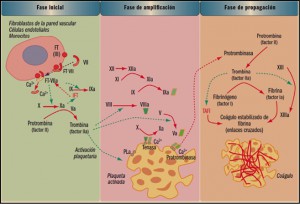
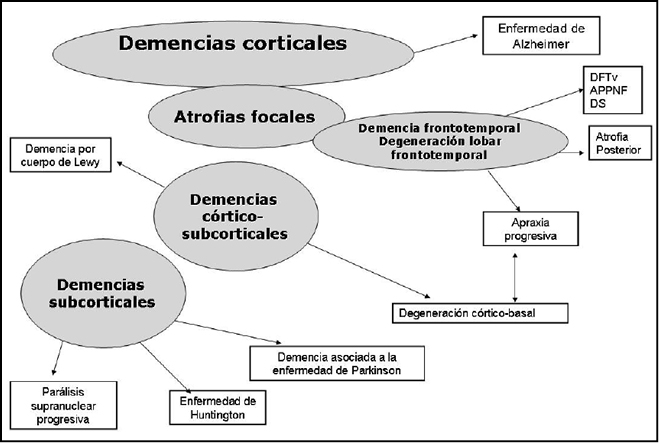
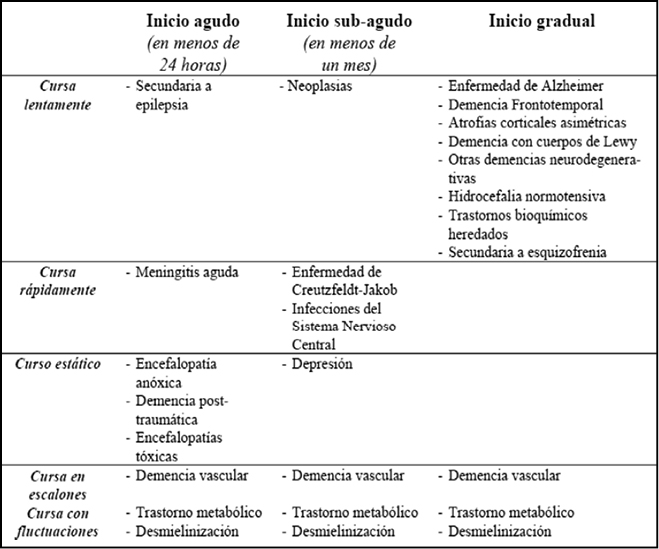
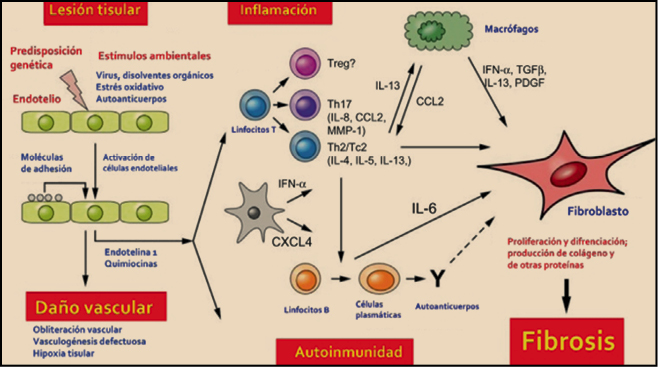
TERAPIAs avanzadas
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.
|
CLASIFICACIÓN DE NUEVOS PRODUCTOS DE TERAPIAS AVANZADAS EN LOS ÚLTIMOS DOCE MESES, |
|||
|
Medicamento |
Indicación |
Clasificación |
Fecha |
|
Células citotóxicas naturales transducidas para expresar el receptor de Fc CD16 (hnCD16) no fraccionable de alta afinidad |
Tumores neoplásicos sólidos |
Terapia génica |
12/05/2017 |
|
Células enteras de tumor colorrectal no proliferativo, haptenizado, estimulado e irradiado alogénico derivado de 3 líneas celulares colorrectales |
Cáncer colorrectal |
Terapia celular somática |
12/05/2017 |
|
Virus adenoasociado recombinante que expresa la globulina humana de unión a tiroxina uridina difosfato glicuronosiltransferasa 1A1 |
Síndrome de Crigler-Najjar |
Terapia génica |
21/04/2017 |
|
Sistema implantable de monitorización continua de glucosa basado en células modificadas genéticamente |
Monitorización de glucosa en pacientes diabéticos |
Terapia génica |
21/04/2017 |
|
Adenovirus oncolítico recombinante |
Cáncer de páncreas |
Terapia génica |
21/04/2017 |
|
Células mesenquimatosas pluripotenciales derivadas de médula ósea autóloga |
Tratamiento del como (lesiones cerebrales, ictus) |
Terapia cellular somática |
21/04/2017 |
|
Leucocitos alogénicos almacenados |
Adenocarcinoma ductal pancreático |
Terapia cellular somática |
21/04/2017 |
|
Células mesenquimoatosas pluripotenciales derivadas del cordón umbilical alogénico |
Regeneración del disco intervertebral |
Ingeniería tisular |
21/04/2017 |
|
Linfocitos autólogos de infiltración tumoral |
Tratamiento del melanoma metastásico |
Terapia celular somática |
22/03/2017 |
|
Médula ósea derivada de células mesenquimales |
Tratamiento de la enfermedad aguda del injerto contra el huésped |
Terapia celular somática |
05/01/2017 |
|
Antígeno quimérico anti-BCMA del receptor de células T |
Tratamiento del mieloma múltiple y el linfoma de células B |
Terapia génica |
18/11/2016 |
|
Suspensión autóloga de células de la piel |
Tratamiento de quemaduras y heridas de la piel |
Ingeniería tisular |
18/11/2016 |
|
Tejido adiposo autólogo derivado de células madre mesenquimales |
Tratamiento de la reparación cardiaca tras un infarto de miocardio |
Ingeniería tisular |
18/11/2016 |
|
Médula ósea autóloga derivada de células no hematopoyéticas |
Tratamiento de la esclerosis múltiple |
Ingeniería tisular |
18/11/2016 |
|
Rilimogen galvacireped y rilimogen glafolivec |
Tratamiento del cáncer de próstata metastásico resistente |
Terapia génica |
18/11/2016 |
|
Virus Ankara modificado que expresa mucina 1 e interleucina 2 humanas |
Tratamiento del cáncer avanzado de pulmón de células no pequeñas y no escamosas |
Terapia génica |
18/11/2016 |
|
Células T autólogas expresando un receptor NKG2D quimérico |
