Resumen
La tianeptina es un agente antidepresivo, autorizado para el tratamiento de la depresión mayor en adultos. Su mecanismo de acción aún no está completamente dilucidado, aunque no parece estar directamente relacionado con el de otros antidepresivos. Parece un jugar un complejo papel en la interrelación de diversos sistemas de neurotransmisores, facilitando la restauración de la neuroplasticidad del sistema límbico y revirtiendo la alteración de la transmisión glutamatérgica inducida por el estrés bioquímico neurológico, que parecen jugar un papel central en las funciones perturbadas en los estados deprimidos. No es un fármaco reciente, de hecho data de los años 70 del pasado siglo. Los datos clínicos disponibles indican que su eficacia clínica no es inferior a la de los antidepresivos ISRS de referencia (fluoxetina, sertralina, paroxetina); da lugar a la aparición del efecto antidepresivo de forma relativamente rápida (1-2 semanas) y presenta un perfil toxicológico aceptable, comparable a la de los ISRS. Presenta un mecanismo de acción antidepresivo que resulta sugerente aunque no está completamente establecido; en cualquier caso, se ha contrastado en clínica su capacidad para mejorar el estado neurocognitivo de los pacientes deprimidos; además, ha demostrado su eficacia, en combinación a un ISRS, en pacientes con respuesta insatisfactoria a estos últimos. Asimismo, su uso ha sido contrastado en pacientes con edades avanzadas e incluso en pacientes deprimidos con tendencias adictivas al alcohol. Sin embargo, cabe indicar también que la mayoría de los estudios clínicos que avalan su uso en depresión son de corta duración y que la posología puede dificultar la adherencia al tratamiento. En definitiva, una opción más a considerar para el tratamiento de la depresión, que puede resultar de cierto interés en pacientes intolerantes o refractarios a otros tratamientos antidepresivos más establecidos.
DEPRESIÓN
La depresión es un trastorno emocional que se presenta como un estado de abatimiento e infelicidad, que puede ser transitorio o permanente; es una alteración del humor en la que destaca un ánimo deprimido, falta de energía y/o pérdida de interés, o una disminución de la capacidad para disfrutar u obtener placer (anhedonia), que afectan a la vida de la persona durante la mayor parte del día y durante al menos 2 semanas. Los síntomas se relacionan con 3 alteraciones centrales: del estado de ánimo, caracterizado por tristeza y/o irritabilidad, con una pérdida de interés en actividades que previamente agradaban al paciente (anhedonia); de la cognición, que hace que el pensamiento sea lento e ineficiente, apareciendo además un componente de gran autocrítica; y de la actividad, que disminuye, aunque puede ser ocultada por la presencia de ansiedad o agitación.
La depresión afecta a una de cada 5 mujeres y a uno de cada 10 hombres, de manera que se calcula que sólo en España hay entre 1,2 y 1,5 millones de enfermos con depresión. Según un estudio realizado por la Escuela de Salud Pública de la Universidad de Harvard y la Organización Mundial de la Salud (OMS), la depresión será en el año 2020 el segundo mayor problema de salud del mundo, solo superado por los problemas cardiovasculares (Martín Carrasco, 2014).
Por otra parte, tanto por su epidemiología como por la gravedad clínica de los cuadros depresivos, su impacto en la calidad de vida es superior al de enfermedades crónicas como la artritis, la diabetes o las enfermedades cardiacas y respiratorias. La DM es, entre las enfermedades no “fatales”, la mayor causa de años vividos con discapacidad, ocupando el cuarto lugar entre las enfermedades que provocan discapacidad. Asimismo, la depresión unipolar figura como tercera causa de carga de enfermedad, detrás de la isquemia coronaria y de los accidentes cerebrovasculares, explicando el 6% de la carga causada por todas las enfermedades. Entre el 10% y el 20% de los pacientes atendidos en las consultas de atención primaria padece un trastorno afectivo (Álamo, 2012).
