Resumen
El idelalisib es un agente antineoplásico que inhibe selectivamente la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias de linfocitos B, al estar implicada en numerosas vías bioquímicas de señalización que impulsan la proliferación, supervivencia, migración y retención de las células tumorales en los tejidos linfoides y en la médula ósea. El efecto final de todo el proceso es la inducción de la apoptosis y la inhibición de la proliferación de las células tumorales primarias y de las líneas celulares derivadas de los linfocitos B tumorales. El medicamento ha sido autorizado para el tratamiento, en combinación con rituximab, de los pacientes adultos con leucemia linfocítica crónica que han recibido al menos un tratamiento anterior o como tratamiento de primera línea en presencia de la deleción 17p o de la mutación TP53 en pacientes no adecuados para quimioinmunoterapia; también está indicado en monoterapia de los pacientes adultos con linfoma folicular refractario a dos líneas de tratamiento anteriores. El nuevo medicamento incorpora una nueva diana farmacológica antineoplásica: la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias de linfocitos B. En definitiva, un nuevo medicamento que parece tener un papel relevante como tratamiento de segunda línea en la leucemia linfocítica crónica, especialmente para pacientes de altos riesgo y con comorbilidades múltiples y un prometedor agente para los linfomas no-Hodgkin.
ASPECTOS FISIOPATOLÓGICOS
Los linfomas son neoplasias del sistema linfoide que constituyen un grupo heterogéneo de enfermedades neoplásicas definidas por aspectos morfológicos, inmunofenotípicos y genéticos, que tienen su origen en los sistemas mononuclear fagocítico y linfático. Clásicamente, los linfomas se clasificaban en dos grandes grupos, la enfermedad de Hodgkin, que representa el 15-20% de los casos, y los linfomas no Hodgkin. Actualmente, se acepta la clasificación de la Organización Mundial de la Salud (OMS), en la que se definen 3 categorías de neoplasias linfoides: el linfoma de Hodgkin y, dentro de los linfomas no Hodgkin, los de origen B o T/NK. Los linfomas de Hodgkin consisten en una proliferación, localizada o diseminada, de células tumorales que se originan en el sistema linforreticular y que afecta principalmente los ganglios linfáticos y la médula ósea.
Los linfomas no-Hodgkin (LNH) incluyen a todos los linfomas que no encajan dentro de la definición de linfoma de Hodgkin; por tanto, son neoplasias linfoides que pueden presentar fenotipo de células B ó T/NK. Los LNH representan el 4-5% de los nuevos casos de cáncer diagnosticados al año, ocupando el quinto lugar en frecuencia; los de linfocitos B representan el 80-85% de los LNH y los T el 15-20%, mientras que los de células NK (Natural Killer; citotóxicas) tienen una frecuencia marginal.
Entre los linfomas de células B, los más comunes son el linfoma difuso de células B grandes (30-35%) y el linfoma folicular (20-25%); menos prevalentes son el linfoma de tejido linfoide asociado a mucosas (TLAM) (7-10%), el linfoma linfocítico pequeño o leucemia linfocítica crónica (6-8%), el linfoma de células del manto (5-7%), el linfoma de Burkitt (2-3%) y el linfoma mediastínico (tímico) de células B grandes (2-3%). Menos del 2% de los linfomas de células B corresponden al linfoma linfoplasmacítico (macroglobulinemia de Waldenström), el nodal de células B de la zona marginal, el esplénico de zona marginal, el extranodal de células B de zona marginal, el intravascular de células grandes B, el de efusión primaria y la granulomatosis linfomatoide.
Por su parte, los linfomas de células T, que suponen aproximadamente el 12% de todos los LNH, se clasifican en: linfoma extranodal T, linfoma cutáneo de las células T (Síndrome de Sézary y Micosis fungoide), linfoma anaplásico de células grandes y linfoma angioinmunoblástico de las células T. La incidencia de linfomas de células NK es notablemente inferior.
En las últimas décadas se ha registrado un aumento en las tasas de incidencia y de mortalidad de los linfomas no Hodgkin, principalmente en países industrializados. Específicamente, se ha observado un aumento especialmente acusado de las tasas de incidencia de los linfomas no-Hodgkin en España e Italia. El aumento afecta a todos los grupos de edad adulta, aunque el mayor aumento se registra en los sectores de edad más avanzada de la población. En España, según datos del Instituto Nacional de Estadística, murieron en 2013 un total de 4.832 personas (en 2008, fueron 4.451) por tumores malignos del tejido linfático, excepto leucemias, de las que un 53% eran varones y un 47% mujeres.
