Resumen
Secukinumab es un anticuerpo monoclonal humano de tipo IgG1/κ que se une, neutralizándola, a la interleucina 17A (IL-17A), una citocina proinflamatoria considerada como uno de los principales inductores de la hiperplasia epidérmica y modificadores de la diferenciación epidérmica observadas en la psoriasis, a través de la formación de factor nuclear κB (NFκB). El medicamento ha sido autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
PSORIASIS
La psoriasis es una enfermedad inflamatoria de la piel de carácter crónico, aunque fluctuante. Puede afectar a la piel, uñas, articulaciones (artritis psoriásica) y, menos frecuentemente, a las mucosas. La lesión característica es una placa de color rojo oscuro, con escamas no adherentes de un peculiar tono blanco-nacaradas y con borde bien delimitado. Se manifiesta habitualmente de forma bilateral, siendo las localizaciones más frecuentes las superficies de extensión articular (codos y rodillas), la zona sacra y el cuero cabelludo. La afectación de las mucosas es muy rara, aunque se han citado casos localizados en los labios y en el pene. Las uñas están afectadas en un 20-50% de los casos, especialmente las de las manos. Es aún más frecuente si hay afectación articular y en la forma eritrodérmica de psoriasis. Las lesiones más características son los hoyuelos o pits (depresiones puntiformes), manchas amarillentas debajo de la placa ungueal (mancha de aceite), fragilidad (onicolisis) e hipertrofia subungueal.
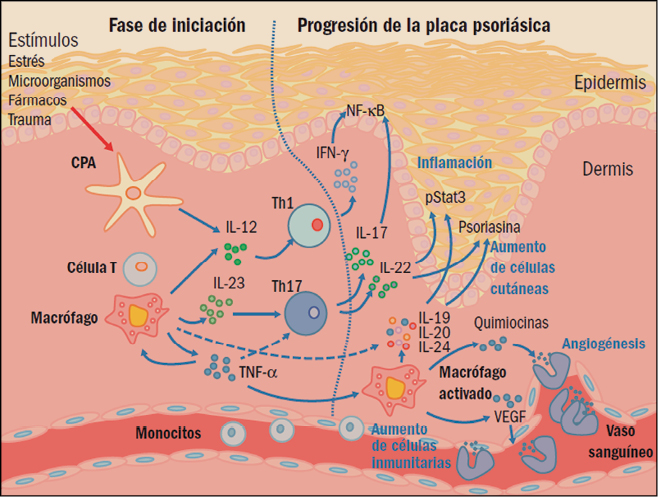
CPA: célula presentadora de antígenos; IFN-γ: interferón gamma; IL: interleucinas; NF-κβ : factor nuclear κß ; Th: linfocitos T helper (facilitadores); TNFα: factor de necrosis tumoral α; VEGF: factor de crecimiento endotelial vascular.
La psoriasis es la más común de las enfermedades cutáneas crónicas humanas, con una incidencia del 2% en la población mundial. La prevalencia en niños varía desde el 0% en Taiwán al 2,1% en Italia, mientras que en los adultos oscila entre el 0,9% de Estados Unidos y el 8,5% de Noruega, con una incidencia entre las 78,9/100.000 persona-año en Estados Unidos y las 230/100.000 persona-año en Italia. Los datos sugieren que la aparición de la psoriasis varía según la edad y la región geográfica, siendo en general más frecuente en los países más distantes desde el ecuador (Parisi, 2013). En España la prevalencia es del 1,4%. Puede comenzar a cualquier edad, pero es rara en menores de 5 años. Presenta dos picos de máxima incidencia: la segunda década (de origen generalmente familiar) y los 55-60 años. Evoluciona con remisiones y recaídas espontáneas. Puede persistir toda la vida o durar sólo unos meses. Aunque muy raramente llega a poner en peligro la vida del paciente, puede ser muy discapacitante, limitando considerablemente la calidad de vida.
Entre las principales características histológicas de la psoriasis pueden citarse la hiperplasia epidérmica, definida como una diferenciación anormal y la maduración incompleta de los queratinocitos; un engrosamiento de la epidermis y una capa granular reducida o ausente. Todo ello es debido a la hiperproliferación y diferenciación de queratinocitos epidérmicos de evolución acelerada, cuyo ciclo vital es mucho más rápido de lo normal: tardan 7-10 días en lugar de 50-75 días. También se puede apreciar una infiltración epidérmica de células del sistema inmune (linfocitos T) y de células dendríticas CD11c+ en la dermis; por otro lado, se encuentran células CD8+ y neutrófilos en la epidermis.
Además de estas presencias celulares anómalas, también se puede observar un aumento en el proceso de formación de nuevos vasos sanguíneos (angiogénesis) y la inflamación de la piel. En definitiva, aunque durante mucho tiempo se había pensado que la psoriasis es causada simplemente por la hiperproliferación de queratinocitos, actualmente se admite que el sistema inmune es un factor decisivo en el desarrollo de la enfermedad. En definitiva, hoy se considera que la psoriasis es una enfermedad inflamatoria crónica de origen autoinmune en la cual células dendríticas, linfocitos T, macrófagos y neutrófilos inducen hiperproliferaciones locales de queratinocitos que, en última instancia, son las responsables de las lesiones de la piel.
Las células presentadoras de antígenos (antigen presenting cells, APC) son un elemento clave del sistema inmunológico, implicado en la captación, procesamiento y presentación de moléculas de carácter antigénico sobre la membrana. Estas células permiten dar a conocer dichos antígenos al sistema inmune, especialmente por los linfocitos T, iniciando la cadena de respuestas inmunitarias antigénicas específicas. En concreto, la presentación del antígeno y la formación de la sinapsis inmunológica en las APC provoca la secreción de diversas citocinas e induce la diferenciación de los linfocitos T en células efectoras específicas (linfocitos T facilitadores o helper, Th), particularmente las Th1, Th2 y Th17, cada una de ellas secretando sus propias citocinas.
