de respuesta viral sostenida a las 12 semanas (RVS12) – una variable subrogada robusta de la eficacia clínica, considerada como estándar en hepatitis C – que oscilan entre el 90% y el 100% para todos los genotipos virales (salvo el 5, para el que no hay datos) en tratamientos de ocho semanas de duración, y del 95,3% al 100% para todos los genotipos en tratamientos de 12 semanas. No se observaron diferencias relevantes entre las tasas alcanzadas en pacientes con o sin hepatitis, así como en función de su estatus terapéutico previo (naïve o pretratados con interferón alfa y/o ribavirina). La combinación presenta un perfil toxicológico benigno, equiparable al del placebo. Los eventos adversos más comúnmente descritos en relación con el tratamiento son fatiga, cefalea y náusea. El porcentaje de pacientes que suspendieron el tratamiento por este motivo fueron del 0,4%.
Por lo que respecta a los datos clínicos correspondientes sofosbuvir/velpatasvir/voxilaprevir, también resultan altamente positivos, con tasas de RSV12 que oscilan entre el 91% y el 100% para todos los serotipos de VHC, tanto en pacientes naïve (no tratados previamente con antivirales de acción directa, AAD) con tratamientos de 8 semanas de duración, como en aquellos con historial de tratamiento previo en tratamiento de 12 semanas. La eficacia en 8 semanas con sofosbuvir/velpatasvir/voxilaprevir es equiparable a la obtenida en pacientes naïve tratados durante 12 con sofosbuvir/velpatasvir. Como la combinación anterior, el perfil toxicológico es benigno.
Debe destacarse que el pibrentasvir es el primer inhibidor de NS5A con una actividad conservada contra cepas virales que contienen la mutación Y93H, cuya presencia es responsable de las menores tasas de respuesta observadas en pacientes afectados con VHC de genotipo 3 tratados con los inhibidores NS5A actuales, lo que hace de la combinación pibrentasvir/glecaprevir 3 una opción particularmente interesante para los pacientes con hepatitis C infectados por este genotipo viral, donde se alcanzan tasas de respuesta viral sostenida del 95,5% incluso en pacientes previamente tratados. Asimismo, el voxilaprevir es el primero inhibidor de NS3/4A de carácter pangenotípico (activo sobre todas las cepas de VHC).
En el caso de la combinación pibrantasvir/glecaprevir, son relevantes las altas tasas de respuesta viral sostenida con tratamientos de tan solo ocho semanas en pacientes infectados por el genotipo 1 – el más común en la Unión Europea – en pacientes no cirróticos (97%), así como del tratamiento de 12 semanas sin ribavirina en los cirróticos (97%), a lo que cabe agregar las elevadas tasas encontradas en el resto de genotipos virales. Esta forma abreviada de tratamiento es valorada por la EMA como una parte importante para el objetivo de erradicación del VHC (objetivo para 2030 por la OMS). Asimismo, es destacable la eficacia mostrada (98%) en pacientes con insuficiencia renal grave (más del 80% de los pacientes estaban sometidos a diálisis).
Por lo que respecta a la combinación sofosbuvir/velpatasvir/voxilaprevir, las tasas de RSV12 son muy elevadas en todos los serotipos de VHC, tanto en pacientes cirróticos como no cirróticos. Asimismo, destaca la alta eficacia de los tratamientos de 8 semanas de duración en pacientes naïve.
Actualmente (MSSSI, 2017), las tasas de respuesta viral sostenida registradas en España con los medicamentos actualmente comercializados oscilan, según el grado de fibrosis entre un mínimo del 94,3% (F4) y un máximo del 96,7% (F1), siendo la media del 95,5%; asimismo, según el genotipo viral, van desde un 93,3% (genotipo 3), hasta el 96,2% (1b). Es decir, las tasas actuales de curación son muy elevadas y, por tanto, el margen de mejora por parte de los nuevos medicamentos es, afortunadamente, pequeño; no obstante, la combinación pibrentasvir/glecaprevir – farmacológicamente similar a las de otros inhibidores selectivos de NS5A/NS3/4a como ombitasvir/paritaprevir y elbasvir/grazoprevir – presenta algún aspecto innovador desde el punto de vista de las resistencias virales – relevante especialmente para el genotipo viral 3 – y de su utilidad en pacientes con insuficiencia renal grave, sin olvidar la alta tasa de respuesta incluso con formas abreviadas (8 semanas) de tratamiento en los pacientes no cirróticos. Asimismo, es resaltable la elevada eficacia y la actividad sobre todos los tipos virales de VHC de la combinación sofosbuvir/velpatasvir/voxilaprevir.
|
VALORACIÓN |
|
GLECAPREVIR/PIBRENTASVIR
MAVIRET® (AbbVie) SOFOSBUVIR/VELPATASVIR/VOXILAPREVIR
VOSEVI® (Gilead) |
|
Grupo Terapéutico (ATC): J05AP. TERAPIA ANTIINFECCIOSA SISTÉMICA. Antivirales para el tratamiento del VHC |
|
Indicaciones autorizadas: Tratamiento de la hepatitis C crónica en adultos. |
|
INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |
|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Boceprevir |
Victrelis |
Merck Sharp Dohme |
2011 |
|
Telaprevir |
Incivo |
Janssen Cilag |
2011 |
|
Simeprevir |
Olysio |
Janssen Cilag |
2014 |
|
Sofosbuvir |
Sovaldi |
Gilead |
2014 |
|
Daclatasvir |
Daklinza |
Bristol-Myers Squibb |
2015 |
|
Ledipasvir/Sofosbuvir |
Harvoni |
Gilead |
2015 |
|
Ombitasvir/Paritaprevir/Ritonavir |
Viekirax |
Abbvie |
2015 |
|
Dasabuvir |
Exviera |
Abbvie |
2015 |
|
Elbasvir/Grazoprevir |
Zepatier |
Merck Sharp Dohme |
2017 |
|
Sofosbuvir/Velpatasvir |
Epclusa |
Gilead |
2017 |
|
Glecaprevir/Pibrentasvir |
Maviret |
Abbvie |
2017 |
|
Sofosbuvir/Velpatasvir/Voxilaprevir |
Vosevi |
Gilead |
2017 |
BIBLIOGRAFÍA
Olaratumab ▼ Lartruvo® (Lilly) en sarcoma de tejidos blandos
Resumen
El olaratumab es un anticuerpo monoclonal que se une al receptor alfa del factor de crecimiento derivado de plaquetas (PDGFRα), impidiendo la unión con su ligando (PDGFα). El fármaco ha sido autorizado como medicamento huérfano, en combinación con doxorubicina, para el tratamiento de pacientes adultos con sarcoma de tejidos blandos en estado avanzado que no son susceptibles de tratamiento curativo con radioterapia o cirugía y que no han sido tratados previamente con doxorubicina. La combinación de olaratumab más doxorubicina supone una nueva opción terapéutica de primera línea para los pacientes con sarcoma de tejidos blandos. En definitiva, un fármaco relativamente innovador en términos mecanísticos, que se incorpora a la primera línea de quimioterapia junto con la doxorubicina, a la que añade en cuanto eficacia 2,5 meses de supervivencia libre de enfermedad y 11,8 meses de supervivencia global. Aunque la toxicidad es relevante, sin embargo es manejable clínicamente.
ASPECTOS FISIOPATOLÓGICOS
Se define como sarcoma a cualquier neoplasia de carácter invasivo que se origine en un tejido conjuntivo (vasos sanguíneos, músculos, huesos, cartílagos, etc.). Difiere de los carcinomas en que estos derivan de células de origen epitelial, mientras que los sarcomas derivan de células que durante la fase embrionaria constituyen el mesodermo; es decir, proceden de células mesenquimatosas.
Tradicionalmente se divide a los sarcomas en sarcomas de partes blandas y en osteosarcomas. Los primeros se desarrollan en tejidos blandos, tales como el conectivo, músculos lisos o estriados, nervios, vasos sanguíneos o tejido adiposo, entre otros. Es más común en adultos jóvenes (15-35 años) y suele presentarse más comúnmente en las extremidades superiores e inferiores. Hay multitud de tipos: condrosarcoma extraesquelético, dermatofibrosarcoma, fibrosarcoma, hemangiosarcoma, histiocitoma fibroso maligno, leiomiosarcoma, linfangiosarcoma, liposarcoma, neurofibrosarcoma, rabdomiosarcoma, sarcoma de Kaposi, etc. Por su parte, el osteosarcoma se origina en las células osteoclásticas de la cubierta externa del hueso, especialmente en las epífisis de los huesos largos (80-90%), en la región de la rodilla (fémur y tibia, 50%) o en el brazo, específicamente el húmero proximal (25%), aunque también se han descrito osteosarcomas de pelvis y de cráneo.
De las 3 capas embrionarias – ectodermo, mesodermo y endodermo – el mesodermo es el origen de los músculos, los tejidos cartilaginosos, las articulaciones, los huesos, los tejidos conectivos, los vasos sanguíneos y linfáticos y los tejidos grasos. Dentro del término de tumores de partes blandas se incluye un grupo heterogéneo de lesiones benignas y malignas originadas en las partes carnosas del cuerpo, que pueden localizarse en cualquier parte entre el esqueleto y la epidermis. Las lesiones malignas son denominadas sarcomas de partes blandas, a partir del término griego sarcoma, que significa tumor carnoso.
Los sarcomas de partes blandas pueden aparecer en los tejidos mesodérmicos de las extremidades (más de la mitad de los casos), del tronco y del abdomen e incluso del cuello y la cabeza. También surgen en tracto gastrointestinal; como los sarcomas del estroma gastrointestinal (GIST, gastrointestinal stromal tumours). En realidad, existen tantos tipos de sarcomas de partes blandas, como tipos de células de las que se originan. En España, los tipos histológicos más frecuentes en el adulto son el histiocitoma fibroso maligno (25-30%), rabdomiosarcoma (10-15%), liposarcoma (10%), leiomiosarcoma (5-10%), fibrosarcoma (5-10%) y sarcoma sinovial (5-7%). Con menor incidencia aparecen el dermatofibrosarcoma protuberans, sarcoma de células claras y sarcoma epiteloide.