La depresión mayor (DM) es un síndrome o agrupación de síntomas en el que predominan los síntomas afectivos (tristeza patológica, decaimiento, irritabilidad, sensación subjetiva de malestar e impotencia frente a las exigencias de la vida) aunque, en mayor o menor grado, también están presentes síntomas de tipo cognitivo, volitivo o incluso somático, por lo que podría hablarse de una afectación global de la vida psíquica, haciendo especial énfasis en la esfera afectiva. La gravedad y persistencia de estos síntomas los diferencia de cambios ordinarios en el estado de ánimo que no llegan a ser patológicos ni llegan a provocar deterioro funcional ni social, hechos característicos en la depresión.
La asociación entre trastorno depresivo y ansiedad es casi más la norma que la excepción. La comorbilidad de la depresión se observa también con el abuso del alcohol o consumo de sustancias y con algunas enfermedades orgánicas cerebrales y sistémicas. Los síndromes depresivos causados por enfermedades médicas y por medicaciones o sustancias psicoactivas no se consideran trastornos primarios del estado de ánimo y no se los atribuye el diagnóstico de trastorno depresivo mayor.
El humor depresivo y la pérdida de interés son los síntomas clave. Los pacientes comentan que se encuentran tristes, desesperanzados, inútiles y con un dolor emocional que es una agonía. Unos dos tercios de los pacientes deprimidos se plantean el suicidio y entre un 10 y un 15% llega a cometerlo. Casi todos los pacientes depresivos manifiestan una pérdida de energía, un empeoramiento del rendimiento escolar y laboral y una disminución de la motivación para iniciar nuevos proyectos. Un 80% de los pacientes tiene dificultades para dormir, sobre todo para mantener el sueño, con múltiples despertares que aprovecha para rumiar sus problemas. Muchos pacientes han perdido el apetito, con la consecuente pérdida de peso, tienen irregularidades con la menstruación y han perdido el interés por las relaciones sexuales. La ansiedad es un síntoma común en la depresión, que afecta a un 90% de los pacientes y, entre los síntomas cognitivos, destacan la sensación subjetiva de dificultades para la concentración y otras alteraciones en los procesos del pensamiento.
Por su parte, el trastorno distímico o distimia es el prototipo de depresión crónica e insidiosa que no alcanza la intensidad suficiente para cumplir los criterios diagnósticos de episodio depresivo mayor. Por lo tanto, las diferencias con la DM son de intensidad y duración más que categoriales, siendo práctica habitual el doble diagnóstico en un mismo paciente. El trastorno distímico puede iniciarse a cualquier edad. Finalmente, el trastorno ciclotímico es un trastorno bifásico caracterizado por oscilaciones bruscas de una fase a otra (hipomanía o depresión), donde cada fase dura días, con ánimo normal poco frecuente. Entre un 3 y un 10% de los pacientes psiquiátricos tratados ambulatoriamente puede presentar un trastorno ciclotímico; en la población general, la prevalencia a lo largo de la vida del trastorno se ha estimado en un 1%, igual que con el trastorno bipolar. El trastorno psicológico coexiste con frecuencia con el trastorno límite de la personalidad.
Hay datos que avalan una participación genética en la depresión mayor, en especial en las formas más graves, lo que supone un factor de vulnerabilidad a padecerla ante determinados desencadenantes, como por ejemplo el estrés. El riesgo es mayor en los parientes de primer grado y, de hecho, éstos responden generalmente al mismo antidepresivo. No obstante, no todo el mundo con este componente genético de vulnerabilidad frente a la DM desarrollará la enfermedad. De hecho, no se ha identificado ninguna variación genética única que sea responsable de un aumento sustancial del riesgo de depresión. Se piensa que son necesarios múltiples factores genéticos, junto con factores ambientales, para el desarrollo de DM. Otras patologías psiquiátricas, como la depresión bipolar, la esquizofrenia o el trastorno por déficit de atención, tienen mayor carga genética.