Los linfomas no hodgkinianos pueden aparecer en cualquier edad de la vida, pero la mediana de presentación se sitúa en torno a los 50 años, siendo más frecuentes en los varones. Tanto en las neoplasias linfoides B como en las T se distinguen dos tipos de transformación neoplásica, uno que se origina a partir de las células precursoras y el otro a partir de las células periféricas.
La etiopatogenia de los LNH varía en los distintos tipos, pero presentan factores de riesgo comunes, tales como la existencia de un sistema inmune debilitado (ya sea por una enfermedad hereditaria o tras un trasplante de órganos), edad elevada, antecedentes familiares, exposición a agentes tóxicos (herbicidas) e infecciones por algunos virus (virus linfotrópico de células T del ser humano tipo 1 –HTLV-1–, virus de la inmunodeficiencia humana –VIH–, virus de Epstein-Barr –EBV–) y bacterias (Helicobacter pylori).
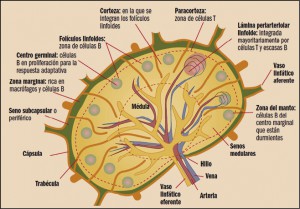
Los linfomas se pueden clasificar por la célula maligna de origen: centro germinal o no centro germinal; también tenemos la zona del manto y la zona marginal. También es muy importante la manera en que se infiltra el ganglio, por ejemplo: el linfoma difuso de célula grande (LDCG) es un linfoma de linfocitos B de tamaño grande (por su estado de maduración) que infiltra el ganglio de forma difusa y puede infiltrarlo tanto en el centro germinal como fuera del mismo (no centro germinal).
El tratamiento de los linfomas de grado bajo o indolentes no está estandarizado, aunque la terapia inicial suele consistir en el empleo de antineoplásicos de tipo de los agentes alquilantes (ciclofosfamida,bendamustina) o antimetabolitos (fludarabina) en monoterapia o en combinaciones de 2, 3 o 4 agentes: vincristina, ciclofosfamida, doxorubicina y prednisona más rituximab (protocolo CHOP+R), ciclofosfamida, vincristina, prednisona + rituximab (CVP+R), fludarabina, ciclofosfamida, mitoxantrona, rituximab (FCMR) o fludarabina, mitoxantrona, dexametasona, rituximab (FND-R). Este tipo de tratamientos permite alcanzar tasas de respuesta global de hasta un 90%, que son completas hasta en el 60% de los casos primarios. La duración media de la respuesta oscila entre uno y cuatro años. En caso de recaída, la opción más básica consiste en repetir el tratamiento, generalmente añadiendo un escalón más en combinación.
Los receptores CD20 están presentes en la superficie celular de los linfocitos B, tanto normales como malignos (incluyendo las formas maduras, proliferantes y diferenciadas). Actúan como receptores moleculares del antígeno Bp35, una proteína fosforilada responsable de la restricción de la diferenciación de los linfocitos B que es expresada durante las fases más precoces. Esta circunstancia ha conducido al desarrollo de anticuerpos específicos, como el rituximab, capaces de provocar la destrucción selectiva de linfocitos B (aunque sin distinción entre fisiológicos y malignos) como consecuencia de una combinación, en diferentes grados, de efectos celulares de citotoxicidad y fagocitosis mediados por anticuerpos, así como apoptosis independiente de la cascada de caspasas (inducción directa de la muerte celular) y citotoxicidad dependiente del complemento; no obstante, se considera que la citotoxicidad y la fagocitosis celulares mediadas por anticuerpos son los mecanismos antineoplásicos principales de este tipo de antucuerpos anti-CD20.