Se ha demostrado la participación directa de varias citocinas en el incremento de la proliferación de los queratinocitos en la psoriasis. Particularmente, el factor de necrosis tumoral alfa (tumor necrosis factor α, TNFα) activa el desarrollo de las lesiones mediante el aumento del número de moléculas que participan en la respuesta inflamatoria a las moléculas de adhesión. Los queratinocitos así activados producen citocinas y quimiocinas, que atraen a los linfocitos al sitio de la inflamación y que potencian su proliferación. De hecho, no es difícil encontrar subpoblaciones de linfocitos Th1 y Th17 en las lesiones psoriásicas de la piel, además de queratinocitos, células dendríticas y células de Langerhans y, como consecuencia de todo ello, un aumento de la concentración de TNFα en las zonas de la piel afectadas. Es más, se ha observado que una disminución del TNFα, tanto en suero como en las lesiones, se relaciona con una mejora clínica, lo que sugiere un importante rol en la enfermedad.
Asimismo, se ha observado que las interleucinas (IL) 12 e IL-23 pueden tener también un importante papel patológico en la psoriasis, ya que la IL-12 – producida por las células presentadoras de antígenos – es capaz de inducir la producción de nuevas poblaciones de linfocitos T e incrementar las respuestas de los linfocitos Th1, sobre todo en la producción de interferón (IFN). Estas células también parecen estimular la inmunidad mediada por células y la síntesis de anticuerpos fijadores del complemento. Por su parte, la IL-23 facilita la adquisición de memoria inmunológica por los linfocitos T, en especial de las subpoblaciones de linfocitos Th17, y parece tener un papel crítico en la patogénesis de la psoriasis. En este sentido, datos procedentes de modelos inflamatorios de la piel sugieren que los linfocitos Th17, que producen IL-17 e IL-22, podrían ser los inductores principales de la hiperplasia epidérmica, modificando la diferenciación epidérmica en la psoriasis (Cuéllar, 2014).
Se ha observado que las anomalías en la regulación de IL-12 e IL-23 no sólo se asocian a psoriasis, sino también a otras patologías de índole autoinmune, como la enfermedad de Crohn, la artritis reumatoide y la colitis ulcerosa, entre otras. De hecho, entre el 5% y el 7% de todos los pacientes con psoriasis y hasta un 40% de aquellos con la forma más grave (>10% de superficie corporal afectada) desarrollan artritis psoriásica, usualmente entre 5 y 10 años tras el inicio de la enfermedad cutánea. La afectación articular es más frecuente en los pacientes con 40-50 años, siendo la forma más común (50-70%) la oligoarticular asimétrica seronegativa, que afecta a las pequeñas articulaciones de algunos dedos de las manos.
También parece que las interacciones entre el antígeno asociado a la función leucocitaria de tipo 1 (LFA-1) y las moléculas de adhesión intercelular facilitan la patogenia de la psoriasis. En concreto, favorecen la migración de los linfocitos T desde la circulación sistémica a los tejidos de la dermis y de la epidermis, y su consiguiente reactivación. Todo ello conduce a una infiltración de las células T activadas en el tejido y a una proliferación anormal de los queratinocitos. Por su parte, la alta producción de factores de crecimiento endoteliales vasculares (vascular endothelial growth factors, VEGF) en los queratinocitos psoriásicos promueve la angiogénesis, lo que provoca un aumento de la vascularización y la inflamación local. Los neutrófilos se encuentran en grandes cantidades en las lesiones psoriásicas; de hecho, se ha demostrado que algunas citocinas, tales como la IL-8, causan la acumulación de neutrófilos en la piel.
Sin embargo, a pesar de todos los mecanismos bioquímicos mencionados, el origen específico de la enfermedad sigue siendo desconocido, toda vez que se ignora qué es lo que provoca la respuesta inmunológica y desencadena la hiperqueratosis y el resto de manifestaciones patológicas de la psoriasis. En la aparición de la psoriasis influyen significativamente algunos factores genéticos, como lo demuestra la marcada agregación familiar, así como la concordancia en gemelos y la asociación a determinados antígenos principales de histocompatibilidad (HLA), es más frecuente en presencia del alelo Cw6 y en pacientes con HLA B27 el inicio de la psoriasis es más precoz y la evolución más grave.
Entre los factores externos desencadenantes pueden citarse traumatismos externos, determinadas infecciones (la forma “en gotas” aparece poco después de una faringitis estreptocócica), el uso de determinados fármacos (litio, betabloqueantes, antipalúdicos, antiinflamatorios no esteroideos (AINE), supresión del tratamiento con esteroides, etc.), bebidas alcohólicas, factores psicógenos (especialmente el estrés, que puede actuar como desencadenante o agravante), el clima (el clima cálido y la luz solar son beneficiosos, el frío empeora las lesiones), factores metabólicos (hipocalcemia, alcoholismo, diálisis, etc.) y factores endocrinos (mayor incidencia en la pubertad y la menopausia, mejora en el embarazo).
La forma clínica más frecuente (hasta el 80% de los casos) es la psoriasis vulgar o en placas. Este último término hace referencia a las formaciones escamosas y de color rojizo presentes en las zonas de extensión (codos y rodillas, principalmente), así como en el cuero cabelludo. Se trata de placas bien delimitadas con una distribución simétrica en la mayoría de los casos, aunque pueden confluir y formar figuras policíclicas. El porcentaje del cuerpo afectado por las placas psoriásicas puede variar desde una forma leve (<2%) hasta las formas más graves (>10%), pasando por la forma moderada (2-10%). La enfermedad es considerada como crónica, aunque su curso puede ser muy variable, con recaídas y remisiones de duración diversa. Suele manifestarse por vez primera en dos grupos de edad: 16-22 y 57-60 años.