La incidencia anual de los sarcomas de tejidos blandos en la Unión Europea es de 4,7 casos por cada 100.000 habitantes y representan el 0,8-1% de las neoplasias malignas, aunque en los jóvenes de menos de 15 años esta tasa asciende al 5,5%. En España, el 40% afecta a mayores de 55 años; un 46% afecta a extremidades inferiores, un 13% a superiores, un 31% en tronco y un 9% a la región de cabeza y cuello. Son responsables del 2% de las muertes por cáncer.
Son lesiones que no presentan preferencia familiar, de sexo, de raza o geográfica. Los rabdomiosarcomas son más frecuentes en la infancia y en la adolescencia, mientras que los demás tipos histológicos lo son en la edad adulta. Se ha relacionado a los sarcomas de tejidos blandos con diversos factores, pero no existe evidencia clara de ello, salvo para algunos muy específicos. No obstante, se relaciona a los traumatismos como un antecedente frecuente en este tipo de tumores, también se les ha relacionado con agentes químicos carcinogénicos – tales como hidrocarburos policíclicos, asbestos y dioxinas – y radiaciones, especialmente la radioterapia. Entre los virus oncogénicos, el VIH está estrechamente relacionado con el sarcoma de Kaposi.
Los estados de inmunodeficiencia, patológica o iatrogénica, se relacionan con sarcomas, así como la aparición de angiosarcomas en regiones limfadenectomizadas. Por su parte, algunos sarcomas tienen una base genética, tales como la enfermedad de Von Reklinghausen – neurofibromas que degeneran en un 1-5% de los casos a Schwanoma maligno – o el síndrome de Gardner. Lipomas, leiomiomas, tumores glómicos, xantomas, paragangliomas y varias formas de fibromatosis se relacionan con factores genéticos.
La clasificación por estadio (de I al IV) contempla diversas variables: tamaño, grado histológico de malignidad, afectación ganglionar regional y metástasis a distancia. El grado del sarcoma indica el comportamiento biológico y determina la estrategia terapéutica. El tamaño tumoral y la extensión de la necrosis tienen especial importancia en el pronóstico para cada tumor en concreto. El tamaño también determina la relación con estructuras circundantes: los tumores confinados a un grupo muscular (lesiones intracompartimentales) tienen mejor pronóstico que las que rompen la barrera fascial (lesiones extracompartimentales).
La supervivencia media a los 10 años del 90% para el estadio I, del 60% para el II, del 20% para el III y del 3% para el estadio IV. La presencia de metástasis ganglionares, infrecuente, equivale a la existencia de metástasis a distancia, e implica una evolución desfavorable en todos los casos. La mediana de supervivencia en pacientes con enfermedad metastásica es de 11-15 meses.
El control local de la enfermedad se logra hasta en un 90 % de los casos en localizaciones favorables como el tronco y, en especial, en las extremidades. No obstante, un 30-40% de estos enfermos acabará por desarrollar metástasis a distancia, la mayoría durante los primeros dos años de seguimiento.
La base del tratamiento curativo de los sarcomas de partes blandas es la cirugía, y su objetivo es la resección de la lesión en bloque, sin exponerla y sin dejar tumor macroscópico ni microscópico residual. Este tratamiento quirúrgico se puede combinar con la radioterapia y la quimioterapia. Esto permite controlar la enfermedad tumoral, evitar recidivas locales y reducir las posibilidades de difusión metastásica. No obstante, incluso con las mejores técnicas quirúrgicas, las cifras de recurrencia son del 15 al 30%, según el tipo de tumor.
Los sarcomas de partes blandas crecen de forma centrífuga y comprimen el tejido circundante aparentando estar encapsulado; sin embargo, el tumor se puede extender a través de la pseudocápsula formando lesiones satélites. Por este motivo, la cirugía debe ser amplia, ya que la simple enucleación del tumor presenta un 90% de recidivas locales. Aproximadamente el 80% de las recurrencias locales después de tratamiento quirúrgico solo, se producen dentro de los dos años de la resección.
La radioterapia asociado a la cirugía mejora el control local de la enfermedad. Puede administrares con anterioridad a la cirugía, durante la intervención o de forma postoperatoria, la modalidad más habitual. La radioterapia ha sido eficaz en eliminar la enfermedad microscópica residual en los sarcomas de partes blandas y ha contribuido a asegurar el control local del tumor cuando la cirugía ha sido marginal o a conservar estructuras como vasos, nervios o huesos que, de otra manera, deberían haberse resecado. Por lo tanto, este tratamiento permite la realización de cirugías menos extensas con un mejor resultado funcional que una cirugía radical. También se puede aplicar, de manera exclusiva, en dosis altas en los tumores irresecables.
La quimioterapia se administra con intención coadyuvante o paliativa. Rara vez se utiliza este recurso para intentar disminuir el tamaño de lesiones demasiado extensas como para intentar cirugía. Los datos clínicos disponibles sugieren que puede haber una mejoría en la supervivencia libre de enfermedad, aunque esta diferencia es solo significativa en algunos estudios y parece confinada a los sarcomas de alto grado de las extremidades. En general, la quimioterapia adyuvante se aplica a los pacientes con sarcomas localizados en las extremidades, con grados histológicos II/III y tumores de 5 cm de diámetro máximo.
La quimioterapia paliativa está destinada a aquellos pacientes de enfermedad diseminada o inoperable que no sean ancianos (<70 años), con buena capacidad funcional y ausencia de enfermedad grave concomitante.
Los sarcomas de partes blandas son tumores poco sensibles a la quimioterapia, y el número de agentes citotóxicos disponibles con un grado significativo de actividad frente a este tumor es limitado, donde las antraciclinas y, en particular, la doxorubicina, es el fármaco de referencia, aunque también han mostrado utilizad ifosfamida, ciclofosfamida y dacarbazina, aunque esta última es generalmente relegada debido a su elevada toxicidad. El tipo histológico o el grado histológico de malignidad no parecen influir en la respuesta a la quimioterapia, y son factores desfavorables la presencia de metástasis óseas o hepáticas y, favorables, un buen estado general o la edad joven del enfermo. No existe un tratamiento que pueda considerarse de elección para los sarcomas de partes blandas metastásicos, y se trata de un área de activa investigación. Una combinación de doxorubicina e ifosfamida suele ser el tratamiento de elección en pacientes jóvenes con un buen estado general. En éstos se ha mostrado especialmente eficaz el empleo de dosis altas de ifosfamida (12-14 g/m2).
La trabectedina es un producto de origen natural obtenido originalmente a partir de un tunicado o ascidia marina, la Ecteinascidia turbinata y desarrollado inicialmente en España. No está relacionado ni química ni farmacológicamente con otros agentes antineoplásicos y actúa uniéndose selectivamente a la guanina de la cadena de ADN, provocando la deformación de la cadena y, con ello, la alteración de sus procesos de transcripción y reparación del ADN. Está indicada para el tratamiento de sarcomas de tejidos blandos en estadio avanzado en los que haya fracasado el tratamiento con antraciclinas e ifosfamida, o bien que no sean candidatos a recibir dichos productos (también está indicada en combinación con doxorubicina liposomal pegilada en el tratamiento de pacientes con cáncer de ovario recidivante sensible al platino).
El último agente autorizado para el sarcoma de tejidos blandos ha sido el pazopanib, autorizado para el tratamiento de pacientes adultos con determinados subtipos de sarcoma de tejidos blandos avanzado que hayan recibido previamente tratamiento con quimioterapia para tratar su enfermedad metastásica o en aquellos pacientes adultos cuya enfermedad ha progresado en los 12 meses siguientes tras recibir tratamiento neo-adyuvante y/o adyuvante.
La administración de tasonermina (Factor de Necrosis Tumoral), junto con melfalán, por perfusión regional arterial con hipertermina moderada, ha demostrado ser relativamente eficaz para sarcomas de tejidos blandos en las extremidades, pero no parece influir significativamente en la supervivencia de los pacientes. Hasta el momento, la única utilidad de la tasonermina es como coadyuvante en la cirugía para la extirpación posterior del tumor, con el fin de evitar o retrasar la amputación o como medida paliativa, en caso de sarcoma de tejidos blandos inextirpable de las extremidades, utilizado en asociación con melfalán por perfusión regional arterial (ILP) con hipertermia moderada.
También se ha descrito una elevada actividad con los taxanos, en particular con el paclitaxel, en el tratamiento de sarcomas vasculares localizados en el cuero cabelludo o en la cara, así como en el sarcoma de Kaposi.
En pacientes tratados con cirugía seguida de radioterapia y quimioterapia, más de un 50% de las recurrencias son pulmonares, frente a un 20% de recurrencias locales. La recurrencia local es más frecuente en los sarcomas de tronco y cabeza y cuello, que en los de extremidad.
ACCIÓN Y MECANISMO
El olaratumab es un anticuerpo monoclonal que se une al receptor alfa del factor de crecimiento derivado de plaquetas (PDGFRα), impidiendo la unión con su ligando (PDGFα). El medicamento ha sido autorizado, en combinación con doxorubicina, para el tratamiento de pacientes adultos con sarcoma de tejidos blandos en estado avanzado que no son susceptibles de tratamiento curativo con radioterapia o cirugía y que no han sido tratados previamente con doxorubicina.
La interacción entre olaratumab y PDGFRα previene la unión del receptor por los ligandos PDGF-AA y -BB ligandos, así como la activación del receptor inducida por PDGF-AA, -BB y -CC y de la vía de señalización PDGFR-α. La señalización bioquímica en la que participa este receptor juega un papel en el crecimiento celular, la quimiotaxia y la diferenciación de las células pluripotentes (stem cell) mesenquimatosas. El receptor también se ha detectado en algunas células tumorales y del estroma, incluyendo sarcomas, donde la señalización puede contribuir a la proliferación celular, la metástasis y el mantenimiento del microambiente del tumor.