Asimismo, existen evidencias de que las alteraciones a nivel de varios neurotransmisores y de sus vías de comunicación podrían estar implicadas en la fisiopatología de la depresión, especialmente la noradrenalina (NA) y la serotonina (5-HT). En concreto, existe una disminución de los niveles del 3-metoxi-4-hidroxifenilglicol (un metabolito de la NA) en sangre y orina en los pacientes depresivos, además de una disminución de la bomba de recaptación de la 5-HT en las plaquetas. Se han observado, además, niveles bajos de ácido 5-hidroxiindolacético (metabolito de la 5-HT) en pacientes que cometieron suicidio. Aunque de forma menos relevante que NA y 5-HT, también se ha observado que la depleción de dopamina (DA) aumenta el riesgo de depresión.
Asimismo, se ha sugerido la existencia de diferencias en la actividad del transportador de 5-HT (5-HTT) en la depresión unipolar. Concretamente, el gen que codifica el 5-HTT presenta en su región promotora un polimorfismo (5-HTTLPR-s) que aumenta la vulnerabilidad de sufrir depresión unipolar, en presencia de factores estresantes. Asimismo, el polimorfismo del gen que codifica el 5-HTT (forma “l”) se ha asociado a una mejor respuesta al tratamiento con antidepresivos frente a la forma “s” del transportador. Por otra parte, parece existir una correlación entre el aumento de los niveles del 5-HTT en plaquetas y linfocitos y una mejoría en las escalas clínicas de depresión. Además del 5-HTT, varios receptores serotoninérgicos también muestran cambios en la depresión. El receptor 5-HT1A muestra una disminución en la depresión y se sospecha de un papel importante de los receptores 5-HT2 a la luz de su papel en otros trastornos como la esquizofrenia y el trastorno bipolar y de la eficacia antidepresiva de algunos antipsicóticos que actúan sobre dichos receptores.
Varias estructuras prefrontales y límbicas y los circuitos que las interconectan parecen estar implicados en la regulación afectiva. Las áreas implicadas incluyen a la corteza prefrontal ventromedial (VMPFC), la corteza prefrontal orbital lateral (LOPFC), la corteza prefrontal dorsolateral (DLPFC), la corteza cingulada anterior (CAC), el estriado ventral, incluyendo el núcleo accumbens, la amígdala y el hipocampo. En todas estas zonas se han hallado anomalías en pacientes con DM en comparación con controles sanos que sugieren participar en la expresión sintomática de la DM. En el trastorno depresivo mayor estaría afectada la conectividad dinámica entre las estructuras neuroanatómicas involucradas en la regulación del humor y la respuesta al estrés. En este sentido, en la depresión existe una hiperactividad de la VMPFC, lo que se asocia con mayor sensibilidad al dolor, ansiedad, depresión y tensión; mientras que la hipoactividad de la DLPFC puede producir retraso psicomotor, apatía y déficit en atención y memoria de trabajo. Asimismo, se ha detectado una disminución de la comunicación entre la amígdala y regiones de la corteza cingulada, por lo que ésta perdería su capacidad inhibitoria, importante para la regulación emocional, provocándose una mayor disfunción afectiva y motivacional. En consecuencia, las conexiones entre estructuras cognitivas y ejecutivas son hipofuncionantes, por lo que no controlan las áreas límbicas y éstas estimulan el hipotálamo, lo que conduce a una disregulación neuroendocrina y una hiperactividad simpática, características presentes en la depresión.
Finalmente, existen numerosos factores de riesgo psicológicos y sociales reconocidos para la depresión. Entre los más reconocidos se encuentra una baja autoestima, experiencias adversas en la infancia, patrones de pensamiento negativo y un exceso de acontecimientos vitales recientes no deseables, como los que suponen algún tipo de pérdida: un divorcio, la muerte de un ser querido, etc. Asimismo, la existencia de dificultades importantes persistentes, como el desempleo, la pobreza, etc., son factores de riesgo para padecer depresión. Determinados rasgos de personalidad, como hiperresponsabilidad, honestidad, autoexigencia, poca tolerancia, inseguridad, escrupulosidad, escasa flexibilidad, pesimismo, dependencia, baja autoestima e influenciabilidad, se consideran también factores de riesgo depresivo. Y sin olvidar que determinadas enfermedades orgánicas o psiquiátricas asociadas pueden inducir depresión.