La denominada psoriasis en gotas suele cursar con numerosas lesiones puntiformes (menores de 1 cm), sobre todo en el tronco. Es más común en niños y adolescentes, siendo típica su erupción aguda 10-14 días tras una infección estreptocócica, habitualmente de garganta, y que desaparece espontáneamente en 2-3 meses. Por su parte, la psoriasis invertida afecta a grandes pliegues (axilar, ingles, submamario, interglúteo). Presenta placas rojas lisas y brillantes, de color vivo, sin descamación y ocasionalmente con fi suras. La psoriasis pustulosa es una forma aguda y poco frecuente. Puede ser generalizada (tipo von Zumbusch), como la forma de comienzo de una psoriasis, o aparecer en el curso de una psoriasis crónica. Cursa con una brusca fiebre elevada, malestar general, eritema con pústulas en pocas horas, piel de color rojo escarlata seca y no descamativa. Sin tratamiento puede ser mortal. La forma localizada palmoplantar cursa con brotes repetidos de pústulas estériles sobre una base eritematosa en las palmas y las plantas, simétricas. Suelen secarse, dejando escamas y costras marrones. Finalmente, la psoriasis eritrodérmica consiste en una forma generalizada y grave. Se instaura generalmente sobre cuadros de psoriasis crónica. Se presenta como una eritrodermia exfoliativa seca, que afecta todo el tegumento, incluyendo el pelo y sobre todo las uñas.
La psoriasis se asocia con un aumento del riesgo de la aterosclerosis y del riesgo de enfermedad cardiovascular, que se asocia con mayores tasas de morbilidad y mortalidad, especialmente en los pacientes de psoriasis más jóvenes y con formas más graves de la enfermedad, reduciendo su esperanza de vida (Pietrzak, 2013). Los datos epidemiológicos también parecen apoyar una asociación de la psoriasis y de la artritis psoriásica con una mayor prevalencia de hipertensión (Armstrong, 2013a). Además de las complicaciones vasculares, la psoriasis ha sido relacionada con un incremento de la incidencia de algunas metabolopatías de alta incidencia, especialmente diabetes mellitus de tipo 2 y síndrome metabólico (Armstrong, 2013b).
El tratamiento de la psoriasis es complejo, ya que no sólo se lucha contra una enfermedad de etiología desconocida y con formas clínicas muy diversas, sino que está condicionada por diversos factores sociales. En principio, deben evitarse los factores desencadenantes y favorecedores conocidos: infecciones, golpes, tabaquismo y estrés. El sol es beneficioso, siendo capaz de producir una mejoría significativa de las lesiones; sin embargo, no existen evidencias sobre la posible eficacia de otros tratamientos no farmacológicos.
No existe un tratamiento curativo para la psoriasis, pero en la mayoría de los casos puede controlarse satisfactoriamente, aplicando diferentes tratamientos en función de la gravedad del caso. No obstante, la calificación de los resultados del tratamiento depende en buena medida de la aceptación de los pacientes, de sus criterios estéticos y de su propia personalidad. De hecho, se ha comprobado que el estrés del paciente tiende a agravar y a hacer más frecuentes las recaídas. Por otro lado, las lesiones ungueales asociadas con la psoriasis son difíciles de tratar.
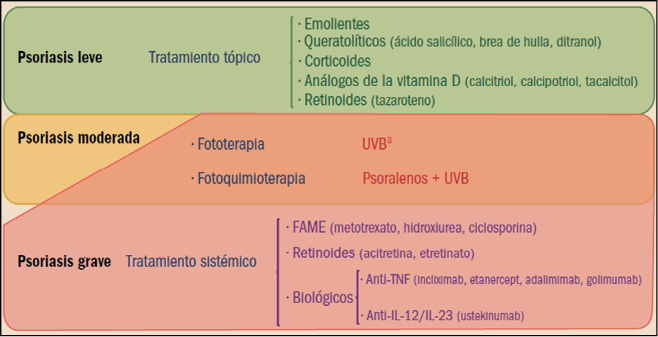
Algoritmo general de tratamiento en la psoriasis. FAME: fármacos antirreumáticos modificadores de la enfermedad; IL: interleucina; TNF: factor de necrosis tumoral; UVB: ultravioleta B. a puede sensibilizarse previamente con brea de hulla.
Los tratamientos tópicos son empleados en los casos más leves (afectación menor del 25% de la superficie corporal) y constituyen la forma más común de tratamiento de la psoriasis en placas, pero también es la menos eficaz en los casos graves. Carecen de utilidad en la artritis psoriásica o en las formas pustulosa o eritrodérmica.
Los agentes emolientes y queratolíticos son utilizados habitualmente como adyuvantes a otros tratamientos para hidratar, evitar la aparición de fisuras y eliminar las escamas. No deben aplicarse en pliegues. Entre los agentes queratolíticos, el ácido salicílico es el menos eficaz de todos los tratamientos disponibles, pero también el más barato y el mejor aceptado por los pacientes, por lo que constituye un paso indispensable en la terapéutica de la psoriasis en placas. La brea de hulla (coal tar) es algo más potente como queratolítico que el anterior. Presenta el inconveniente del olor desagradable. Sus efectos son lentos y de baja potencia, aunque produce remisiones generalmente prolongadas en los pacientes sensibles al tratamiento. El ditranol (antralina) es uno de los componentes activos de la brea de hulla. Debido a su poder irritante para la piel y a su capacidad para manchar la ropa y teñir las uñas y la piel, muchos pacientes tienden a rechazar este tratamiento. Sin embargo, se trata de uno de los tratamientos tópicos más eficaces (más que los anteriores). Sus efectos aparecen lentamente, aunque no tanto como los de la brea de hulla y producen remisiones algo más cortas que ésta.