ASPECTOS MOLECULARES
El olaratumab (Lartruvo®) es un anticuerpo monoclonal de tipo IgG1 que se une al receptor alfa del factor de crecimiento derivado de plaquetas (PDGFR-α). Se trata de una glicoproteína con dos cadenas peptídicas pesadas (γ1) – cada una de ellas con 457 aminoácidos – y otras dos ligeras (κ). Hay un total de 12 enlaces por puente disulfuro (-S-S-) intracatenarios y 4 intercatenarios. Presenta dos puntos de glicosilación en las cadenas pesadas (Asn30 y Asn307).
EFICACIA Y SEGURIDAD CLÍNICAS
Figura 1. Factores que favorecen la aproximación de células mesenquimatosas (MSC) a las células tumorales. Las células cancerosas secretan una serie de quimiocinas (CCL2, CCL15, etc.) que atraen las MSC a través de los receptores de quimiocinas específicos en su superficie. Las quimiocinas pueden ser liberadas directamente en el medio extracelular o incorporadas en vesículas. Otras citocinas, incluyendo VEGF, HGF, TGF, PDGFF-BB, NT-3, MIF y factores como la LL-37, uPA y ciclofilina B liberadas por las células tumorales afectan el tropismo de las MSC. En interacción con las células cancerosas, las MSC secretan citocinas tales como CXCL1, CXCL2, CXCL12 o IL-6 y metaloproteinasas (MMP); éstas últimas tienen la capacidad de facilitar la degradación de la matriz extracelular y la migración.
La eficacia y la seguridad clínicas del olaratumab han sido evaluadas en un único estudio clínico (Tap, 2016) que tuvo una fase abierta (fase 1b), de seguridad, y otra aleatorizada (fase 2) en el que se analizó la eficacia clínica de la combinación de doxorubicina más olaratumab en pacientes con sarcoma de partes blandas irresecable o metastásico, en 16 centros clínicos en Estados Unidos. Tanto para la fase 1b como para la fase 2 del estudio, los pacientes elegibles tenían 18 años o más, un diagnóstico confirmado histológicamente de sarcoma de partes blandas localmente avanzado o metastásico no tratado previamente con una antraciclina, así como tejido tumoral disponible para determinar la expresión de PDGFRα mediante inmunohistoquímica. En la fase 2 del estudio, los pacientes fueron asignados aleatoriamente en una proporción 1:1 para recibir olaratumab (15 mg/kg) por vía intravenosa el día 1 y día 8 más doxorubicina (75 mg/m2) o doxorubicina sola (75 mg/m2) el día 1 de cada ciclo de 21 días, durante hasta ocho ciclos. Durante la fase 1b la variable principal fue la seguridad, en tanto que durante en la fase 2 (aleatorizada), fue de eficacia clínica expresada como supervivencia libre de progresión de la enfermedad, valorándose también la supervivencia global y la tasa de respuesta objetiva.
Durante la fase 1b se incluyó a 15 pacientes tratados con olaratumab más doxorubicina, mientras que 133 pacientes fueron aleatorizados (66 a olaratumab más doxorrubicina; 67 a doxorubicina sola) en la fase 2, de los cuales 129 (97%) recibieron al menos una dosis del tratamiento del estudio (64/65). Al final de la fase 2 del estudio, la mediana de supervivencia libre de progresión fue de 6,6 meses (IC95% 4,1 a 8,3) con olaratumab más doxorubicina y 4,1 meses (IC95% 2,8 a 5,4) con doxorubicina sola (cociente de riesgos estratificados, CRI=0,67; IC95% 0,44 a 1,02; p=0,0615). Por su parte, la mediana de supervivencia global fue de 26,5 meses (IC95% 20,9 a 31,7) vs 14,7 meses (IC95% 9,2 a 17,1) (CRI=0,46; IC95% 0,30 a 0,71; p=0,0003). La tasa de respuesta objetiva fue de 18,2% (IC95% 9,8 a 29,6) vs 11,9% (IC95% 5,3 a 22,2) (p=0,3421).
Los eventos adversos fueron más frecuentes con olaratumab más doxorubicina que con doxorubicina sola e incluyeron neutropenia (58 vs 35%), mucositis (53 vs 35%), náuseas (73 vs 52%), vómitos (45% vs 18%) y diarrea (34 vs 23%); no obstante, la neutropenia febril de grado 3 o superior fue similar en ambos grupos (13 vs 14%). Los eventos adversos que condujeron a la interrupción permanente del tratamiento con olaratumab ocurrieron en el 8% de los pacientes y estuvieron relacionadas fundamentalmente con la perfusión IV del medicamento (3%). Por otra parte, un 25% de los pacientes requirieron una reducción de la dosis para paliar los eventos adversos graves; en este sentido, las reacciones adversas más comunes que resultaron en ajustes posológicos fueron neutropenia (33%), trombocitopenia (8%) y anemia (5%).
ASPECTOS INNOVADORES
El olaratumab es un anticuerpo monoclonal que se une al receptor alfa del factor de crecimiento derivado de plaquetas (PDGFRα), impidiendo la unión con su ligando (PDGFα). El fármaco ha sido autorizado como medicamento huérfano, en combinación con doxorubicina, para el tratamiento de pacientes adultos con sarcoma de tejidos blandos en estado avanzado que no son susceptibles de tratamiento curativo con radioterapia o cirugía y que no han sido tratados previamente con doxorubicina.
La combinación de olaratumab más doxorubicina supone una nueva opción terapéutica de primera línea para los pacientes con sarcoma de tejidos blandos. Los datos clínicos proceden de un estudio de fase 1b/2 en pacientes con sarcoma avanzado de tejidos blandos demostraron que la adición de olaratumab a la doxorubicina prolongó la supervivencia libre de progresión en 2,5 meses y la supervivencia global en 11,8 meses en comparación con la doxorubicina sola. Es relevante que este aumento clínicamente significativo en la supervivencia global no se produjo a expensas de un número significativamente mayor de toxicidades (Tobias, 2017). Está en marcha un ensayo de fase 3 (confirmatorio de eficacia y seguridad), que está previsto que se complete en 2020.
Desde el punto de vista farmacológico, el olaratumab representa el primer anticuerpo monoclonal comercializado cuya diana farmacológica es el receptor alfa del factor de crecimiento derivado de plaquetas (PDGFR-α). Sin embargo, el pazopanib – un inhibidor multicinasa – también es capaz de bloquear efectivamente los efectos de la activación de este receptor por sus ligandos; una propiedad que comparte con otros inhibidores de proteína cinasas, tales como imatinib, lebvatinib, nintedanib, regorafenib y sorafenib (AEMPS, 2015). En cualquier caso, según el Informe de Posicionamiento Terapéutico (AEMPS, 2017), la combinación de olaratumab con doxorubicina seguida de mantenimiento de olaratumab se posiciona como opción preferente frente a doxorrubicina sola en pacientes con sarcoma de tejidos blandos avanzado que no sean candidatos para tratamiento con cirugía o radioterapia y que no hayan sido tratados previamente con antraciclinas, en primera y sucesivas líneas.
En resumen, un fármaco relativamente innovador en términos mecanísticos, que se incorpora a la primera línea de quimioterapia junto con la doxorubicina, a la que añade en cuanto eficacia 2,5 meses de supervivencia libre de enfermedad y 11,8 meses de supervivencia global. Aunque la toxicidad es relevante, sin embargo es manejable clínicamente.
|
VALORACIÓN |
|
OLARATUMAB
LARTRUVO® (Lilly) |
|
Grupo Terapéutico (ATC): L01XC. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Anticuerpos monoclonales |
|
Indicaciones autorizadas: Tratamiento en combinación con doxorubicina de pacientes adultos con sarcoma de tejidos blandos en estado avanzado que no son susceptibles de tratamiento curativo con radioterapia o cirugía y que no han sido tratados previamente con doxorubicina. |
|
INNOVACIÓN importante. Aportación sustancial a la terapéutica estándar |
|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Olaratumab |
Lartruvo |
Lilly |
2017 |
BIBLIOGRAFÍA
Parálisis facial periférica
Resumen
La parálisis de Bell es la patología más frecuente del nervio facial. Esta patología tiene un buen pronóstico, apareciendo una “restitutio ad integrum” generalmente entre la primera y tercera semana de iniciada la enfermedad. Las parálisis faciales periféricas, clínicamente evidentes, de diagnóstico claro, precisan de un examen complementario correcto que debe valorar la intensidad de la lesión, pero también la topografía de la misma. La etiología de la parálisis facial periférica es muy diversa. Los cuadros más frecuentes son debidos a traumatismos e infecciones víricas. Las posibilidades terapéuticas bien codificadas, permiten una rehabilitación social muy satisfactoria, ellas deben incitar al médico a adoptar una actitud diagnóstica y terapéutica activa. Los fármacos corticosteroides son antiinflamatorios. Hay pruebas insuficientes acerca de los efectos de los corticosteroides utilizados en pacientes con parálisis de Bell, aunque su efecto antiinflamatorio quizá prevenga el daño nervioso. La parálisis de Bell es causada por la inflamación del nervio facial. La reducción de la inflamación podría limitar el daño nervioso.
CONCEPTO
Entendemos como parálisis facial periférica cuando se produce la afectación del séptimo par craneal o nervio facial, desde la salida del encéfalo en el surco bulboprotuberancial hasta sus regiones de inervación periférica, pudiéndose lesionar en cualquier punto a lo largo de dicho recorrido. Su incidencia aproximada es de 23 casos por cada 100.000 personas en un año, afectándose de forma similar tanto mujeres como hombres. El pico etario de máxima incidencia se encuentra entre los 10 y 40 años.
RECUERDO ANATÓMICO
El nervio facial es un nervio mixto formado por dos raíces, la motora (el facial propiamente dicho) y otra raíz sensitiva (nervio intermediario de Wrisberg), ambas raíces contienen fibras vegetativas. La raíz motora sale de la parte lateral del surco bulboprotuberancial, mientras que el nervio intermediario de Wrisberg penetra también por el surco bulboprotuberancial, por fuera del facial y por dentro del nervio auditivo.