El tratamiento de la DM incluye la fase aguda (dirigida a obtener la remisión), la fase de continuación (se mantiene la remisión y se previenen las recaídas) y la fase de mantenimiento (orientada a prevenir la recidiva). Los objetivos generales del tratamiento son:
- Reducir y eliminar los síntomas depresivos.
- Recuperar el funcionamiento biopsicosocial y laboral del paciente.
- Disminuir el riesgo de suicidio.
- Minimizar la morbilidad reduciendo recaídas o cronicidad.
- Prevenir la comorbilidad.
- Prevención de recaídas depresivas.
- Mejorar la relación beneficio-riesgo de la medicación en relación con la patología.
En el tratamiento de la depresión se utiliza una familia de psicofármacos, los antidepresivos, heterogénea en cuanto a su estructura química y efectos sobre la neurotransmisión cerebral. En líneas generales, la eficacia de los antidepresivos varía poco entre los diferentes grupos, diferenciándose entre ellos fundamentalmente por sus perfiles de seguridad, tolerabilidad e interacciones.
En depresiones graves, los antidepresivos constituyen el único tratamiento para el que se ha encontrado una clara evidencia de efectividad, ya sean solos o en combinación con psicoterapia. Sin embargo, en depresiones más leves o moderadas, los antidepresivos, pese a ser eficaces, invitan a pensar en la posibilidad de emplear altetrnativamente estrategias de psicoterapia, que también gozan de eficacia.
Existen algunos factores que pueden orientar la elección del antidepresivo. Así, por ejemplo, si un paciente –o un familiar de primer grado– ha respondido previamente a un determinado antidepresivo, se recomienda emplear el mismo fármaco que ha demostrado utilidad. Por otro lado, hay autores que apoyan una mayor eficacia de los antidepresivos tricíclicos (ADT) y, posiblemente, de los antidepresivos duales (venlafaxina y duloxetina) en la DM melancólica, cuadro caracterizado por presentar una mayor endogeneidad. En el caso de una DM con síntomas atípicos o una distimia, los inhibidores selectivos de la recaptación de serotonina (ISRS) podrían ser preferibles a los ADT. Por otro lado, algunos efectos secundarios de los antidepresivos, como la sedación y el aumento del apetito, pueden resultar útiles si el episodio cursa con agitación, insomnio y pérdida de apetito.
Los antidepresivos en general aumentan la tasa de monoaminas en la hendidura sináptica, actuando dichas monoaminas sobre distintos receptores, que actuarían a modo de interruptores poniendo en marcha los mecanismos de transducción responsables del efecto antidepresivo. En este sentido, es probable que todos los antidepresivos, independientemente de que “toquen” el interruptor noradrenérgico o serotoninérgico, desarrollen su efecto molecular a través de una vía final común. Sin embargo, se han descrito cuadros depresivos específicamente relacionados con un déficit funcional serotoninérgico, que cursa con ánimo deprimido, ansiedad, crisis de pánico, fobias, obsesiones-compulsiones y bulimia, y otros que guardan relación con un déficit noradrenérgico, caracterizado por ánimo deprimido, déficits cognitivos (atención, memoria de trabajo y velocidad de procesamiento de la información), inhibición psicomotriz, cansancio y apatía. Cuando estos cuadros son muy claros, se podría intentar de inicio abordar el tratamiento deficitario serotoninérgico con potenciadores de estos mecanismos, por ejemplo, ISRS (fluoxetina, paroxetina, etc.) y, por el contrario, en caso de déficit noradrenérgico se podría iniciar el tratamiento con un inhibidor de la recaptación de NA (IRNA) como, por ejemplo, reboxetina. No obstante, se ha observado que estas vías se estimulan mutuamente, lo que dificulta predecir la eficacia específica a través del mecanismo de acción de cada fármaco.