Los corticosteroides tópicos producen efectos rápidos y potentes, pero la duración de las remisiones es más bien corta. Se pueden considerar de primera elección en la psoriasis leve que no responde a otros tratamientos tópicos y en determinadas localizaciones como la cara, el cuero cabelludo, los pliegues, los genitales (localizaciones que no toleran otros tratamientos tópicos). Presentan el inconveniente de que tras la suspensión del tratamiento la enfermedad puede reactivarse. No es infrecuente la combinación de corticosteroides tópicos con agentes queratolíticos, de efectos menos potentes y rápidos, pero considerablemente más prolongados.
El calcipotriol y el tacalcitol son análogos hormonales de la vitamina D de aplicación tópica, similares al calcitriol. Su empleo en la psoriasis en placas se debe a la observación de que los análogos hormonales de la vitamina D son capaces de inhibir la proliferación y la diferenciación de los queratinocitos. Su eficacia es similar a la de los corticosteroides e incluso inducen periodos de remisión algo más largos que aquéllos.
El empleo de lámparas de radiación ultravioleta (UV) constituye uno de los puntales en el tratamiento de la psoriasis. Sin embargo, la aplicación de radiación UV sólo resulta útil en los casos de psoriasis en placas, resultando ineficaz en el resto de formas de psoriasis (artritis, etc.). Según la longitud de onda de la radiación se distinguen dos tipos básicos de radiación. La de longitud de onda más larga (UVA) tiene una menor capacidad de penetración en la piel; por este motivo, requiere la administración previa de sustancias que sensibilicen la piel (generalmente psoralenos), en lo que se conoce como PUVA (psoralenos + UVA). Esta forma de tratamiento es conocida como fotoquimioterapia. Por su parte la radiación de longitud de onda más corta (UVB) tiene una mayor capacidad de penetración y no requiere ninguna sustancia sensibilizante, aunque se suele emplear brea de hulla previamente); este método es conocido como fototerapia.
El método PUVA o fotoquimioterapia es el tratamiento más eficaz disponible para la psoriasis en placas. Su acción es lenta, pero produce periodos prolongados de remisión. Debido al riesgo de efectos adversos cutáneos se está empleando de forma mucho más restringida, para casos graves refractarios en pacientes de edad media (no en niños ni en jóvenes). Algo menos eficaz es la fototerapia.
En pacientes con psoriasis en el cuerpo y del cuero cabelludo, el tratamiento combinado con vitamina D y corticosteroides funciona mucho mejor que cualquiera de estos solos. Los análogos de la vitamina D producen por lo general mejores resultados que el alquitrán de hulla, pero los resultados en relación con el ditranol son dispares. Los corticosteroides son, como mínimo, igual de eficaces que los análogos de la vitamina D, pero se asocian con una menor incidencia de efectos adversos locales. El tazaroteno es un retinoide que se utiliza por vía tópica. En los pacientes con psoriasis en placas presenta un eficacia similar a la de los corticosteroides tópicos en lo que se refiere a la elevación de las placas psoriásicas, pero su efecto es algo menor en cuanto a la reducción del eritema. La combinación de tazaroteno y corticosteroides produce mejores resultados que el tazaroteno solo.
En el tratamiento sistémico se emplean agentes con efectos antiproliferativos sobre la epidermis. Se trata de fármacos inmunosupresores y derivados retinoides aromáticos. Son considerados como el segundo nivel de tratamiento, estando indicados en psoriasis extensas que no responden a otros tratamientos, formas eritrodérmicas y pustulosas y formas incapacitantes.
Los denominados fármacos antirreumáticos modificadores de la enfermedad (FAME o DMARD, disease modifiying antirheumatic drugs) son ampliamente utilizados como primera opción en el tratamiento de las formas activas moderadas o graves de la psoriasis, en particular en los pacientes con artritis psoriásica. Se trata de potentes inmunosupresores, entre los cuales el más utilizado es, sin duda, el metotrexato, considerado como el tratamiento de elección en las formas graves de psoriasis en placas, así como en la artritis psoriásica, la psoriasis pustulosa y la psoriasis eritrodérmica. Tiene efecto antiproliferativo y antiinflamatorio. Por su parte, la ciclosporina es un inmunosupresor que actúa inhibiendo especialmente la producción de anticuerpos dependientes de células T colaboradoras, aunque también inhibe la producción y liberación de linfocinas, sobre todo de IL-2. La ciclosporina tiene una eficacia clínica similar a la del metotrexato en la psoriasis en placas y en la psoriasis pustulosa, pero algo menor en la psoriasis eritrodérmica y en la artritis psoriásica.
En general, el metotrexato, la ciclosporina, los UVB y los PUVA son consideradas como las formas más eficaces de tratamiento de los casos graves o moderadamente graves de psoriasis, facilitando la desaparición prácticamente completa de las manifestaciones clínicas en gran parte de los pacientes. Una vez alcanzado este objetivo, el tratamiento puede ser reducido o incluso suspendido, al menos hasta que se produzca una recidiva (si es que llega a producirse).