Ibritumomab es otro anticuerpo monoclonal que une de forma específica al receptor CD20; está ligado a un agente quelante, el tiuxetano, que actúa como anclaje de un radioisótopo del itrio (90Y), un radionúclido emisor de radiación beta (electrones), de baja penetración (5-10 mm) y con una vida media de 64 h. El medicamento está indicado en el tratamiento de pacientes adultos con linfoma no Hodgkin (LNH) folicular de células B CD20+ en recaída o refractario a rituximab. Normalmente, el tratamiento con ibritumomab-tiuxetano-itrio(90) va precedido por un tratamiento con bajas dosis de rituxumab, con el fin de eliminar los linfocitos B circulantes, facilitando con ello la acción más selectiva del ibritumomab-tiuxetano-itrio(90). Se trata, por consiguiente, de un agente inmunoradioterapéutico, en el que la fracción inmunológica (ibritumomab) es un anticuerpo específico para los receptores CD20, presentes en la gran mayoría de los linfocitos B humanos, tanto fisiológicos como malignos. Esta fracción tiene como misión localizar los linfocitos B, que es la población celular cuya malignificación conduce al desarrollo del linfoma folicular no-Hodgkin. Esta forma de inmunoradioterapia con ibritumumab-tiuxetano- 90Y para las fases avanzadas de linfoma folicular no Hodgkin da lugar a tasas relativamente altas de respuesta, superiores al rituxumab, incluso en pacientes refractarios a otros tratamientos antineoplásicos (incluyendo al propio rituxumab), con respuestas que llegan a alcanzar en una minoría de pacientes hasta más de tres años, aunque dada la fase avanzada de desarrollo del linfoma y la refractoriedad a otras terapias, previsiblemente no puede considerarse éste como un parámetro especialmente relevante (Cuéllar, 2016).
Por su parte, la leucemia linfática crónica o leucemia linfocítica crónica (LLC) se considera un linfoma de bajo grado, caracterizado por la acumulación de linfocitos B maduros en la sangre, médula ósea y órganos linfáticos. En función de la citogenética, tiene mal pronóstico si presenta la traslocación t(11q; v) o las deleciones del(11q) o del(17p); esta última confiere resistencia a la fludarabina. Por el contrario, el pronóstico es favorable si presenta la deleción del(13q) como única anormalidad. Los linfocitos circulantes son morfológicamente similares a los normales pero funcionalmente anormales; expresan marcadores de superficie CD5, CD20 y CD23. La acumulación se inicia frecuentemente en la médula ósea, diseminándose posteriormente hacia los ganglios linfáticos y bazo, pudiendo haber esplenomegalia.
Es la leucemia más frecuente en los países occidentales, constituyendo el 30 % de todas las formas de leucemia y 75 % de las leucemias crónicas. Su incidencia en la Unión Europea es de 4,2 casos/100.000 habitantes/año, aunque aumenta con la edad, siendo rara antes de los 40 años. A la edad de 50 años alcanza los 5 casos y a los 80 años llega a los 30 casos; de hecho, la mediana de la edad en el momento del diagnóstico es 72 años. Existe un predominio en el sexo masculino (2:1).Afecta a más de 300.000 personas en el mundo y a más de 15.000 en España; concretamente, se diagnostican alrededor de 1.800 nuevos casos cada año en nuestro país.
Los pacientes con LLC activa se caracterizan por una acumulación progresiva de linfocitos B (el diagnóstico requiere la presenta de al menos 5000 linfocitos B en sangre periférica durante al menos tres meses), a veces con linfadenopatía, hepatoesplenomegalia, anemia y trombocitopenia. Asimismo, produce un estado de inmunosupresión que incrementa el riesgo de infecciones, que en última instancia es la principal causa de muerte en estos pacientes. El subtipo más frecuente de la LLC es la que afecta a células (linfocitos) B, que representa más del 97% de los casos. En restante el 2-3%, la proliferación clonal anormal se produce a partir de células T. También se han incluido otros patrones leucémicos crónicos dentro de la leucemia linfocítica crónica, tales como la leucemia prolinfocítica, la fase leucémica del linfoma cutáneo de células T (síndrome de Sézary), la leucemia de células peludas (tricoleucemia) y el linfoma leucemizado.
El origen de la LLC sigue siendo desconocido, aunque se apuntan varios hipotéticos, como las radiaciones ionizantes, los agentes alquilantes o ciertos productos leucemógenos, que parecen aumentar el riesgo de desarrollar LLC. La acumulación de linfocitos parece deberse un funcionamiento erróneo en la apoptosis (muerte celular programada); no obstante, se han descrito otros mecanismos que posiblemente colaboren de alguna manera en la acción proliferativa, como ciertas interleucinas o sus receptores, como el factor de necrosis tumoral (TNF) o las interleucinas IL-4 e IL-6. Aproximadamente la mitad de los pacientes, y aún más en estadios avanzados, presentan algún tipo de alteración citogenética. La más frecuente (25-30%) es la trisomía del par cromosómico 12; otras alteraciones menos frecuentes afectan a los cromosomas 11, 12, 13, 14 y 17 (Cuéllar, 2015).