Tratamiento de varios tipos de tumores |
Terapia génica |
20/10/2016 |
|
Combinación de dos plásmidos codificados por los antígenos E6 y E7A del virus 16 y 18 del papiloma humano |
Tratamiento del HPV-16 y 18 |
Terapia génica |
20/10/2016 |
|
Lactobacillus reuteri vivo genéticamente modificado con un plásmido conteniendo el gen humano CXCL12 |
Tratamiento de las lesiones crónicas de piel en pacientes diabéticos |
Terapia génica |
20/10/2016 |
|
Fracción de células humanas autólogas de estroma vascular y células madre autólogas derivadas del mesénquima adiposo |
Tratamiento de la piel laxa senil |
Ingeniería tisular |
20/10/2016 |
|
Vector viral adenoasociado serotipo 8 codificador de gen humano de glucosa-6 |
Tratamiento de la enfermedad tipo Ia de almacenamiento de glucógeno (enfermedad de von Gierke) |
Terapia génica |
14/09/2016 |
|
Vector viral adenoasociado serotipo 2 codificador del gen humano de L-aminoácido decarboxilasa |
Tratamiento de la enfermedad de Parkinson |
Terapia génica |
14/09/2016 |
|
Cultivo ex-vivo de células progenitoras de sangre de cordón umbilical humano |
Tratamiento de pacientes bajo trasplante de células progenitoras hematopoyéticas |
Ingeniería tisular |
14/09/2016 |
|
Células progenitoras heterólogas derivadas de hígado humano adulto |
Tratamiento de las enfermedades fibroinflamatorias del hígado |
Ingeniería tisular |
14/09/2016 |
|
Fibroblastos autólogos expandidos in vitro |
Tratamiento de cicatrices de diferentes etiologías (postraumáticas, postquirúrgicas, etc.) |
Ingeniería tisular |
15/07/2016 |
|
Concentrado de médula ósea autóloga |
Tratamiento de la isquemia de extremidades sin opción quirúrgica |
Ingeniería tisular |
15/07/2016 |
|
Virus de Epstein-Barr alogénico de linfocitos T citotóxicos |
Tratamiento del virus de Epstein-Barr. Virus asociado al postransplante en trastornos linfoproliferativos |
Terapia celular somática |
23/06/2016
|
|
Células autólogas expandidas ex vivo reguladores de linfocitos T |
Tratamiento de la diabetes tipo 1 |
Terapia celular somática |
23/06/2016
|
|
Virus adenoasociado codificador de genes de los canales de rodopsina en algas |
Tratamiento de la rinitis pigmantosa |
Terapia génica |
23/06/2016
|
|
Células madre mesenquimales aisladas de la médula ósea autóloga, |
Tratamiento de enfermedades neurológicas en niños (encefalopatías, epilepsia y lesión de médula espinal). |
Ingeniería tisular |
23/06/2016 |
|
Evolución cronológica de las clasificaciones y evaluaciones de las terapias avanzadas |
||||
|
AÑO |
CLASIFICACIÓN DE MEDICAMENTOS |
MEDICAMENTOS EVALUADOS |
||
|
REMITIDOS |
ADOPTADOS |
REMITIDOS |
FAVORABLES |
|
|
2017 |
27 |
16 |
1 |
1 |
|
2016 |
60 |
87 |
1 |
2 |
|
2015 |
61 |
31 |
1 |
1 |
|
2014 |
28 |
29 |
2 |
1 |
|
2013 |
20 |
23 |
2 |
2 |
|
2012 |
22 |
16 |
3 |
1 |
|
2011 |
12 |
12 |
2 |
1 |
|
2010 |
19 |
27 |
1 |
0 |
|
2009 |
22 |
12 |
3 |
1 |
|
TOTAL |
271 |
253 |
16 |
10 |