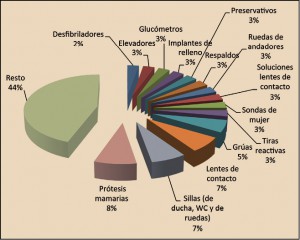
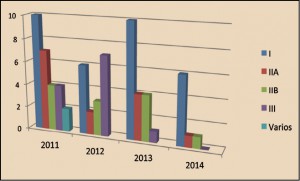
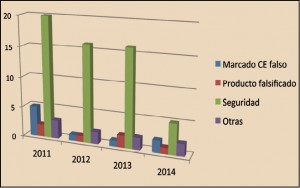
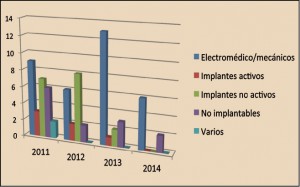
Respecto al protagonismo asistencial de las farmacias comunitarias en estos países, España está en las antípodas del país austral. El Gobierno australiano acaba de firmar con el Pharmacy Guild of Australia (equivalente al Consejo General de Colegios de Farmacéuticos) el 6º Acuerdo con la Farmacia Comunitaria para el periodo 2015-2020, en el que, más allá de recogerse los cuestiones ligadas a la remuneración profesional (se suprime el margen comercial por un pago fijo de 10,42 euros por dispensación) se plasma una partida de 1.260 millones de dólares (unos 856 millones de euros) para financiar en el próximo lustro programas sanitarios en farmacias comunitarias, estudios clínicos y servicios profesionales.
En concreto, 600 millones se asignan a la continuación de programas y servicios ya implantados y otros 600 millones a nuevos proyectos. Mientras tanto, en España la mera asignación de una partida presupuestaria estatal para potenciar el papel asistencial de la farmacia comunitaria, mucho menos en las cantidades que recoge el acuerdo australiano, parece aún una quimera. De hecho, los esfuerzos actuales de la profesión pasan porque, al menos, se les permita ampliar sus funciones asistenciales, dejando de momento a un lado las cuestiones económicas.
Ya antes de este acuerdo, Australia era considerada referente en la apuesta por los nuevos servicios en farmacias centrados en el paciente —como lo demuestra el éxodo de jóvenes farmacéuticos españoles a trabajar en proyectos relacionados con la atención farmacéutica becados por la Universidad Tecnológica de Sidney— y este nuevo acuerdo lo reafirma. Incluso, asignando partidas concretas a programas y servicios concretos. Como ejemplos, el Gobierno australiano destinará en los próximos cinco años 189,2 millones de dólares a programas de adherencia, a programas de reducción de efectos adversos (176,3 millones), a atención farmacéutica domiciliaria (14,5), a atención farmacéutica en residencias (14,2), etc. En una época en la que, en España, los medicamentos de diagnóstico hospitalario están saliendo de las farmacias para su dispensación en los servicios de Farmacia Hospitalaria —justificándolo en su mejor control y seguridad, aunque los colegios y sociedades científicas denuncian criterios económicos además del perjuicio que supone al paciente los desplazamientos a los hospitales—, Australia ha vuelto a dar ejemplo de cómo situar al paciente como ‘centro’ del sistema: así, desde el pasado 1 julio, los pacientes australianos con sida pueden elegir dónde prefieren retirar sus medicamentos: en los hospitales (como ocurría hasta la fecha) o en las farmacias. Con esta medida, la Administración australiana responde a una petición de las asociaciones de pacientes, que incluso presentaron encuestas cuyos resultados reflejaban que el 72% de los pacientes con sida preferían recibir su tratamiento en alguna farmacia.
- El Global, 24 de julio de 2015