Globalmente, la mediana de supervivencia desde el diagnóstico varía entre los 18 meses y más de 20 años, dependiendo de la presencia de factores de riesgo. En pacientes asintomáticos en estadios iniciales, la mediana de supervivencia es de más de 10 años, mientras que en pacientes con enfermedad avanzada, sintomática o progresiva, la mediana de supervivencia oscila entre 18 meses y 3 años. Debe tenerse en cuenta que los pacientes con LLC también son más propensos a desarrollar una segunda neoplasia. Se han descrito tres grupos pronósticos en función de la citogenética, siendo peor para los casos relacionados con una mutación TP 53, una translocación t(11q; v) o una deleción del(11q) o del(17p); particularmente, esta última confiere resistencia a la fludarabina y se considera como de muy alto riesgo, junto con la mutación TP 53. En ambos casos la mediana de supervivencia es de 2-3 años y aunque son relativamente infrecuentes (7% para del(11q) y 8-12% para TP 53) en el diagnóstico inicial, suponen prácticamente el 50% de los casos recidivantes de leucemia linfocítica crónica. La trisomía del par 12 (+12) se asocia con un pronóstico de gravedad intermedia, mientras que los casos con mejor pronóstico son aquellos cuya anomalía citogenética implica una deleción del(13q).
En ninguno de los casos la terapia es curativa y no está indicada en los pacientes con LLC hasta que aparezcan síntomas o progrese la enfermedad. El tratamiento específico incluye inmunoquimioterapia (fludarabina, ciclofosfamida y rituximab), corticoides, cirugía (trasplante alogénico de precursores hematopoyéticos) y radioterapia, aunque la cirugía o la radioterapia sólo son útiles en situaciones especiales. El último fármaco autorizado para esta indicación es el ibrutinib, que actúa inhibiendo de forma irreversible y selectiva a la tirosina cinasa de Bruton (BTK), un miembro de las familia de las tirosina cinasas Tec que participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados en la patogenia de diversas neoplasias de linfocitos B; dicha inhibición impide la adhesión y migración dependientes de integrinas de los linfocitos B. Ha sido autorizado, como medicamento huérfano, para el tratamiento de pacientes adultos con leucemia linfática crónica que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP 53 en pacientes en los que la inmuno-quimioterapia no se considera apropiada (Cuéllar, 2016).
En cualquier caso, se considera que la LLC es, por el momento, una enfermedad incurable; incluso, no se ha demostrado que ninguno de los tratamientos disponibles actualmente disponibles prolongue sustancialmente la supervivencia y teniendo en cuenta que el sobretratamiento es más peligroso que el infratratamiento.
ACCIÓN Y MECANISMO
El idelalisib es un agente antineoplásico que inhibe selectivamente la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias de linfocitos B, al estar implicada en numerosas vías bioquímicas de señalización que impulsan la proliferación, supervivencia, migración y retención de las células tumorales en los tejidos linfoides y en la médula ósea. El efecto final de todo el proceso es la inducción de la apoptosis y la inhibición de la proliferación de las células tumorales primarias y de las líneas celulares derivadas de los linfocitos B tumorales.
El medicamento ha sido autorizado para el tratamiento, en combinación con rituximab, de los pacientes adultos con leucemia linfocítica crónica que han recibido al menos un tratamiento anterior o como tratamiento de primera línea en presencia de la deleción 17p o de la mutación TP53 en pacientes no adecuados para quimioinmunoterapia. También está indicado en monoterapia de los pacientes adultos con linfoma folicular refractario a dos líneas de tratamiento anteriores.
La fosfatidilinositol 3-cinasa p110δ (PI3Kδ) forma parte de la clase I de fosfatidilinositol 3-cinasas (PI3K), una familia de cinasas de lípidos que están implicadas en numerosos procesos intracelulares de señalización que regulan varias funcionales celulares esenciales, entre ellas la supervivencia, la proliferación y la motilidad. Los enzimas de esta clase están formados por una subunidad reguladora y por otra subunidad catalítica, esta última con actividad cinasa, que reciben el nombre (en cada uno de los tipos de PI3K) de p110α, p110β, p110γ y p110δ; estas subunidades catalíticas definen las diferentes isoformas de PI3K: PI3Kα, PI3Kβ, PI3Kγ y PI3Kδ.
