Los corticosteroides incrementan el riesgo de fractura ósea también en jóvenes
La utilización de corticosteroides en pacientes jóvenes con artritis reumatoide de reciente aparición incrementa significativamente el riesgo de fractura ósea, de forma forma proporcional a las dosis diarias y acumuladas. Este riesgo se normaliza al cabo de un año de haber suspendido la administración de los corticosteroides.
El riesgo de fractura ósea asociado al empleo sistémico continuado de corticosteroides es conocido desde hace tiempo. Sin embargo, los datos epidemiológicos proceden de estudios realizados con pacientes con edades relativamente elevadas y con cuadros crónicos diagnosticados hacía tiempo. Por ello, se ha llevado a cabo un estudio con el fin de determinar si tal riesgo de fractura ósea aumenta también en pacientes jóvenes con un diagnóstico reciente de artritis reumatoide. El estudio se hizo a partir de registros procedentes de bancos de datos administrativos, incluyendo a un total de 42.127 pacientes diagnosticados de artritis reumatoide con edades comprendidas entre los 18 y los 64 años.
Un 74% de los pacientes incluidos eran mujeres y la media global de edad era de 49 años; un 1% había padecido anteriormente una fractura ósea. El análisis de los registros estableció que la mayor parte de los pacientes (85%) habían recibido corticosteroides, comprobándose que la tasa de incidencia