Los retinoides son análogos estructurales de la vitamina A (ácido retinoico), pero de carácter aromático. Actúan sobre receptores específicos, reduciendo la producción de estímulos inflamatorios y la diferenciación y proliferación de los queratinocitos. Revierten los cambios típicos hiperqueratósicos de la psoriasis en placas, pero son potentes teratógenos, por lo que su uso debe ser estrictamente vigilado en mujeres. Actualmente solo está disponible la acitretina. Actúan sobre diversas actividades biológicas en la piel, y entre ellas sobre la proliferación y diferenciación celular, la función inmunológica, la inflamación y la producción de sebo. Los efectos de los retinoides son debidos a la activación de receptores específicos del ácido retinoico, conocidos como RAR (retinoic acid receptors).
Como ya se ha indicado, el TNFα es una citocina extremadamente proinflamatoria y muy relevante en el desarrollo de la inflamación en la psoriasis. De hecho, el TNFα estimula la producción de citocinas y la adhesión de moléculas por los queratinocitos y, por lo tanto, aumenta el reclutamiento de células inmunes. La terapia anti-TNFα fue desarrollada precisamente para bloquear el TNFα e impedir o limitar su actividad y, en consecuencia, reducir las interacciones entre las células inmunes y los queratinocitos.
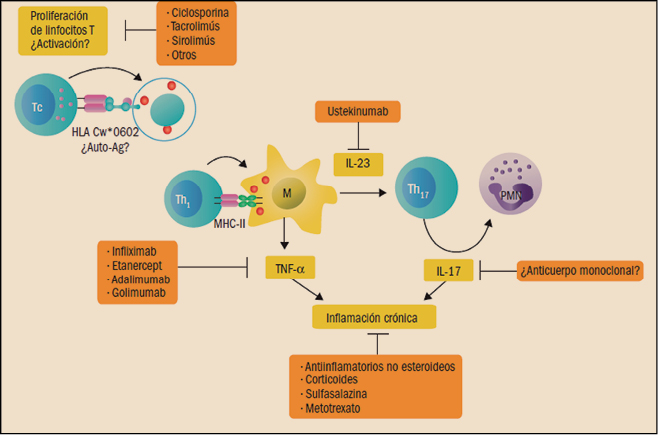
Dianas de los medicamentos antipsoriásicos actuales.
Auto-Ag: autoantígeno; IL: interleucina; M: macrófago; MHC: complejo principal de histocompatibilidad; PMN: polimorfonuclear; TNF
α: factor de necrosis tumoral (Cuéllar, 2014).
La neutralización del TNFα impide su interacción con sus receptores (TNFR1) y, con ello, la subsiguiente cascada bioquímica que, entre otras, desemboca en la activación del factor nuclear kappa B (NF-κB; factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas), un complejo proteico que se encuentra en la mayoría de los tipos de células animales y está implicado en la respuesta celular frente a estímulos como el estrés, las citocinas, la radiación UV y antígenos diversos. El NFκB juega un papel clave en la regulación de la respuesta inmune, dado que las cadenas ligeras kappa son componentes básicos de las inmunoglobulinas. Asimismo, otra de las consecuencias del bloqueo del TNFα es el cambio en los niveles de las moléculas de adhesión responsables de la migración leucocitaria (ELAM-1, VCAM-1 e ICAM-1). Igualmente, se puede apreciar una disminución de los niveles de metaloproteinasas matriciales (MMP-1 y MMP-3), responsables de la remodelación tisular.
Los fármacos anti-TNFα actualmente comercializados en España que están indicados expresamente en la psoriasis son infliximab, etanercept y adalimumab. Otros agentes anti-TNF disponibles en nuestro país son el golimumab, que está indicado en la artritis psoriásica, la artritis reumatoide, la colitis ulcerosa y la espondilitis anquilosante, y el certolizumab pegol, que está indicado para la artritis psoriásica, la artritis reumatoide, la espondilitis anquilosante y la espondiloartritis.
ACCIÓN Y MECANISMO
Secukinumab es un anticuerpo monoclonal humano de tipo IgG1/κ que se une, neutralizándola, a la interleucina 17A (IL-17A), una citocina proinflamatoria considerada como uno de los principales inductores de la hiperplasia epidérmica y modificadores de la diferenciación epidérmica observadas en la psoriasis, a través de la formación de factor nuclear κβ (NFκβ). El medicamento ha sido autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
La interleucina 17A es una citocina proinflamatoria soluble que forma parte de la familia de las IL-17, que juega un importante papel en la patogenia de diversas enfermedades autoinmunes. Es producida por los linfocitos Th17, los cuales forman parte de las respuesta inmune adaptativa; también es producida por linfocitos T CD8+ y γδ, así como por algunas subpoblaciones de linfocitos T citotóxicos (Natural Killers, NK). En algunas condiciones patológicas, varios tipos de células del sistema inmunológico humano – macrófagos, astrocitos, mastocitos y neutrófilos – también pueden producir IL-17A. Las interleucinas IL-12 e IL-23 son producidas fundamentalmente por macrófagos; la IL-12 induce la liberación de interferón gamma (IFN γ) por poblaciones de linfocitos Th1, mientras que la IL-23 actúa sobre poblaciones de linfocitos Th17, que producen IL-17A y IL-22; la IL-17A y el IFN γ inducen la formación de factor nuclear κB (NFκB), implicado directamente en los procesos hiperproliferativos de la epidermis en la progresión de la placa psoriásica.
ASPECTOS MOLECULARES
Secukinumab es un anticuerpo monoclonal humano de tipo IgG1/κ, con una estructura característica en forma de Y, formada por dos cadenas peptídicas ligeras y otras dos pesadas; estas últimas se encuentran glucosiladas, con restos glucídicos de tipo oligosacárido ligados a la posición Asn307 constituidos por estructuras complejas sin galactosa o con uno o dos residuos de ésta. La secuencia de las cadenas ligeras está formada por 215 aminoácidos, mientras que las pesadas contienen 457, totalizando un peso molecular aproximado 148 kDa (sin glucosilación).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del secukinumab han sido adecuadamente contrastadas en las indicaciones autorizadas mediante seis ensayos clínicos principales de fase 3 (confirmatorios de eficacia y seguridad), aleatorizados, multicéntricos, doblemente ciegos y controlados con placebo y, en algunos casos, con tratamientos activos (etanarcept).