La tolerabilidad a los efectos secundarios es otro criterio – y con seguridad más empleado que el del mecanismo de acción – para la elección del antidepresivo. Los ISRS y los antidepresivos más modernos parecen tener un perfil de seguridad algo mejor y se toleran generalmente mejor que el de los más antiguos, por lo que suelen ser los agentes de primera elección en el tratamiento de la depresión leve y moderada. Para la elección del antidepresivo se debe considerar también la posibilidad de interacciones con otros fármacos. Casi todos los antidepresivos, con la excepción de la agomelatina, inhiben en mayor o menor medida diversos isoenzimas del citocromo P450 (CYP), como la CYP2D6, mientras que los ISRS, con la excepción del escitalopram, inhiben además otras como CYP3A4, CYP1A2, CYP2C9 o CYP2C19.
La variabilidad interindividual en la respuesta terapéutica a los fármacos antidepresivos es muy amplia. En el tratamiento de la DM se suelen distinguir 3 fases: aguda, de continuación y de mantenimiento, durante las que el tratamiento muestra su eficacia produciendo inicialmente una “respuesta”, que se considera cuando se observa una mejoría en la intensidad de los síntomas de un 50% en las escalas clínicas empleadas (las más comunes son la Escala de Hamilton – HDRS: Hamilton Depression Rating Scale – y la de Montgomery-Asberg – MADRS: Montgomery-Asberg Depression Rating Scale). Posteriormente, se produce la “remisión” de la enfermedad, caracterizada por que el paciente presenta un estado afectivo normal y muy parecido al que exhibía antes de presentarse la depresión. Cuando la remisión se mantiene en el tiempo, se puede hablar de “recuperación”. Sin embargo, en ocasiones se producen “recaídas”, cuando el sujeto vuelve a mostrar síntomas depresivos antes de la recuperación, o se produce una “recurrencia”, cuando el sujeto vuelve de nuevo a la depresión tras haberse producido la recuperación.
El objetivo durante la fase aguda es suprimir todos los signos y síntomas del episodio en curso, restaurando la funcionalidad psicosocial y ocupacional. Esta fase abarca las primeras semanas de tratamiento, hasta que el paciente alcanza una “respuesta” clínica significativa, que usualmente es de al menos un 40-60% de mejoría en los síntomas. Se considera que aproximadamente las dos terceras partes de los pacientes con depresión responden aceptablemente al tratamiento con antidepresivos. Un tercio tiene una respuesta gradual y suele manifestarse entre la segunda y la sexta semana del inicio del tratamiento, mientras que el otro tercio tiene una respuesta más lenta, parcial o con síntomas residuales que sugieren replantearse el tratamiento aumentando la dosis o añadiendo otro antidepresivo. Finalmente, otro tercio de los pacientes no tiene ninguna respuesta al tratamiento inicial, lo que obliga a un cambio del antidepresivo. No se puede hablar de falta de respuesta o resistencia hasta transcurridas 8 semanas de tratamiento y habiéndose alcanzado la dosis máxima.
El objetivo de la fase de continuación se centra en prevenir las recaídas, manteniendo la respuesta inicial. Es importante destacar que tras el cese de los síntomas agudos de depresión, si se interrumpe la terapia de forma inmediata, una importante proporción de pacientes, hasta casi el 70%, tendrá una recaída. Se considera al paciente recuperado cuando ha permanecido asintomático durante al menos 4 a 9 meses tras el episodio depresivo. Una vez alcanzada la recuperación, el tratamiento de continuación puede interrumpirse, recomendándose un periodo de entre 6 y 12 meses para evitar las recurrencias. En definitiva, cuando la remisión se mantiene de 6 a 12 meses, se habla de recuperación y el paciente entra en la fase de mantenimiento, cuyo objetivo es prevenir un nuevo episodio de depresión. Su duración podría oscilar desde 1 año a toda la vida del sujeto, dependiendo de la probabilidad de recurrencias.