La PI3Kδ actúa fosforilando el fosfatidilinositol para producir fosfatidilinositol 3,4,5-trifosfato (PI3P), un segundo mensajero bioqímico lípidico con importantes funciones intracelulares. La actuación de la PI3Kδ es determinante para la señalización del receptor CD40 de linfocitos B (implicado en la supervivencia celular), del receptor BCR de linfocitos B (implicado en la supervivencia y proliferación celular, así como en la secreción de quimiocinas), de los receptores de quimiocinas CXCR4 y CXCR5 (cuya activación por los quimiocinas CXCL12 y CXCL13, respectivamente, induce la migración y retención de los linfocitos B tumorales en el microambiente tumoral) y de las integrinas (implicadas en la retención y la adhesión celular).
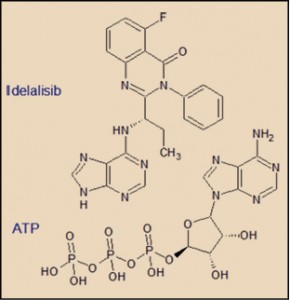
El efecto del idelalisib sobre la PI3Kδ se debe al efecto competitivo con el ATP, impidiendo a éste su unión con el dominio catalítico del enzima. Este efecto inhibitorio se traduce en el bloqueo del proceso de fosforilación del fosfatidilinositol y del enzima Akt, una proteína cinasa B.
ASPECTOS MOLECULARES
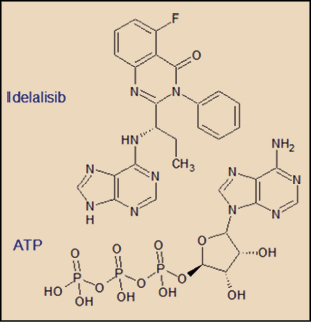
El idelalisib presenta una estructura química que recuerda abiertamente al núcleo purínico del ATP, lo que justifica su acción sobre la PI3Kδ, compitiendo con el ATP e impidiendo a éste su unión con el dominio catalítico del enzima.
Las particularidades estructurales del dominio catalítico de cada una de las isoformas de PI3K ha permitido el desarrollo de moléculas capaces de actuar de forma selectiva sobre determinadas isoformas, como es el caso del idelalisib sobre la PI3Kδ. En este sentido, en ensayos in vitro, el idelalisib es unas 450 veces más potente inhibiendo la PI3Kδ que la PI3Kα, 210 que la PI3Kβ y 110 que PI3Kγ (EMA, 2014).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del idelalisib en las dos indicaciones autorizadas han sido demostradas mediante sendos ensayos clínicos que, en leucemia linfocítica crónica fue de fase 3, multicéntrico, aleatorizado y controlado con placebo, mientras que en linfoma folicular fue de fase 2 y abierto.
En el caso de la leucemia linfocítica crónica, se incluyó a pacientes con diagnóstico clínico y citológico confirmado, con linfadenopatía patente (al menos una lesión nodular con al menos 2 cm en el diámetro mayor y al menos 1 cm en el menor), que hubieran recaído tras un tratamiento previo con anticuerpos ant-CD20 (rituxumab, ofatumumab) o dos tratamientos con antineoplásicos citotóxicos, con progresión de la enfermedad en los dos últimos años.
Todos los pacientes recibieron 8 infusiones IV rituximab (la primera de 375 mg/m2 y el resto de 375 mg/m2), distribuidas cada dos semanas las cuatro primeras y cada cuatro semanas las siguientes cuatro infusiones IV. El idelalisib se administró de forma continua por vía oral, en dosis de 150 mg/12 h de forma continua; en algunos pacientes la dosis fue reducida a 100 mg/12 h.