Ambas raíces se dirigen hacia el conducto auditivo interno, por el que penetran. Recorren el acueducto de Falopio, donde se unen en la primera porción del mismo formando el ganglio geniculado. Durante su recorrido por dicho acueducto, el facial da sus ramas colaterales intrapetrosas y sale por el agujero estilomastoideo. Posteriormente penetra en la parótida donde al dividirse aparecen sus ramas terminales. En este último trayecto, anterior a la parótida, nacen sus ramas colaterales extrapetrosas.
Las ramas terminales son la rama temporofacial (que formará el plexo carotideo junto con el auriculotemporal) y la rama cervicofacial (inervando los músculos cutáneos de la cara y del cuello).
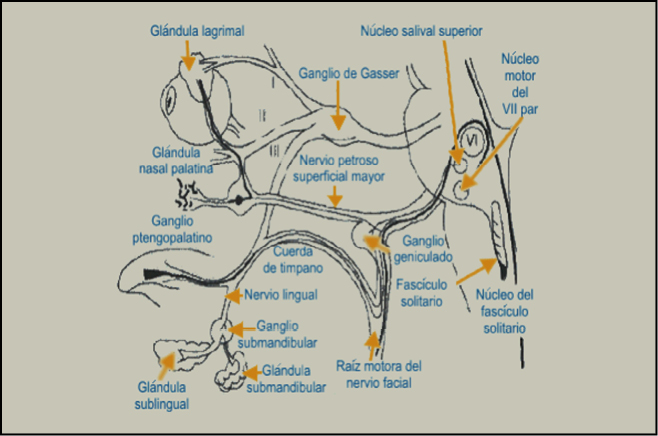
Esquema de anatomía del recorrido del nervio facial.
Las funciones que realiza el nervio facial son:
- Motora: inerva la musculatura estapedial, facial y cervical.
- Sensitiva: inerva el velo palatino y el área de Ramsay Hunt.
- Sensorial: recoge la sensibilidad de los dos tercios anteriores de la lengua (salado y ácido), controla la secreción salivar y nasal y participa en la regulación del tono vestibular.
- Refleja: participa en el reflejo palpebral, de amenaza, acústico y de succión.
ETIOLOGÍA Y ClASIFICACIÓN
La causa más frecuente, aproximadamente el 70% de los casos, es la parálisis de Bell. Sin embargo la propia etiología de esta entidad es desconocida, aunque la concepción actual es que es de origen vírico. La segunda causa corresponde a las traumáticas, aproximadamente un 15% de los casos; mientras que el tercer escalón lo integra el herpes zoster ótico con un 8%. Cabe destacar, a pesar de su infrecuencia, la posible causa neoplásica de la afectación del facial, ya sea por un tumor intracraneal o extracraneal.
La parálisis facial puede ser:
- Parálisis central o supranuclear: se produce por lesión en una zona anterior a la localización del núcleo facial.
Se diferencia de la periférica porque se alteran otras estructuras del SNC y se preserva la musculatura frontal y orbicular de los parpados (ya que poseen inervación bilateral).
Causas:
- Enfermedad cerebrovascular isquémica o hemorrágica.
- Procesos neoformativos tumorales.
- Parálisis periférica: por afectación del nervio facial en su núcleo(situado en la protuberancia), hasta las fibras periféricas ya sean intra o extracraneales.
Causas a destacar (Tabla 1):
|
tabla 1 |
|
|
Primarias |
Parálisis facial idiopática o de Bell |
|
Secundarias |
– Traumática – Infecciosa • Vírica: herpes simple, Herpes Zoster • Bacteriana – Neurológica • Guillen-Barre • Neuropatía hipertrófica hereditaria • Sídrome de Moebius – Tumoral • Parotideo • Colesteatoma • Neurinoma del acústico – Enfermedades sistémicas • Diabetes • Sarcoidosis • Hipertiroidismo • Autoinmunes • Leucemias |
- La parálisis de Bell es la forma más común de parálisis facial y es de etiología desconocida. SE presenta con mayor frecuencia en enfermos hipertensos, diabéticos y en el tercer trimestre del embarazo.
- Virus herpes zoster o Ramsay-Hunt: que cursa con parálisis, otalgia intensa, hipoacusia y vesículas en conducto auditivo externo, faringe y paladar.
CLÍNICA
La manifestación clínica más relevante es la afectación de la musculatura facial, imposibilidad para cerrar el ojo, fenómeno de Bell (desviación del globo ocular en sentido craneal, cuando el paciente intenta cerrar los ojos) y desviación de la comisura labial al lado contra lateral, sin embargo es muy importante realizar el diagnóstico topográfico de la lesión (Figuras 1 y 2).
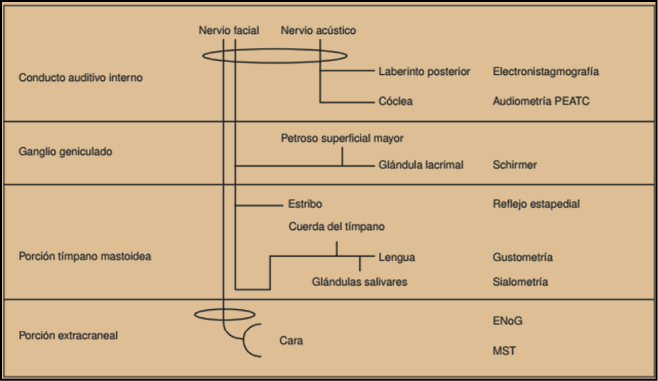
Figura 1. Anatomía del nervio facial junto con la clínica acompañante. PEATC: potenciales evocados auditivos del tronco cerebral; ENoG: electroneurografía; MST: test de la máxima estimulación. Figura extraída de M. Maños Pujol, J. Nogues Orpi, R. Jiménez Montoya y M. Dicenta Sousa.
- Distal a la salida por el agujero estilomastoideo: parálisis de los músculos ipsilaterales de la cara.
- Proximal a la salida por el agujero estilomastoideo: además de la clínica previa encontramos ageusia y/o disgeusia en los dos tercios anteriores de la lengua acompañado de disminución de la salivación.
- Proximal al ganglio geniculado: hiperacusia. Disminución del lagrimeo. En ocasiones puede acompañarse de lesión del nervio acústico, en cuyo caso aparecería sordera unilateral.
- Lesión a nivel de la porción timpánica y mastoidea: en ocasiones afectación del VIII par. Lagrimeo conservado.
Es importante determinar la intensidad de la parálisis facial, ya que va a influir notablemente en el pronóstico. Según el método de House se establecen 6 grados:
- Grado I: normal.
- Grado II: disfunción leve, con ligera debilidad de la musculatura facial.
- Grado III: disfunción moderada, con asimetría facial evidente sin ser desfigurante.
- Grado IV: disfunción moderadamente intensa con asimetría facial desfigurante.
- Grado V: disfunción intensa con caída de la comisura labial.
- Grado VI: parálisis total con pérdida completa del tono muscular.
Otro punto importante es el examen otoscópico que puede variar desde ser una exploración normal hasta presentar signos de otitis media crónica supurativa, hemotímpano o lesiones vesiculosas en pabellón auricular y conducto auditivo externo, ayudándonos así a aclarar la etiología de la entidad.
Además se deben explorar todos los pares craneales con el fin de excluir una tumoración del tronco encefálico. Otro punto importante es la exploración del gusto, principalmente debemos valorar si existe ageusia o disgeusia en los dos tercios anteriores de la lengua. Por último se puede acompañar de otra sintomatología, como otalgia o dolor en mastoides, audiofobia o hiperacusia.
DIAGNÓSTICO
El diagnóstico de una parálisis facial se basa en la existencia de hallazgos en tres aspectos: clínico, topográfico y eléctrico.
Diagnóstico clínico
- Consiste en la exploración de las funciones propias y de la motilidad voluntaria dependientes del VII par.
- Otoscopia: para descartar patología del oído medio y la presencia de vesículas en el síndrome de Ramsay-Hunt.
- Exploración cervical: Valorando cuidadosamente la región parotídea y la posible palpación de adenopatías que nos hagan sospechar la existencia de un proceso tumoral.
PRUEBAS COMPLEMENTARIAS: Su realización va dirigida a determinar la severidad de la parálisis, y el descarte de una posible etiología no idiopática.
- Rx tórax: ES útil para descartar la presencia de infiltrados y adenopatía hiliares.
- Analítica de sangre: Hemograma, bioquímica y estudio de coagulación.
Diagnóstico topográfico
El estudio de la funcionalidad de las diversas estructuras inervadas por las ramas del nervio facial nos permite localizar el área del nervio lesionada. Su valor estriba en que son indicadores parciales de la intensidad de la lesión y por ende indicadores relativos de pronóstico.
- Prueba de la lacrimación o test de Schrimer. Consiste en el estudio de la función secretora de la glándula lacrimal del lado afecto comparándola con la del lado sano. Para ello, se secan las conjuntivas oculares y se coloca un papel de filtro en ambos fórnix conjuntivales inferiores durante 5 minutos, leyéndose posteriormente la longitud de papel mojado en cada ojo. La reducción de un 30% o más unilateralmente, o la reducción unilateral a menos de 25 mm se consideran significativas. Topográficamente indicaría que la lesión se localizaría a nivel suprageniculado.
- Sialometría. Consiste en el estudio de la función secretora de la glándula submaxilar. Se compara la secreción en ambas glándulas cateterizando los conductos de Wharton y recogiendo la saliva en un tubo durante 1 a 5 minutos, previa estimulación con zumo de limón. Indicaría la presencia de una lesión situada por encima del tercio medio de la porción mastoidea.
- Reflejo estapedial. Estudia la contracción refleja del músculo estapedial inducida por un estímulo sonoro intenso mediante técnicas de impedanciometría. La reaparición de dicho reflejo si se encontraba previamente abolido antes de la primera semana desde el inicio de parálisis facial (PF) indica la presencia de una parálisis facial incompleta. Este hecho conlleva un pronóstico excelente.