de fracturas fue de 5-9 por 1000 personas-año con dosis de corticosteroides (en dosis equivalentes de prednisona) inferiores a 15 mg/día, mientras que con dosis diarias iguales o mayores de 15 mg/día tal incidencia aumentaba hasta 16,0 por 1.000 personas-año (IC95% 11,0 a 22,6); igualmente, la incidencia de fracturas óseas fue de 13,4 por 1.000 personas-año (IC95% 10,7 a 16,7) con dosis totales acumuladas del tratamiento ≥5.400 mg. En términos comparativos, el riesgo de fractura fue doble entre los pacientes tratados con las dosis más altas de corticosteroides que entre aquellos no tratados con estos (o con dosis acumuladas inferiores a 675 mg).
El riesgo de fractura ósea fue paulatinamente descendiendo con el paso del tiempo tras finalizar el tratamiento con los corticosteroides; en este sentido, tal riesgo descendió un 29% durante los 60-182 días después y acabó por normalizarse al cabo de un año. Los datos de incidencia fueron similares entre todos los grupos de edad, incluyendo los menores de 50 años.

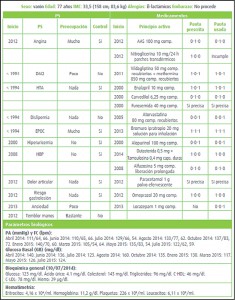
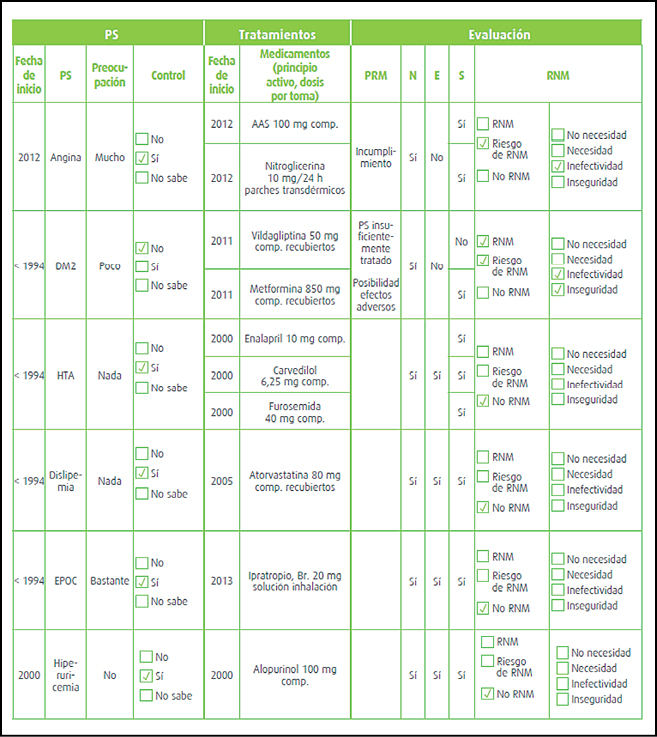
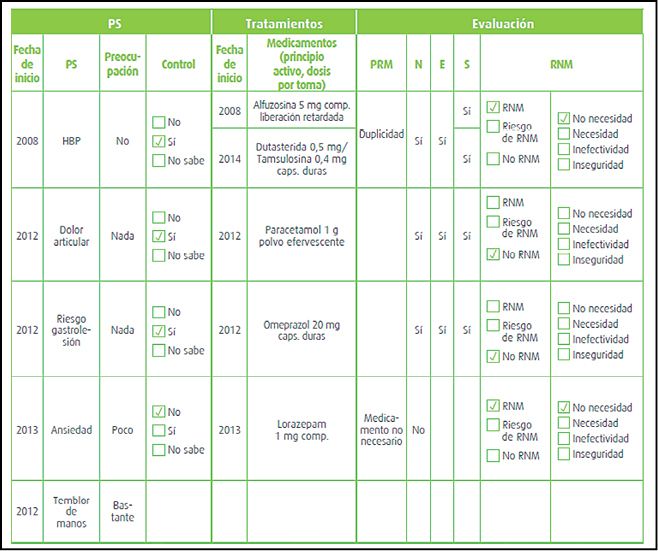
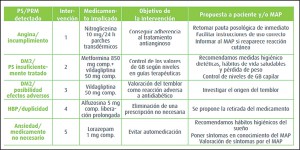
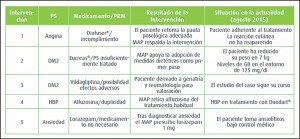
{kind=link}
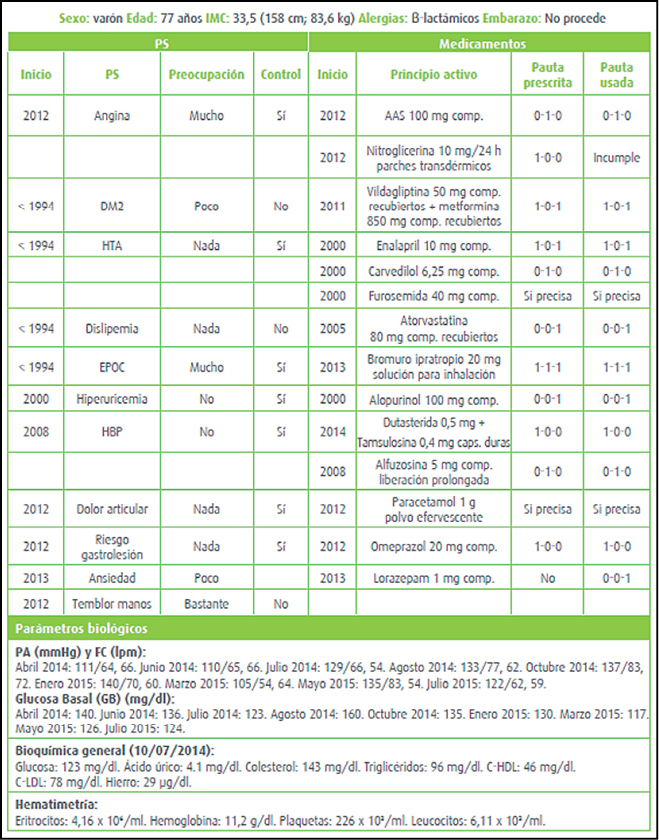{kind=link}
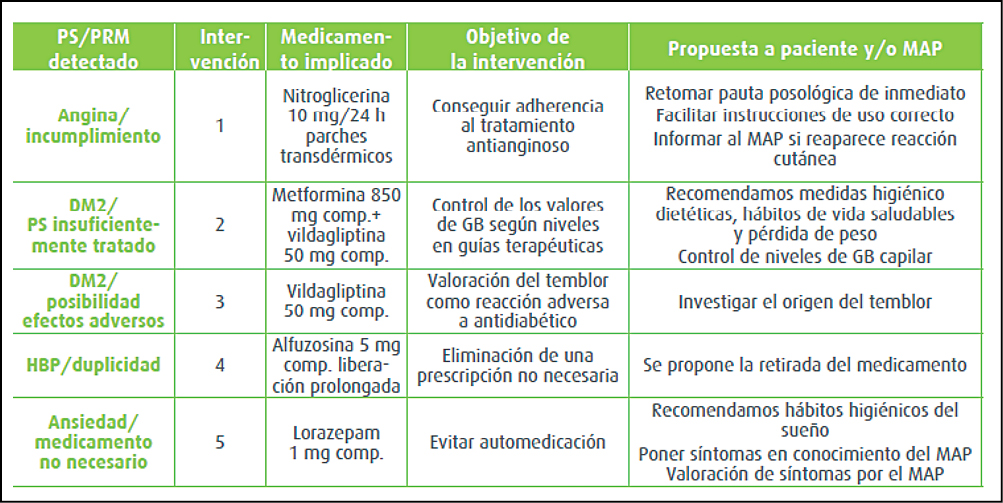{kind=link}
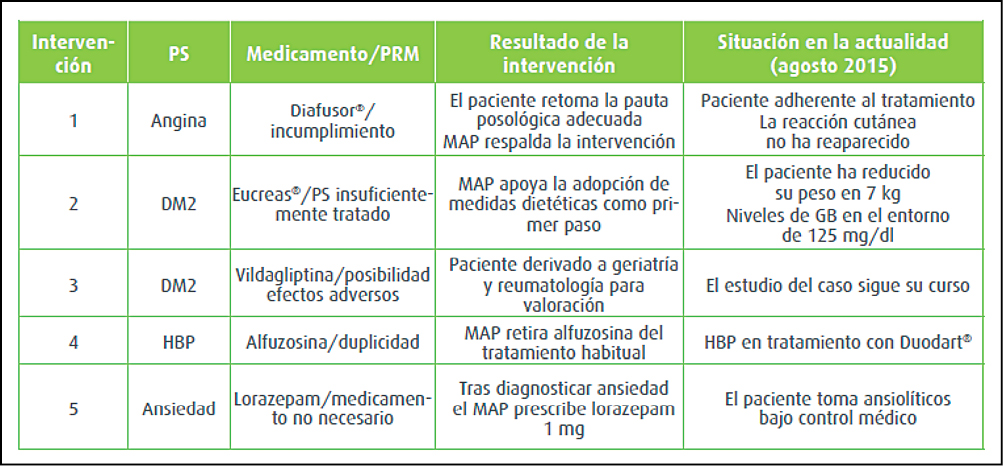{kind=link}