Las variables primarias de eficacia utilizadas en estos estudios fueron el porcentaje de pacientes que alcanzan el PASI 75 (mejoría de al menos un 75% en la puntuación del Índice de Área y Gravedad de la Psoriasis, Psoriasis Area and Severity Index, PASI)1. Asimismo, se utilizó como variable coprimaria la tasa de pacientes respondedores según la IGA (Investigator’s Global Assessment)2. Se valoraron secundariamente los porcentajes de pacientes con PASI-50, PASI-90 y PASI-100, y la tasa de mantenimiento de PASI75 e IGA 0/1 en los periodos de extensión.
Los estudios tuvieron una duración que osciló entre 12 y 52 semanas, totalizando más de 3000 pacientes adultos aleatorizados, con psoriasis crónica (≥6 meses) en placas de moderada a grave (PASI≥12 e IGA≥3), e inadecuadamente controlados con terapia tópica, fototerapia o terapia sistémica previa. Se utilizaron, en general, dosis semanales de secukinumab de 150 o 300 mg durante el primer mes (4 semanas) y posteriormente cada cuatro semanas, por vía subcutánea.
En un análisis combinado (EMA, 2014; EPAR) de los datos de los cuatro primeros ensayos clínicos (ERASURE, FEATURE, FIXTURE y JUNCTURE), las variables primarias y secundarias mostraron tasas de respuestas PASI 75 a las 12 semanas fueron del 69,2% con secukinumab 150 mg, 79,4% con secukinumab 300 mg, del 44,0% con etanercept y 4,2% con placebo; por lo que respecta a la IFA 0/1 fueron 51,4% con secukinumab 150 mg, 65,0% con secukinumab 300 mg, 27,2% con etanercept y 2,2% con placebo; finalmente, la PASI 90 se alcanzó en 41,1% con secukinumab 150 mg, 56,6% con secukinumab 300 mg, 20,7% con etanercept y del 1,2% con placebo. Todas las diferencias entre las dos dosis de secukinumab, con etanercept y con placebo fueron estadísticamente significativas (p<0,05).
Otros estudios (SCULPTURE y STATURE) analizaron el efecto de utilizar una pauta a demanda vs. una pauta fija (SCULPTURE) o el empleo inicial (cuatro primeras semanas) de la administración intravenosa cada dos semanas, en lugar de la subcutánea cada cuatro (STATURE). Estos dos últimos estudios están resumidos en la tabla 2.
Desde el punto de vista de la seguridad, el secukinumab presenta un perfil toxicológico benigno, con eventos adversos asociados generalmente leves o moderados, transitorios y tratables fácilmente. Los más frecuentes son infecciones del tracto respiratorio superior (18,6% con la dosis de 150 mg; 17,0% con la de 300 mg y 10,4% con placebo), diarrea (2,6/4,1/1,4%), candidiasis (3,6/1,9/1,0%), urticaria (1,2/0,6/0,1%), rinorrea (0,3/1,2/0,1%) y herpes oral (0,1/1,3/0,3%), sin que se encontrasen diferencias significativas entre ambas dosis de secukinumab; en el estudio comparativo con etanercept, ambas dosis de secukinumab fueron mejor toleradas que aquél. La tasa de eventos adversos graves a lo largo de periodos de un año de tratamiento fue del 6,8% (150 mg), 7,4% (300 mg), 7,5% (placebo) y 7,0% (etanercept), en tanto que la tasa de descontinuación del tratamiento motivada por eventos adversos fue del 3,1/3,3/1,4/3,7%.
Debido al potencial incremento del riesgo de infecciones, no se aconseja su uso en pacientes con infecciones activas graves, como la tuberculosis; sin embargo, en los estudios clínicos se utilizó el secukinumab en pacientes con tuberculosis tratados correctamente, en los que no se apreció ninguna reactivación de la infección o pérdida de la eficacia antituberculosa. Por el contrario, sí se han observado en algunos pacientes con enfermedad de Crohn una reactivación o exacerbación de la misma, eventualmente grave (EMA; EPAR, 2014).