La variable primaria de eficacia fue la supervivencia sin progresión tumoral, definida como el intervalo entre el inicio del estudio y la primera identificación documentada de progresión definida o la muerte por cualquier motivo. Como variables secundarias de eficacia se emplearon la supervivencia global (intervalo entre el inicio del estudio y la muerte por cualquier causa), la tasa de respuesta global (porcentaje de pacientes que alcanzaron una respuesta completa, RC, o parcial, RP) y la tasa de respuesta de los ganglios linfáticos (porcentaje de pacientes que alcanzaron una reducción de al menos un 50% en la suma de los productos de los diámetros perpendiculares máximos en las lesiones linfáticas).
|
Tabla 1. Estudios clínicos con idelalisib |
||
|
Indicación |
Leucemia linfocítica crónica |
Linfoma no-Hodgkin de células B indolente |
|
Tipo de estudio |
Fase 3, doblemente ciego, controlado con placebo |
Fase 2, abierto |
|
Tratamientos comparados |
Idelalisib + Rituximab Placebo + Rituximab |
Idelalisib |
|
Pacientes aleatorizados |
220 |
125 |
|
Edad (mediana) |
71 años (22% <65 años) |
64 años (55% <65 años) |
|
Sexo (% varones) |
66% |
64% |
|
Raza (% blancos) |
90% |
89% |
|
IMC (mediana) |
25,4 kg/m2 |
25,9 kg/m2 |
|
Esplenomegalia (% pacientes) |
69% |
|
|
Hepatomegalia (% pacientes) |
53% |
|
|
Tipo histológico (% pacientes) |
– |
Folicular: 58% Linfocítico pequeño: 22% |
|
Tiempo desde el diagnóstico (mediana) |
102 meses |
|
|
Presencia de mutación IGHV* (% pacientes) |
16% |
|
|
Presencia de deleción 17p y/o TP53 (% pacientes) |
43% |
|
|
Número de tratamientos previos (mediana) |
3 |
4 |
|
Tratamiento previo con anticuerpos anti-CD20 (% pacientes) |
96% |
|
|
Tiempo desde el último tratamiento (mediana) |
– |
3,9 meses |
|
Supervivencia sin progresión tumoral (mediana) |
Idelalisib + Rituxumab: NA (IC95% 10,7 a NA) Placebo + Rituximab: 5,5 meses (IC95% 3,8 a 7,1) |
Global: 11,0 meses Folicular: 8,5 meses Linfocítico pequeño: 11,4 meses |
|
Supervivencia sin progresión tumoral (razón de riesgo) |
0,18 (IC95% 0,10 a 0,32) |
|
|
Supervivencia global (mediana) |
– |
Global: 20,3 meses Folicular: NA Linfocítico pequeño: 20,3 meses |
|
Supervivencia global (razón de riesgo) |
0,28 (IC95% 0,11 a 0,69) |
|
|
Supervivencia global a las 48 semanas (% pacientes) |
– |
Global: 82,0% Folicular: 88,8% Linfocítico pequeño: 70,0% |
|
Tasa de respuesta global (% pacientes) |
Idelalisib + Rituxumab: 74,5% Placebo + Rituximab: 14,5%) |
Global: 57,0% Folicular: 54,2% Linfocítico pequeño: 60,7% |
|
Tasa de respuesta global (% pacientes con deleción 17p y/o TP53) |
Idelalisib + Rituxumab: 78,3% Placebo + Rituximab: 12,2%) |
|
|
Tasa de respuesta global (% pacientes con IGHV no mutado) |
Idelalisib + Rituxumab: 73,6% Placebo + Rituximab: 15,1%) |
|
|
Tasa de respuesta global (% pacientes ≥65 años) |
Idelalisib + Rituxumab: 74,2% Placebo + Rituximab: 15,7%) |
|
|
Tasa de respuesta de los ganglios linfáticos (% pacientes) |
Idelalisib + Rituxumab: 92,2% Placebo + Rituximab: 5,9%) |
Global: 54,9% Folicular: ND Linfocítico pequeño: ND |
|
Tiempo hasta la respuesta (mediana, meses) |
– |
Global: 1,9 meses Folicular: ND Linfocítico pequeño: ND |
|
Duración de la respuesta (mediana, meses) |
– |
Global: 12,5 meses Folicular: 7,4 meses Linfocítico pequeño: ND |
|
Referencia |
312-0116 (Furman, 2014) |
101-09 (Gopal, 2014) |
Notas: NA: No alcanzada. ND: No disponible. Todas las diferencias en las variables descritas para cada brazo de tratamiento fueron estadísticamente significativas.