- Gustometría. Consiste en el estudio por comparación de la sensibilidad gustativa de los dos tercios anteriores de la lengua.
Electrodiagnóstico
Son las pruebas consideradas como más fiables para determinar de forma objetiva el grado de lesión neuronal, emitir un pronóstico adecuado y considerar la posibilidad de modificar el tratamiento instaurado. Aún así, no permiten detectar lesiones antes del tercer día ya que, al ser imposible la evaluación directa del nervio en su trayecto intratemporal, la afectación neural sigue un trayecto evolutivo en sentido distal-descendente que puede llegar a tardar hasta 3 días en hacer posible la positividad de dichas pruebas diagnosticas; sólo se estimulan las fibras neuropráxicas, y no nos permite distinguir entre axonotmesis y neurotmesis. Los resultados permanecen anormales durante días o semanas. Las pruebas eléctricas no son buenas para monitorizar la remielinización.
- Prueba de la máxima excitabilidad. Es una prueba sencilla, fiable y económica para la evaluación de la degeneración del nervio facial poco después del inicio de la parálisis. Es una prueba comparativa que estudia la intensidad de la contractura muscular producida por una estimulación eléctrica supramáxima sobre las siguientes ramas del facial: temporal, orbicular ocular, zigomática, bucal, mandibular y cervical.
- Electroneuronografía. Es el estudio del potencial de acción obtenido tras la estimulación percutánea supramáxima del tronco del nervio facial. El hallazgo de una diferencia superior al 85%-90% entre el lado sano y el afecto indica la presencia de una axonotmesis y mal pronóstico, mientras que si es menor al 85%-90% correlaciona con buen pronóstico y neuroapraxia.
- Electromiografía. Es el estudio de los potenciales de acción musculares durante la actividad voluntaria. Carece de utilidad en la fase aguda, pero es la prueba más fiable para mostrar signos de reinervación precoces.
- Reflejo palpebral. Consiste en el estudio de las respuestas aparecidas al provocar el reflejo orbicular tras la estimulación del nervio orbitario (reflejo del parpadeo). Permite la evaluación del trayecto craneal sobre todo, y también intrapetroso del nervio facial.
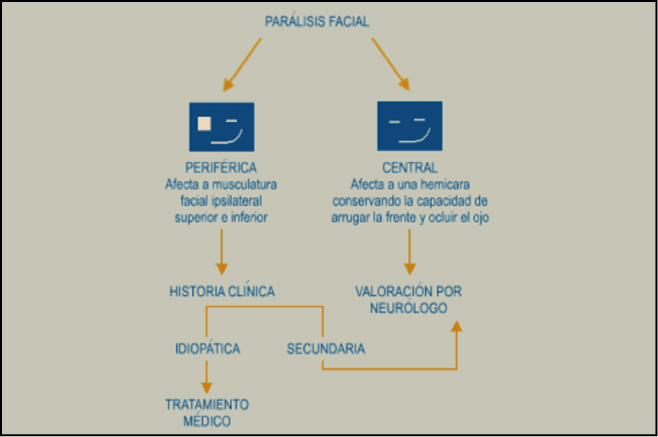
Figura 2.
CRITERIOS DE DERIVACIÓN
El manejo de la parálisis facial idiopática puede ser iniciado en el ámbito de atención primaria. En caso de sospecha de parálisis central o secundaria, o no idiopática (Tabla 2) se remitirá al neurólogo (Figura 2).
|
tabla 2. signos de sospecha de parálisis no idiopática o secundaria |
|
– Instauración lenta (>48 h). – Presencia de espasmos previo a la parálisis. – Mayor paresia de la musculatura facial inferior. – Afectación bilateral. – Falta de recuperación en 6 semanas. |
PRONÓSTICO
Los factores que más inciden en el pronóstico del paciente afecto de parálisis facial son la clínica y el electrodiagnóstico:
- Clínica
Existen una serie de factores clínicos que presentan incidencia en el pronóstico como son: la edad, cuanto más joven mejor pronóstico; la presencia de enfermedades sistémicas asociadas como la diabetes o la hipertensión que ensombrecen el pronóstico; y por supuesto la intensidad y el grado de afectación.
- Electrodiagnóstico
Tienen un alto índice de error estándar. Sólo se deben realizar si el clínico considera variar su actitud terapéutica en función del resultado de las mismas.
TRATAMIENTO
Va a depender de la etiología. El tratamiento de las secuelas y la prevención de las mismas, va a ser en muchos casos semejante. Las secuelas más frecuentes son las sincinesias, sobre todo la bucal al cerrar el ojo, que se debe tratar con toxina botulínica tipo A en dosis bajas y rehabilitación neuromuscular.
PARÁLISIS FACIAL IDIOPÁTICA AGUDA O PARÁLISIS DE BELL (PB)
Aparece de forma aguda, espontánea y es de causa desconocida.
- Diagnóstico. Es fundamentalmente clínico, de sospecha. Se trata de un diagnóstico realizado por exclusión de cualquier otra causa.
- Pronóstico Puede emitirse evaluando dos aspectos, la situación clínica del paciente y los resultados de las pruebas complementarias.
- Clínico. Se basa en tres factores:
- La edad de presentación: a menor edad, mejor pronóstico.
- Grado de afectación.
- Presencia de enfermedades sistémicas asociadas: el pronóstico es más desfavorable en hipertensos y diabéticos.
- Eléctrico. No se pueden obtener datos electrodiagnósticos antes del cuarto día.
Tratamiento
Entre el 73% y el 84% de las parálisis de Bell (PB) se recuperan sin tratamiento alguno, por lo que algunos facultativos recomiendan no tratarla. Sin embargo, la instauración de un tratamiento precoz y correcto puede disminuir por un lado el dolor y por otro el grado e intensidad de la denervación y sus secuelas.
- Tratamiento médico:
- Corticosteroides. Es un tratamiento excelente en las enfermedades desmielinizantes inmunomediadas, si bien no existe unanimidad sobre si su utilización mejora la recuperación de la PB. Disminuyen de forma significativa la denervación, así como la intensidad del dolor de modo que permiten disminuir la administración de analgésicos. La dosis utilizada de prednisona es de 1 mg/kg de peso y día durante 15 días, realizando una pauta descendente de dosis hasta la retirada completa del esteroide.
- Aciclovir. Se ha mostrado efectivo como tratamiento antivírico, reduciendo el grado de denervación y el dolor, y haciendo desaparecer las vesículas. Las dosis aconsejadas son de 200-400 mg, 5 veces al día durante 10 días. La asociación prednisona-aciclovir ha presentado unos mejores resultados que la prednisona más placebo.
- Protección ocular. Debe tratarse con lágrimas artificiales el ojo afecto, 5 veces al día, y con oclusión palpebral nocturna evitando que la córnea tenga contacto con el aire libre.
- Rehabilitación. Las secuelas de la PB son de dos tipos:
- Neurofisiológicas: sincinesias, movimientos asociados y parálisis flácida.
- Psicosociales: como consecuencia psíquica de la falta de motilidad en una hemofacies. Esta alteración de los patrones del funcionamiento del nervio facial puede ser compensados mediante el conocimiento y posterior modificación de la organización del sistema nervioso central (SNC) creando nuevos patrones motores.
- Descompresión quirúrgica del nervio facial. Si bien fue un tratamiento indiscutible hace unos años, hoy el papel de la cirugía en la PB es muy discutido cuando no rechazado.
HERPES ZOSTER ÓTICO
Síndrome caracterizado por parálisis facial, aparición de vesículas herpéticas en el pabellón auricular y alteraciones cocleovestibulares.
- Diagnóstico. El diagnóstico es fundamentalmente clínico.
- Tratamiento. El tratamiento clásico han sido los esteroides a 1 mg/kg/ de peso/día, con pauta descendente posterior durante 3 semanas; añadiendo aciclovir 800 mg cada 5 horas durante 10 días.
bibliografía
Producción de anticuerpos monoclonales
Resumen
Los anticuerpos monoclonales constituyen en el momento actual el principal grupo de medicamentos biotecnológicos. Un hito importante en relación con la obtención de estos fármacos fue la puesta a punto de la tecnología del hibridoma por Khöler y Milstein en 1975. Desde entonces, la naturaleza molecular de los anticuerpos monoclonales terapéuticos, así como los métodos de producción han evolucionado considerablemente. Actualmente, las estrategias para la producción de estos fármacos pasan inevitablemente por la selección de dianas terapéuticas de interés, hecho que entraña gran dificultad, ya que, a menudo no se conocen completamente las rutas bioquímicas implicadas en la génesis y desarrollo de muchas patologías. Su obtención puede llevarse a cabo utilizando ratones genéticamente modificados en los cuales se introducen los genes que codifican las inmunoglobulinas humanas, utilizando los vectores adecuados. También puede emplearse la tecnología de presentación de proteínas en la superficie de fagos filamentosos o Phage Display, que permite obtener anticuerpos monoclonales sin utilizar ratones. Mediante esta técnica se obtienen bibliotecas de anticuerpos, a partir de las cuales podrán seleccionarse aquellos que contengan la secuencia genética que codifica el anticuerpo con mayor afinidad y especificidad por la proteína diana. La tecnología de ADN recombinante también ha hecho posible la obtención de moléculas recombinantes derivadas de anticuerpos, como los fragmentos de anticuerpo y las proteínas de fusión, así como otros, llamados biespecíficos, en los cuales los dos sitios de unión al antígeno poseen especificidades diferentes.
Introducción
Los medicamentos biotecnológicos constituyen en la actualidad la vanguardia en la innovación terapéutica farmacológica. En las últimas décadas, los grandes avances de la Biotecnología y la Biología Molecular han permitido el desarrollo de gran cantidad de medicamentos biotecnológicos innovadores destinados al tratamiento de enfermedades neoplásicas, autoinmunes, infecciosas, inflamatorias y cardiovasculares, entre otras. Estos medicamentos tienen un enorme potencial en el tratamiento de estas enfermedades graves para las cuales, hasta el momento de su introducción, no existían fármacos eficaces disponibles.