ASPECTOS INNOVADORES
Secukinumab es un anticuerpo monoclonal humano de tipo IgG1/κ que se une, neutralizándola, a la interleucina 17A (IL-17A), una citocina proinflamatoria considerada como uno de los principales inductores de la hiperplasia epidérmica y modificadores de la diferenciación epidérmica observadas en la psoriasis, a través de la formación de factor nuclear κβ (NFκβ). El medicamento ha sido autorizado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
|
Tabla 1. Estudios clínicos de fase 3 con secukinumab |
||||
|
Brazos de estudio |
Secukinumab 150 mg Sekukinumab 300 mg Placebo (a) |
Secukinumab 150 mg Sekukinumab 300 mg Placebo |
Secukinumab 150 mg Sekukinumab 300 mg Etanercept 50 mg (b) Placebo (a) |
Secukinumab 150 mg Sekukinumab 300 mg Placebo (a) |
|
Pacientes aleatorizados |
738 |
177 |
1306 |
1306 |
|
Duración estudio |
52 semanas |
12 semanas |
52 semanas |
12 semanas |
|
Edad (media) |
45 años (92 % <65) |
– |
44 años (94 % <65) |
– |
|
Sexo (varones) |
69 % |
66 % |
71% |
– |
|
Raza (blancos) |
70 % |
92 % |
67% |
– |
|
Peso (media) |
89 kg |
92 kg |
83 kg |
– |
|
Índice de Masa Corporal (IMC; kg/m2) |
30,1 |
30,7 |
28,3 |
– |
|
Fumadores activos |
37 % |
29 % |
35 % |
– |
|
PASI basal (media) |
22,1 (45 %>20) |
20,8 |
23,7 |
– |
|
Área corporal afectada (media) |
32 % |
32 % |
34 % |
– |
|
Tratamiento sistémico |
49 % (77 % fracasos) |
50 % (75 % fracasos) |
61 % (84 % fracasos) |
– |
|
Tratamiento biológico previo (% pac.; % fracasos) |
29 % (33 % fracasos) |
44 % (53 % fracasos) |
12 % (36 % fracasos) |
– |
|
PASI 75 (12 semanas; % pac.) |
Secukinumab 150: 71,6 % Secukinumab 300: 81,6 % Placebo: 4,5 % |
Secukinumab 150: 69,5 % Secukinumab 300: 75,9 % Placebo: 0,0 % |
Secukinumab 150: 67,0 % Secukinumab 300: 77,1 % Etanercept 50: 44,0 % Placebo: 4,9 % |
Secukinumab 150: 71,7 % Secukinumab 300: 86,7 % Placebo: 3,3 % |
|
IGA 0/1 (12 semanas; % pac) |
Secukinumab 150: 51,2 % Secukinumab 300: 65,3 % Placebo: 2,4 % |
Secukinumab 150: 52,5 % Secukinumab 300: 69,0 % Placebo: 0,0 % |
Secukinumab 150: 51,1 % Secukinumab 300: 62,5 % Etanercept 50: 27,2 % Placebo: 2,8 % |
Secukinumab 150: 53,3 % Secukinumab 300: 73,3% Placebo: 0,0 % |
|
PASI 90 (12 semanas; % pac) |
Secukinumab 150: 39,1 % Secukinumab 300: 59,2 % Placebo: 1,2 % |
|
Secukinumab 150: 41,9 % Secukinumab 300: 54,2 % Placebo: 2,8 % |
|
|
Mantenimiento de PASI 75 (52 semanas; % pac) |
Secukinumab 150: 72,4 % Secukinumab 300: 80,5 % |
|
Secukinumab 150: 82,2 % Secukinumab 300: 84,3% Etanercept 50: 72,5 % |
|
|
Mantenimiento de IGA 0/1 (52 semanas; % pac) |
Secukinumab 150: 59,2 % Secukinumab 300: 74,4% |
|
Secukinumab 150: 67,7 % Secukinumab 300: 79,7% Etanercept 50: 56,8 % |
|
|
Referencia |
Langley, 2014 (ERASURE) |
Blauvelt, 2015 (FEATURE) |
Langley, 2014 (FIXTURE) |
Paul, 2015 (JUNCTURE) |
Nota: Todas las diferencias entre los datos correspondientes al secukinumab y los del comparador (placebo y etanercept) fueron estadísticamente significativas (p<0,05). (a) Placebo: durante las 12 primeras semanas; durante el resto del estudio los pacientes inicialmente tratados con placebo que no fueron respondedores para el PASI75 fueron asignados a secukinumab 150 o 300 mg; los respondedores PASI75 iniciales a placebo lo mantuvieron. (b) Etanercept (SC): 50 mg dos veces por semana, hasta la semana 12; luego, 50 mg/semana hasta la semana 52.
La superioridad clínica del secukinumab ha sido demostrada frente a placebo y etanercept, mediante estudios amplios y metodológicamente robustos, tanto en pacientes naïve como tratados previamente con terapias sistémicas (biológicas o no). En este sentido, el índice PASI 75 (mejoría de al menos un 75% en la puntuación del Índice de Área y Gravedad de la Psoriasis, Psoriasis Area and Severity Index, PASI) se alcanzó en 69% con la dosis de 150 mg y en el 79% con la de 300 mg, frente a tan solo un 4,2% con placebo y un 44% con etanercept; igualmente, las tasas de pacientes con IGA 0/1 (puntuación 0 o 1 en la escala Investigator’s Global Assessment) fueron del 51%, 65%, 2,2% y 27%. Las diferencias entre las dos dosis de secukinumab y entre éstas y etanercept y placebo fueron estadísticamente significativas, y clínicamente muy relevantes. El comienzo de la acción con secukinumab es relativamente rápido, alcanzándose una reducción del 50% en la puntuación PASI en 3,0 (300 mg) y 3,7 semanas (150 mg), en término medio. Por otro lado, el efecto clínico se mantuvo durante al menos un año (EMA; EPAR, 2014).