1 El gen IFHV@ codifica la cadena pesada de determinadas inmunoglobulinas. En la leucemia linfocítica crónica, las mutaciones IGHV se asocian con una mejor respuesta al tratamiento y a una mayor supervivencia.
En el caso del linfoma no-Hodgkin de células B indolente, se incluyó a pacientes con diagnóstico clínico y citológico confirmado con alguna de las variedades (folicular, linfocítico pequeño, linfoplasmocitoide o de zona marginal), con linfadenopatía patente (al menos una lesión nodular con al menos 2 cm), que hubieran recaído tras un tratamiento previo con rituxumab y un agente alquilante (bendamustina, ciclofosfamida, etc.) o dos tratamientos con quimio o inmunoterapia. Todos los pacientes recibieron idelalisib por vía oral, en dosis de 150 mg/12 h de forma continua; en algunos pacientes la dosis fue reducida a 100 o incluso a 75 mg/12 h.
La variable primaria de eficacia fue la tasa de respuesta global (porcentaje de pacientes que alcanzaron una respuesta completa, RC, o parcial, RP). Como variables secundarias de eficacia se emplearon la supervivencia sin progresión tumoral, definida como el intervalo entre el inicio del estudio y la primera identificación documentada de progresión definida o la muerte por cualquier motivo, la supervivencia global (intervalo entre el inicio del estudio y la muerte por cualquier causa), la tasa de respuesta nodular (porcentaje de pacientes que alcanzaron una reducción de al menos un 50% en la suma de los productos de los diámetros perpendiculares máximos en las lesiones linfáticas), la duración de la respuesta (intervalo entre la primera respuesta documentada y la aparición documentada de progresión tumoral o muerte por cualquier causa) y el tiempo hasta respuesta (intervalo desde el inicio hasta la primera respuesta documentada).
Desde el punto de vista de la seguridad, el idelalisib presenta un perfil toxicológico importante, aunque mayoritariamente controlable; los eventos adversos más frecuentemente comunicados fueron de tipo digestivo y hematológico. En monoterapia (linfoma folicular), los más comunes fueron diarrea (40%), fatiga (37%), náusea (28%), tos (25%), pirexia (25%) y neutropenia (24%); los más relevantes clínicamente (grado ≥3) fueron neutropenia (15%; febril en el 5,1%), aumento de los niveles de transaminasas (11%), neumonía (11%), diarrea (9,1%), anemia (7,1%) y trombocitopenia (6,3%). Un 23% de los pacientes suspendieron el tratamiento por eventos adversos.
En terapia combinada con rituximab (leucemia linfocítica crónica), los eventos adversos más frecuentes en el grupo tratado con idelalisib fueron náusea (26% con rituximab + idelalisib y 21% con rituximab + placebo), diarrea (19 vs. 15%), estreñimiento (13 vs. 11%), vómitos (13 vs. 8,3%), neutropenia (27 vs. 17%), pirexia (35 vs. 17%), escalofríos (21 vs. 16%), cefalea (10 vs. 5,0%) y erupciones exantemáticas (10 vs. 5,0%). Entre aquellos valorados como de grado ≥3, los más comunes fueron neutropenia 22 vs. 12%; febril en 4,5 vs. 3,7%), sepsis (3,6 vs. 2,8%), neumonitis (3,6 vs. 0,9%), fatiga (4,5 vs. 2,8%), pirexia (2,7 vs. 0,9%), aumento de los niveles de transaminasas (2,7 vs. 0,9%) y diarrea (3,6 vs. 0%). Un 15% suspendieron el tratamiento por eventos adversos.
ASPECTOS INNOVADORES
El idelalisib es un agente antineoplásico que inhibe selectivamente la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias de linfocitos B, al estar implicada en numerosas vías bioquímicas de señalización que impulsan la proliferación, supervivencia, migración y retención de las células tumorales en los tejidos linfoides y en la médula ósea. El efecto final de todo el proceso es la inducción de la apoptosis y la inhibición de la proliferación de las células tumorales primarias y de las líneas celulares derivadas de los linfocitos B tumorales. El medicamento ha sido autorizado para el tratamiento, en combinación con rituximab, de los pacientes adultos con leucemia linfocítica crónica que han recibido al menos un tratamiento anterior o como tratamiento de primera línea en presencia de la deleción 17p o de la mutación TP53 en pacientes no adecuados para quimioinmunoterapia. También está indicado en monoterapia de los pacientes adultos con linfoma folicular refractario a dos líneas de tratamiento anteriores.