Pero, ¿qué es un medicamento biotecnológico?
Un medicamento biotecnológico es aquel cuyo principio activo, de origen biológico, ha sido obtenido utilizando tecnología de ADN recombinante. Y una sustancia biológica es aquella que se produce o se extrae a partir de una fuente biológica y que necesita, para su caracterización y determinación de su calidad, una combinación de ensayos fisicoquímicos y biológicos junto con el proceso de producción y su control.1
Según la Agencia Europea de Medicamentos (EMA) un medicamento biológico es el que contiene uno o más principios activos sintetizados o derivados de una fuente biológica (como microorganismos, órganos y tejidos de origen vegetal o animal, células o fluidos de origen humano o animal y diseños celulares biotecnológicos). Es decir, aquel cuyo principio activo es producido por un organismo vivo o a partir de él.2
Actualmente se habla de fármacos biológicos o biofármacos para hacer referencia, en general, a todos los productos de origen biológico, ya que la mayor parte de ellos se obtienen mediante biotecnología, aunque existen algunas excepciones2.
Si bien los medicamentos biológicos pueden ser de naturaleza química muy diversa, incluyendo polisacáridos, hoy en día la mayor parte de estos productos la constituyen las proteínas o, en general, moléculas con un importante componente peptídico. La incorporación de estos fármacos a la terapéutica se produjo paralelamente a la introducción de las técnicas de ADN recombinante. En 1953, Watson y Crick descubrieron la estructura de doble hélice del ADN y a partir de ahí se conoció la estructura de los genes y cómo la información que portaban se traducía en funciones o características.3 Entonces se comenzó a buscar la forma de aislarlos, analizarlos, modificarlos y hasta de transferirlos de un organismo a otro para conferirle una nueva característica. Básicamente, de eso trata la ingeniería genética, que se podría definir como un conjunto de metodologías que permite transferir genes de un organismo a otro y expresarlos (producir las proteínas para las cuales estos genes codifican) en organismos diferentes al de origen.
El ADN que combina fragmentos de organismos diferentes se denomina ADN recombinante. En consecuencia, las técnicas que emplea la ingeniería genética se denominan técnicas de ADN recombinante y los organismos que reciben un gen que les aporta una nueva característica se denominan organismos genéticamente modificados o transgénicos.
Gracias a la aplicación de estas técnicas, que permiten la manipulación del ADN, ha sido posible la obtención de proteínas recombinantes, es decir, proteínas obtenidas a partir de una especie o una línea celular distinta a la original; combinando material genético de diferente origen.
Los biofármacos son proteínas o glicoproteínas de elevado peso molecular, con una estructura tridimensional compleja, que contienen un número elevado de aminoácidos con una secuencia determinada y con especificidad en el número y localización de puentes disulfuro que unen las cadenas proteicas. En ocasiones, contienen moléculas de carbohidratos que matizan su actividad biológica. Además, la cadena proteica puede presentar hélices en número, tamaño y configuración variados. Todo ello les hace poseer determinadas propiedades inherentes a las cadenas peptídicas que las forman y a la estructura tridimensional en que se constituyen tras los diferentes plegamientos y configuraciones espaciales que se pueden producir. En definitiva, son estructuras de tal complejidad que sólo los organismos vivos las pueden producir y cuya obtención por los métodos convencionales de síntesis química sería inabordable.
Los fármacos biológicos son, por tanto, fármacos muy complejos que han sufrido una gran evolución. El primer paso se dio con la identificación de los fragmentos de ADN responsables de la producción de determinadas hormonas, enzimas, factores de crecimiento, etc. Así se obtuvieron los biofármacos más simples, que componen la primera generación de medicamentos biológicos, como la insulina, la hormona de crecimiento, las gonadotropinas, los factores de la coagulación o los factores estimuladores de colonias, cuyo “mecanismo de acción” consistía únicamente en suplir a un factor que se encontraba deficitario en el organismo. Casi inmediatamente después, de forma paralela al avance en el conocimiento de los procesos fisiopatológicos, se realizaron observaciones de las que se extrajeron consecuencias provechosas, de tal manera que, en la actualidad, gracias a la introducción de los biofármacos de segunda generación, los anticuerpos monoclonales, los posibles mecanismos de actuación de los medicamentos biológicos son, teóricamente, ilimitados. En el momento actual los anticuerpos monoclonales se han establecido como el principal grupo de fármacos biotecnológicos.
Los anticuerpos y el sistema inmune
A modo de apunte histórico cabe recordar que en 1900, Ehrlich expuso su teoría de las “cadenas laterales” aplicada a la inmunología.4 Afirmaba que las células del organismo producían unos receptores, unas cadenas laterales, que eran capaces de unirse a las toxinas bacterianas por complementariedad molecular; de tal manera que cuando una célula está en peligro elabora un exceso de tales cadenas, que se separan de la célula y se convierten en antitoxinas circulantes, capaces de actuar frente a las toxinas bacterianas. Es decir, lo que hoy llamamos anticuerpos. Años más tarde, en 1972, Edelman y Porter, obtuvieron el premio Nobel por sus descubrimientos relativos a la estructura química de los anticuerpos.5,6
Como sabemos, los anticuerpos naturales o inmunoglobulinas (Ig) son los principales efectores de la respuesta humoral del sistema inmune. Como parte del sistema inmunológico normal, los linfocitos B y las células plasmáticas derivadas de ellos, producen cientos de miles de anticuerpos (policlonales) que son capaces de reconocer de manera específica componentes antigénicos presentes en bacterias, virus u otros agentes invasores y formar con ellos complejos estables (inmunocomplejos). Las inmunoglobulinas pueden encontrarse en forma soluble (anticuerpos), o bien, tal como decía Ehrlich, ancladas a la membrana de los linfocitos B constituyendo el receptor para antígeno (BCR) de estas células.
En el organismo humano existen cinco tipos diferentes de inmunoglobulinas, G, E, D, M y A. Si bien la estructura general de todas ellas es muy similar, cada tipo de inmunoglobulina tiene una función diferente en el sistema inmunológico. De todas las inmunoglobulinas, las IgG son las que desempeñan un papel preponderante en la respuesta inmunológica, siendo el tipo más abundante en el organismo.7
Estructura básica de una inmunoglobulina
La estructura más sencilla de inmunoglobulina tiene la forma característica de una “Y” (Figura 1). Está constituida por dos cadenas peptídicas largas (H, pesadas) e iguales y dos cadenas peptídicas cortas e iguales (L, ligeras), interconectadas por varios puentes disulfuro en una región conocida como región bisagra. Aproximadamente, los primeros cien aminoácidos de la secuencia de cada cadena son distintos, de ahí el nombre de variable (V) para esta región, mientras que el resto de la cadena es idéntico en cada anticuerpo de una determinada clase (C).
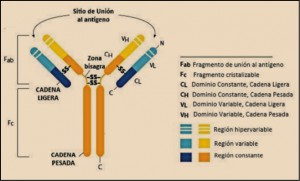
Figura 1. Estructura básica de una inmunoglobulina.
El fragmento Fab (Fragment antigen binding) comprende la región variable de las cadenas H y L y una parte de la región constante. Esta región determina su “especificidad” ya que en ella se encuentra la zona de unión al antígeno, mientras que en la región Fc (Fragment crystallizable) residen las llamadas “propiedades efectoras” de la reacción inmune, que convierten la unión inicial parátopo-epítopo en una reacción biológica en la que participan otras células y moléculas solubles. El resultado es la neutralización de la sustancia tóxica o la destrucción del microorganismo portador de la molécula de antígeno.
Estos segmentos peptídicos se yuxtaponen para formar una superficie o cavidad (parátopo) donde se aloja la región del antígeno reconocida por el anticuerpo (epítopo). El parátopo conforma una imagen especular del epítopo, y los seis segmentos que lo forman reciben el nombre de regiones hipervariables o regiones determinantes de la complementariedad (CDR, Complementary Determining Regions). Las regiones CDR confieren a las inmunoglobulinas una enorme especificidad y diversidad. 7, 8
Origen de los anticuerpos monoclonales
Los anticuerpos monoclonales son inmunoglobulinas modificadas, diseñadas específicamente para actuar frente a dianas concretas, con el objetivo de interrumpir un proceso patológico concreto, estimular una acción celular determinada o desviar un mecanismo celular hacia una vía de interés.
Su historia comienza en 1975, fecha en la que se produjo un hito importante en el descubrimiento de estos fármacos, la puesta a punto de la tecnología del hibridoma, por Khöler y Milstein (Figura 2), hecho por el cual recibieron el premio Nobel de Fisiología y Medicina en 1984.9
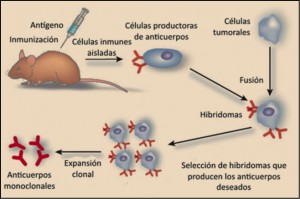
Figura 2. Representación esquemática de la obtención de anticuerpos monoclonales mediante la tecnología del hibridoma. Michnick W and Sidhu S (2008).
Esta tecnología supuso un importante avance, ya que permitió obtener anticuerpos específicos capaces de reconocer a un único epítopo o determinante antigénico y producirlos de forma ilimitada. El procedimiento comienza con la inmunización de un ratón con el antígeno de interés, tras la cual, se extraen los linfocitos B a partir del bazo del animal y se fusionan con células neoplásicas de mieloma. De esta manera se obtiene una célula de fusión, un hibridoma, que combina ciertas ventajas de sus progenitores, la capacidad de producir grandes cantidades de un único anticuerpo, propia de los linfocitos B y la capacidad de multiplicarse indefinidamente, característica de las células neoplásicas. A continuación, se seleccionan los hibridomas capaces de producir los anticuerpos deseados y se produce la expansión del clon de mayor interés. Finalmente se procede a la purificación de los anticuerpos obtenidos. De esta manera se obtiene una fuente casi inagotable de anticuerpos producidos por un clon celular, que derivan de un único linfocito B, y que son por tanto idénticos y específicos de epítopos individuales.