|
Tabla 2. Otros estudios clínicos de fase 3 con secukinumab |
||
|
Brazos de estudio |
Secukinumab 150 mg Sekukinumab 300 mg (a) |
Secukinumab IV mg Sekukinumab SC (b) |
|
Pacientes aleatorizados |
843 |
43 |
|
Duración estudio |
52 semanas |
52 semanas |
|
Edad (media) |
46 años (93 % <65) |
47 años (88 % <65) |
|
Sexo (varones) |
66 % |
67 % |
|
Raza (blancos) |
72 % |
74 % |
|
Peso (media) |
89 kg |
97 kg |
|
Índice de Masa Corporal (IMC; kg/m2) |
29,0 |
– |
|
Fumadores activos |
34 % |
– |
|
PASI basal (media) |
23,6 (54 %>20) |
21,8 (39 %>20) |
|
Área corporal afectada (media) |
35 % |
32 % |
|
Tratamiento sistémico no biológico previo y tasa de fracasos (% pac; % fracaso) |
55 % (80 % fracasos) |
25 % |
|
Tratamiento biológico previo (% pac.; % fracasos) |
26 % (49 % fracasos) |
37 % |
|
PASI 75 (12 semanas; % pac.) |
Secukinumab 150: 84,4 % Secukinumab 300: 91,1 % |
|
|
Mantenimiento de PASI 75 (52 semanas; % pac) |
Secukinumab 150: 62,1 % Secukinumab 300: 78,2% |
|
|
PASI 75 (8 semanas; % pac) |
|
Secukinumab IV*: 90,5 % Secukinumab SC*: 66,7 % |
|
IGA 0/1 (8 semanas; % pac) |
|
Secukinumab IV: 66,7 % Secukinumab SC: 33,3 % |
|
Referencia |
Mrowitz, 2015 (SCULPTURE) |
Traçi, 2015 (STATURE) |
Nota: Todas las diferencias entre los grupos de tratamiento fueron estadísticamente significativas (p<0,05), salvo aquellas notadas con un asterisco (*). (a) A la 12ª semana, los pacientes que alcanzaron un PASI75 fueron realeatorizados para recibir los dos niveles de dosis según fuera necesario a criterio clínico, vs. intervalo fijo. (b) Después de 12 semanas de tratamiento con secukinumab 150 o 300 mg SC, se asignó a los pacientes que habían obtenido una respuesta parcial a este tratamiento a recibir una dosis intravenosa cada dos semanas (0, 2 y 4) de 10 mg/kg durante un mes (4 semanas) o 300 mg SC cada cuatro semanas (0 y 4); tras ello, todos los pacientes recibieron secukinumab SC 300 mg cada cuatro semanas hasta la semana 40.
Desde el punto de vista de la seguridad, el secukinumab presenta un perfil toxicológico benigno, con eventos adversos generalmente leves o moderados, transitorios y tratables fácilmente. Los más frecuentes son infecciones del tracto respiratorio superior, diarrea, candidiasis, urticaria, rinorrea y herpes oral, sin que se encontrasen diferencias significativas entre ambas dosis de secukinumab; en el estudio comparativo con etanercept, ambas dosis de secukinumab fueron mejor toleradas que aquél.
Naturalmente, se echa de menos la comparación clínica directa con el ustekinumab, un anticuerpo monoclonal activo frente a IL-12 e IL-23, con un mecanismo complementario al del secukinumab en el control de la patogénesis de la psoriasis. En este sentido, cabe recordar que las interleucinas IL-12 e IL-23 son producidas fundamentalmente por macrófagos; la IL-12 induce la liberación de interferón gamma (IFN γ) por poblaciones de linfocitos Th1, mientras que la IL-23 actúa sobre poblaciones de linfocitos Th17, que producen IL-17A y IL-22; la IL-17A y el IFN γ inducen la formación de factor nuclear κB (NFκB), implicado directamente en los procesos hiperproliferativos de la epidermis en la progresión de la placa psoriásica.
El ustekinumab produce tasas de PASI 75 del orden del 65-75% en pacientes con psoriasis (Cuéllar, 2009), en líneas con el 69-79% del secukinumab, con tasas del 3-4% de respuesta placebo en ambos casos. Obviamente, esto adolece de todas las limitaciones que toda comparación indirecta implica (diversidad de criterios de inclusión y exclusión de los pacientes, nivel de gravedad, utilización de tratamiento previos, etc.), por lo que estos datos debe ser siempre considerados con la natural reserva. En cualquier caso, permiten ilustrar la eficacia muy relevante en relación al placebo, en ambos casos, y no parece que existan diferencias clínicas notables entre uno y otro anticuerpo; ni siquiera tampoco en sus perfiles de seguridad, relativamente benignos. Igualmente, las pautas de administración (subcutánea) son bastante cómodas (secukinumab: dosis semanales de secukinumab durante el primer mes y posteriormente cada 4 semanas; ustekinumab: cada 12 semanas), frente a otros tratamientos subcutáneos más frecuentes (adalimumab, cada dos semanas; etanercept, dos veces por semana) y, desde luego, con la administración en forma de infusión intravenosa de al menos dos horas de duración que requiere infliximab cada 8 semanas.
En definitiva, el secukinumab puede considerarse como una incorporación relativamente innovadora al tratamiento de la psoriasis en placas, que viene a completar las opciones relacionadas con el control de la formación de factor nuclear κB (NFκB), implicado directamente en los procesos hiperproliferativos de la epidermis en la progresión de la placa psoriásica. Dado su eficacia demostrada tanto en pacientes naïve como en tratados previamente con terapias sistémicas (biológicas o no), es una opción primaria en los cuadros moderados o graves de psoriasis en placas donde se considere necesario recurrir a un tratamiento sistémico.
|
VALORACIÓN |
|
|
SECUKINUMAB  COSENTYX®(Novartis) |
|
|
Grupo Terapéutico (ATC): L04AC. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores: inhibidores de interleucinas. |
|
|
Indicaciones autorizadas: Tratamiento de psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar. |
⇑ |
|
Novedad molecular: Mecanismo de acción innovador. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Basiliximab |
Simulet |
Novartis |
1999 |
|
Anakinra |
Kineret |
Swedish Orphan Biovitrum |
2002 |
|
Canakinumab |
Ilaris |
Novartis |
2009 |
|
Tocilizumab |
Roactemra |
Roche |
2009 |
|
Ustekinumab |
Stelara |
Janssen Cilag |
2010 |
|
Secukinumab |
Cosentyx |
Novartis |
2015 |
BIBLIOGRAFÍA