Los datos clínicos disponibles justifican las dos indicaciones autorizadas. En el caso de los linfomas indolentes no-Hodgkin se trata de un estudio abierto de fase 2 sin brazo comparador, lo que limita la robustez de las conclusiones. Aun así, los datos son contundentes, aportando una tasa de respuesta global del 57% y, específicamente, un 54% para los pacientes con linfoma folicular, con una mediana de supervivencia sin progresión tumoral de 11 meses (8,5 para el linfoma folicular), una tasa de pacientes supervivientes al cabo de un año del 82% (89%) y una mediana de la duración de la respuesta de 12,5 meses (7,4).
En el caso de la leucemia linfocítica crónica, los datos clínicos disponibles son más rigurosos desde el punto de vista metodológico, ya que se trata de un estudio de fase 3 doblemente ciego y controlado con placebo, frente al que se ha demostrado (siempre en asociación a rituximab) una notable superioridad, tanto en términos de supervivencia sin progresión tumoral como de supervivencia global y tasa de respuesta objetiva (75 vs. 15%). Tal superioridad fue manifiesta incluso en los pacientes con mutaciones del17p y/o TP53, como con IGHV no mutado y en personas con ≥65 años.
En cualquier caso, en ambas indicaciones los resultados tienen una elevada relevancia clínica, dado que no dejan mucho lugar a dudas, incluyendo a pacientes de alto riesgo y de mal pronóstico; asimismo, es importante la eficacia mostrada independientemente del grado de refractoriedad a los tratamientos previos. En este sentido, en pacientes con mutaciones del17p y/o TP53 los resultados están en la línea de los conseguidos con fludarabina o alemtuzumab, aunque estos últimos son considerablemente más tóxicos y su uso no es admisible en muchos de los pacientes. Por otro lado, su perfil de toxicidad es relevante, aunque manejable, pero diferente del de los fármacos actualmente utilizados en estas indicaciones. Su perfil de intereacciones es, sin embargo, problemático toda vez que es un potente inhibidor del citocromo CYP3A4, una vía metabólica de numerosos fármacos.
Ciertamente, los datos clínicos de eficacia no son muy robustos en el caso del linfoma folicular y de duración relativamente corta en el de la leucemia linfocítica crónica (debido a que rápidamente se constató la superioridad sobre el placebo). En particular, sería muy interesante la comparación directa con ibrutinib, recientemente incorporado en España (Cuéllar, 2016), aunque la comparación indirecta parece sugerir que no existen diferencias sustanciales en cuanto a eficacia. En esa misma línea se ha expresado el Informe de Posicionamiento Terapéutico (AEMPS, 2016), que no encuentra diferencias clínicamente relevantes con sus alternativas en leucemia linfocítica crónica.
Es importante, por otro lado, hacer constar dos aspectos farmacológicos de interés, como son el hecho de que su administración es oral (como el ibrutinib) y, lo que puede ser mucho más relevante, su mecanismo de acción es completamente innovador, incorporando la diana farmacológica de la fosfatidilinositol 3-cinasa p110δ (PI3Kδ), cuya actividad enzimática está potenciada en las neoplasias de linfocitos B, al ámbito de los agentes antineoplásicos.
En definitiva, un nuevo medicamento que parece tener un papel relevante como tratamiento de segunda línea en la leucemia linfocítica crónica, especialmente para pacientes de altos riesgo y con comorbilidades múltiples (Nair, 2016) y un prometedor agente para los linfomas no-Hodgkin (Hewett, 2016).
|
VALORACIÓN |
|
IDELALISIB
 ZYDELIG® (Gilead) |
|
Grupo Terapéutico (ATC): L01XX. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Otros |
|
Indicaciones autorizadas: Tratamiento, en combinación con rituximab, de los pacientes adultos con leucemia linfocítica crónica (LLC) que han recibido al menos un tratamiento anterior o como tratamiento de primera línea en presencia de la deleción 17p o de la mutación TP53 en pacientes no adecuados para quimioinmunoterapia. También está indicado en monoterapia de los pacientes adultos con linfoma folicular (LF) refractario a dos líneas de tratamiento anteriores. |
|
INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |
|
Novedad clínica: Mejora la eficacia clínica con relación al tratamiento estándar. |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles |
BIBLIOGRAFÍA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}