El primer anticuerpo monoclonal terapéutico producido mediante esta tecnología fue muromonab, de origen murino, dirigido frente al antígeno CD3 que se expresa en la superficie de los linfocitos T. Fue aprobado por la FDA (Food and Drugs Administration) en 1986 para el tratamiento del rechazo en pacientes trasplantados.
Sin embargo, el empleo de anticuerpos monoclonales murinos obtenidos mediante esta tecnología presentaba importantes limitaciones derivadas de su origen no humano, como su corta semivida plasmática o unas funciones efectoras ineficientes, pero sobre todo, la producción de anticuerpos humanos frente a las cadenas murinas, que provocaba importantes efectos adversos que limitaban su uso clínico.
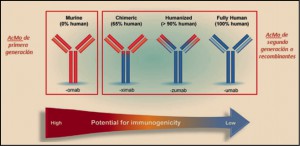
Figura 3. Tipos de anticuerpos monoclonales según su origen (Foltz IN, 2013).
Con el fin de evitar estos problemas, potencialmente graves, y gracias a las técnicas de ingeniería genética, se desarrollaron nuevas técnicas moleculares que dieron lugar a la obtención de los anticuerpos monoclonales de segunda generación, los anticuerpos monoclonales recombinantes (Figura 3), en los cuales se reemplazaron porciones del anticuerpo murino por proteínas humanas. En primer lugar se obtuvieron los anticuerpos monoclonales quiméricos, en los que la región Fab es de origen murino mientras que la fracción cristalizable Fc es de origen humano, y posteriormente los anticuerpos monoclonales humanizados, en los que la región Fc sigue siendo de origen humano y la región Fab es parcialmente humana y parcialmente murina. Los anticuerpos monoclonales quiméricos contienen aproximadamente un 65-90% de cadena humana mientras que los humanizados contienen aproximadamente un 90-95%. En 1997 se aprobó el primer anticuerpo monoclonal quimérico, rituximab, y en 1999 el primero humanizado, daclizumab.
Actualmente, se han desarrollado procedimientos que permiten obtener anticuerpos monoclonales completamente humanos, reduciéndose considerablemente el potencial inmunogénico. El primero de ellos, adalimumab,10 fue autorizado en 2003.
Producción de anticuerpos monoclonales terapéuticos
Un método para obtener anticuerpos monoclonales completamente humanos consiste en la utilización de ratones transgénicos, en los cuales se introducen los genes de las inmunoglobulinas humanas utilizando distintos vectores. Los ratones transgénicos portadores de los genes de las inmunoglobulinas humanas se cruzan con ratones en los cuales se han eliminado los genes de las inmunoglobulinas endógenas murinas, con el fin de conseguir un ratón capaz de producir exclusivamente inmunoglobulinas humanas (Figura 4).11,12
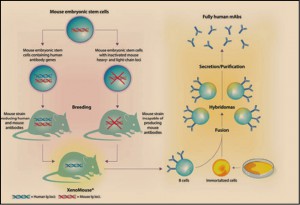
Figura 4. Obtención de anticuerpos monoclonales completamente humanos a partir de ratones transgénicos (Foltz IN, 2013).
Por otra parte, la tecnología Phage Display (presentación de proteínas en la superficie de fagos filamentosos) permite obtener anticuerpos monoclonales sin utilizar ratones. Los fagos son partículas víricas que infectan bacterias, sin producir enfermedades en los seres humanos. Mediante técnicas de ADN recombinante es posible introducir genes de cualquier organismo, sin importar la complejidad, en el genoma de los fagos. Estos fagos modificados genéticamente pueden expresar en su superficie diferentes proteínas terapéuticas. En líneas generales, lo que se pretende con esta tecnología es obtener una molécula que posea la máxima afinidad por una determinada diana terapéutica. Y para ello, el primer paso es obtener todas las posibles combinaciones genéticas que produzcan la molécula que se está buscando y cada una de estas secuencias se fusiona con el gen que codifica las proteínas de la cápside del fago, para lograr que sean expresadas en su superficie. De esta forma se obtienen clones de fagos, cada uno de los cuales produce una variante diferente de la secuencia de la proteína que se desea obtener. Posteriormente, se eligen aquellos fagos que expresen en su superficie la molécula con las mejores propiedades de unión a la molécula diana (Figura 5).13-15
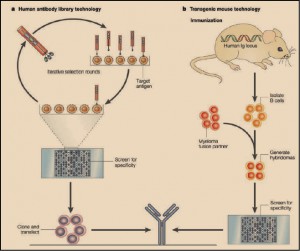
Figura 5. Técnicas de producción de anticuerpos mediante presentación en fagos (in vitro) y mediante la utilización de ratones transgénicos (in vivo). (Brekke and Sandlie, 2003).
A continuación, se describe el proceso de obtención de un anticuerpo monoclonal destinado a bloquear la acción de una proteína diana en un determinado proceso patológico. Dicho proceso se inicia con el aislamiento de los genes que codifican las cadenas ligeras (VL) y pesadas (VH) de la fracción variable, a partir de los linfocitos B aislados de donantes humanos adultos sanos. Estos genes se amplifican mediante un proceso denominado reacción en cadena de la polimerasa (PCR), que permite aumentar su cantidad exponencialmente. Una vez aisladas y copiadas las secuencias genéticas, éstas se someten a un proceso de recombinación, obteniéndose todas las combinaciones posibles, lo que supone la creación de una gran cantidad de anticuerpos diferentes. El siguiente paso es clonar las combinaciones obtenidas, para lo cual se insertan en el ADN del fago junto a los genes que codifican las proteínas estructurales de la cápside. Los fagos se replican en el interior de E. coli y los anticuerpos funcionales son expresados en su cápside, de la misma manera que un linfocito B expresa los anticuerpos en su superficie. Los múltiples fagos originados, con sus correspondientes anticuerpos en la superficie constituyen una biblioteca de anticuerpos, a partir de la cual deberán seleccionarse aquellos que contienen la secuencia génica que codifica el anticuerpo con la mayor afinidad y especificidad por la proteína diana en cuestión.
Una vez seleccionada la combinación VH/VL (de la fracción Fab), ésta se ensambla con el esqueleto estructural del anticuerpo, la fracción Fc. La secuencia genética resultante se introduce en células de ovario de hámster chino (células CHO).10
Seguidamente, se detallan las etapas del proceso de fabricación industrial.
El proceso de producción (Figura 6) consta de dos fases fundamentales:
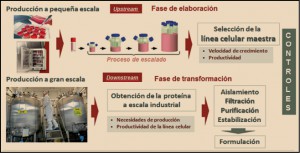
Figura 6. Pasos fundamentales del proceso de producción de un anticuerpo monoclonal.
- Fase de elaboración o upstream o de producción a pequeña escala y
- Fase de transformación o downstream o de producción a gran escala.
La fase de elaboración comienza con el cultivo de las células, que serán las encargadas de producir la proteína terapéutica. Estas células se cultivan a pequeña escala, en frascos o placas Petri, que contienen el medio de cultivo necesario para que las células crezcan. Una vez conseguida la línea celular, se somete a crioconservación en numerosos viales para crear un banco de células. Cuando se va a llevar a cabo el proceso de fabricación de un lote, se descongela un vial del banco de células y se inicia el cultivo celular en un matraz que contiene un pequeño volumen de medio de cultivo. Durante el proceso de escalado, las células se transfieren secuencialmente a recipientes cada vez más grandes, que contienen mayor volumen de medio de cultivo, de manera que las células se dividen constantemente siempre que el entorno de crecimiento siga siendo favorable. Los cambios más pequeños pueden afectar a las células y alterar las proteínas que producen. Por eso se requieren controles estrictos para garantizar la calidad y reproducibilidad del producto final. Hay que controlar variables como: temperatura, pH, nivel de oxígeno, concentración de nutrientes, etc. Además, deben realizarse pruebas para comprobar la ausencia de contaminación con bacterias, levaduras u otros microorganismos, ya que cualquier contaminación de un cultivo estropeará la totalidad del lote. Estos controles se realizan manualmente.
A continuación, las células que han mostrado mayor capacidad de crecimiento de forma estable a lo largo de varios cultivos se cultivan en equipos de volumen superior y se evalúa su capacidad de producción de la proteína de interés y la calidad de la misma, en base a su correcta estructura, glicosilación, funcionalidad, etc.
Esta etapa es muy crítica, ya que debe finalizar con la selección de la línea celular maestra, que será la que definitivamente se traslade a la última etapa del proceso.
La selección de la línea celular maestra se lleva a cabo en función de:
- La velocidad de crecimiento (concentración de células/unidad de tiempo) en el sistema final
- La productividad de la línea celular (cantidad de proteína producida por cada célula).
La siguiente etapa es la fase de transformación o downstream o de producción a gran escala; es decir, a la escala de producción necesaria, que vendrá determinada por las necesidades de producción (en base al volumen de mercado al que va destinado el fármaco) y la productividad de la línea celular.
A continuación, se aísla el anticuerpo monoclonal a partir de las células para ser sometido posteriormente a sucesivos procesos de filtración y purificación (cromatografía y ultrafiltración). Estos procesos también son una parte muy importante del proceso de fabricación, ya que puede tener una influencia crítica en la economía global del proceso en función del rendimiento obtenido.
Finalmente, se formula el producto según las especificaciones vigentes y se acondiciona para su uso clínico.
Cada uno de los pasos que se llevan a cabo para la producción de un anticuerpo monoclonal es específico para cada fármaco. Cualquier mínima alteración puede dar lugar a cambios en la estructura del principio activo, comprometiendo su estabilidad o su comportamiento terapéutico, o incrementando el riesgo de efectos adversos. Los parámetros que definen los procesos de fabricación deben estar bien especificados para una correcta valoración de la calidad del producto.16
Por tanto, actualmente pueden obtenerse de manera específica anticuerpos totalmente humanos, que se dirigen de modo muy específico y con un buen perfil de seguridad hacia una molécula concreta que desempeña una función clave en un proceso patológico. Esta molécula puede ser un receptor de superficie celular, una molécula de señalización, etc. El objetivo es que el anticuerpo reconozca dicha molécula, se una a ella y finalmente, se detenga el proceso patológico.
Todos los anticuerpos aprobados para uso terapéutico pertenecen a la familia de las IgG. Pero, a la hora de desarrollar un anticuerpo monoclonal terapéutico, surge la pregunta de cuál elegir. ¿Existen razones para preferir un isotipo u otro de IgG?
En humanos, existen cuatro isotipos de IgG, que se diferencian estructuralmente entre sí por el tamaño de la región bisagra, en el número de puentes disulfuro entre las cadenas pesadas, en su forma funcional in vivo, en su vida media, en el número de alotipos (polimorfismo alotípico), así como en su capacidad para ejercer funciones efectoras.
A la hora de elegir el isotipo será necesario tener en cuenta los resultados que se pretendan obtener in vivo, sobre todo dependiendo del mecanismo de acción que se busque.
Las funciones efectoras son muy útiles, por ejemplo, cuando lo que se pretende es destruir células tumorales. Los anticuerpos diseñados para tener un mecanismo de acción en enfermedades neoplásicas requieren un isotipo capaz de activar el complemento y que ejerza funciones efectoras para provocar la muerte celular por citotoxicidad dependiente de anticuerpos. Sin embargo, si el objetivo es la neutralización de antígenos solubles, las funciones efectoras son menos relevantes.
Por tanto, a la hora de desarrollar anticuerpos monoclonales terapéuticos deben considerarse las propiedades de los diferentes isotipos, aunque no existen reglas generales para la selección de uno u otro, ya que la elección final debe guiarse por los resultados de los estudios pertinentes.17
Moléculas recombinantes derivadas de anticuerpos monoclonales
Además de los anticuerpos monoclonales completos, también se utilizan en clínica diversas moléculas recombinantes derivadas de ellos para finalidades concretas, considerando la diana farmacológica hacia la que se dirigen.
Estas moléculas derivadas de anticuerpos constituyen un conjunto heterogéneo de proteínas recombinantes entre las que se incluyen los fragmentos de anticuerpos monoclonales, tipo Fab, Fv de cadena sencilla (ScFv), diabodies, triabodies, etc. y las proteínas de fusión.
Antes del desarrollo de las técnicas de ingeniería genética la obtención de fragmentos de anticuerpo (Fab, fragment antigen-binding) (Figura 7) se llevaba a cabo mediante la acción de enzimas proteolíticas. Posteriormente se obtuvieron versiones recombinantes funcionales de Fv y Fab en E. coli. E. coli es en la actualidad el sistema de expresión más accesible para la producción de estos fragmentos debido a las ventajas indiscutibles que ofrece en lo que se refiere a manipulación, transformación, cinética de crecimiento y condiciones de fermentación.
Figura 7. A. Fragmentos de anticuerpos monoclonales. B. Ingeniería de anticuerpos empleando fragmentos variables de cadena única o ScFv como base. (Sanz L, 2009).
La utilización de fragmentos de anticuerpo, como los Fv, formados por las regiones variables de la cadena pesada y ligera, donde se encuentra la zona de reconocimiento del antígeno, se debe a que su bajo peso molecular les confiere unas propiedades óptimas tanto en el campo diagnóstico como en el terapéutico. Sin embargo, las interacciones débiles que unen las regiones V los hacen muy inestables a temperatura fisiológica. Con el fin de mejorar la estabilidad y preservar la conformación de este tipo de fragmentos de anticuerpo recombinantes se han desarrollado diferentes estrategias que incluyen la introducción de puentes disulfuro adicionales y la adición de segmentos peptídicos flexibles (linkers), que conectan los genes de las regiones V de la misma cadena o de cadenas distintas.
De esta manera se han obtenido fragmentos recombinantes en los que las regiones VH y VL están unidas por un linker peptídico. Estas estructuras se denominan scFv (single chain Fv). También se han obtenido fragmentos bivalentes (diabodies) que presentan dos sitios de unión al antígeno, y diabodies biespecíficos que pueden reconocer dos antígenos distintos.
La tecnología de ADN recombinante ha hecho posible el diseño de moléculas artificiales denominadas proteínas de fusión o proteínas recombinantes quiméricas (Figura 8) derivadas de anticuerpos monoclonales, que son el resultado de combinar la porción Fc de una IgG humana con fracciones peptídicas de determinados receptores biológicos o de sus correspondientes ligandos. Las proteínas de fusión constituyen, por tanto, una alternativa para mejorar la expresión de proteínas recombinantes.
Figura 8. Proteínas de fusión. Análogos de receptores solubles.
La tecnología de ADN recombinante también ha hecho posible el diseño de anticuerpos monoclonales biespecíficos. Se trata de inmunoglobulinas construidas artificialmente mediante técnicas de ingeniería genética, en las cuales los dos sitios de unión a antígeno poseen especificidades diferentes. De esta manera, una parte del anticuerpo es capaz de reconocer un tipo de molécula o célula diferente de la otra.
Para ello deben obtenerse dos cadenas, VHA-VLB y VHB-VLA (siendo A y B, los dos antígenos diferentes), que se expresan en el mismo vector y se ensamblan para formar dímeros con dos sitios distintos de unión al antígeno.8,18
En conclusión, podemos afirmar que los anticuerpos monoclonales constituyen una indiscutible herramienta terapéutica, que, junto con los grandes avances en el conocimiento de la Biología Molecular, han permitido la actuación de manera específica sobre dianas clave en la etiopatogenia de muchas enfermedades graves. Por otra parte, la tecnología actual de fabricación de estos fármacos permite cada vez nuevos y mejores diseños capaces de ampliar sus horizontes terapéuticos.
Bibliografía
- Real Decreto 1345/2007, de 11 de octubre por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. https://www.boe.es/boe/dias/2007/11/07/pdfs/A45652-45698.pdf
- http://www.ema.europa.eu/ema/
- Watson JD, Crick FHC. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953; 171: 737-8
- Ehrlich P. Collected papers of Paul Ehrlich. Compiled and edited by F. Himmelweit. EA. London, Pergamon Press, 1956-60
- Edelman GM, Poulik MD. Studies on structural units of the g-globulins. J Exp Med. 1961; 113: 861–84
- Porter RR. The structure of antibodies. Scientific American. 1967; 180: 713–6.
- García Merino A. Anticuerpos monoclonales. Aspectos básicos. Neurología 2011; 26(5): 301-6
- Sanz L, Álvarez-Vallina L. Anticuerpos monoclonales. Biotecnología y Biofármacos. Consejo General de Colegios Oficiales de Farmacéuticos. 2009
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256(5517): 495-7.
- Martínez de Lagrán Z, Pérez-Barrio S, Díaz-Pérez JL. Adalimumab: la molécula y el proceso de obtención. Actas Dermosifiliogr. 2008; 99(Supl 3): 3-9
- Lonberg N. Human antibodies from transgenic animals. Nature biotechnology 2005; 23(9): 117-25
- Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies. What cardiologists need to know. Circulation 2013; 127: 2222-30
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nature 2003; 2: 52-62.
- Marcus RD, Fiorini RN. Different approaches for obtaining antibodies from human B cells. Current Drug Discovery Technologies 2014; 11: 41-7.
- Michnick W, Sidhu S. Submitting antibodies to bindin arbitration. Nature Chemical Biology 2008; 4: 326-9
- http://www.amgenbiotech.es/index.jsp
- Salfeld JG. Isotype selection in antibody engineering. Nature Biotechnology 2007; 25(12): 1369-72.
- http://www.labome.es/method/Antibody-Structure-and-Fragments.html
Actividad física y patología esofágica
Es bien conocido que la actividad física afecta el funcionamiento del sistema gastrointestinal a través de los efectos locales y sistémicos y que puede jugar un papel importante en la reducción del riesgo de adenocarcinoma esofágico. Una reciente revisión ha evaluado los mecanismos biológicos y la evidencia epidemiológica de la relación entre la actividad física y el desarrollo del adenocarcinoma esofágico y sus enfermedades precursoras: enfermedad por reflujo gastroesofágico (ERGE) y esófago de Barrett.
Una amplia revisión bibliográfica sobre estudios analíticos que examinaron las asociaciones entre los niveles recreativos y/o ocupacionales de actividad física y el riesgo de ERGE, esófago de Barrett y adenocarcinoma esofágico, permitió seleccionar un total de siete estudios (dos de cohortes y cinco de casos y controles). Para la ERGE, hubo tres estudios de casos y controles con 10.200 casos entre 78.034 participantes, con una reducción del riesgo del 33% (OR = 0,67; IC95% 0,57 a 0,78) para niveles altos vs bajos de actividad física recreativa. En el esófago de Barrett, hubo un único estudio de casos y controles, que no encontró una asociación estadísticamente significativa (OR = 1,19; IC95% 0,82 a 1,73). Finalmente, para el adenocarcinoma esofágico, hubo tres estudios (dos prospectivos de cohortes y uno de casos y controles) con 666 casos entre 910.376 participantes. El estudio de cohortes más grande encontró una reducción del 32% para niveles altos vs bajos de actividad física recreativa (RR = 0,68; IC95% 0,48 a 0,96), aunque los otros dos estudios no encontraron una asociación estadísticamente significativa con ningún tipo de actividad física, ocupacional y/o recreativa. En opinión de los autores de este estudio, aunque las pruebas son limitadas, apuntan a una cierta evidencia de que los niveles más altos de actividad física recreativa pueden reducir el riesgo tanto de ERGE como de adenocarcinoma esofágico, aunque obviamente se necesitan estudios de cohortes adicionales que examinen el tipo, la intensidad y la duración de las actividades que pueden ser beneficiosas.