ACCIÓN Y MECANISMO
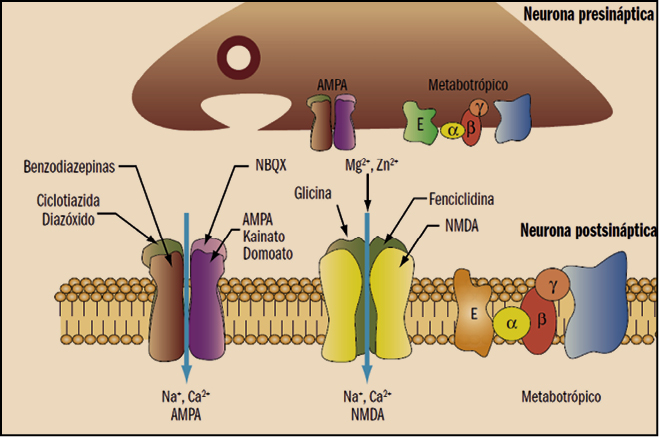
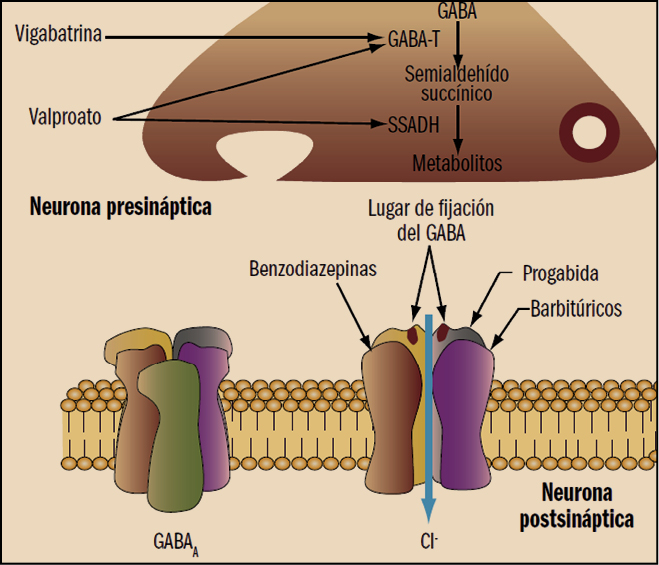
La tianeptina es un agente antidepresivo, autorizado para el tratamiento de la depresión mayor en adultos; su actividad antidepresiva tiene un comienzo relativamente rápido (7-14 días). Su mecanismo de acción aún no está completamente dilucidado, aunque no parece estar directamente relacionado con el de otros antidepresivos. De hecho, no muestra ninguna afinidad hacia la mayoría de los receptores de neurotransmisores conocidos en diversas localizaciones neurológicas (hipocampo, estriado y córtex frontal, sistema límbico), ni parece alterar las concentraciones ni la afinidad de los receptores alfa y beta-adrenérgicos, serotonérgicos, de benzodiazepinas (BZ), GABA o ácido glutámico. No inhibe la recaptación sináptica de serotonina o de noradrenalina en el sistema nervioso central; de hecho, al contrario que los antidepresivos tricíclicos y los inhibidores de la recaptación de serotonina (5-HT), la tianeptinaincrementa la recaptación de este neurotransmisor.
La tianeptina parece un jugar un complejo papel en la interrelación de diversos sistemas de neurotransmisores, facilitando la restauración de la neuroplasticidad del sistema límbico y reviritiendo la alteración de la transmisión glutamatérgica inducida por el estrés bioquímico neurológico, que parecen jugar un papel central en las funciones perturbadas en los estados deprimidos. En este sentido, la modificación de los mecanismos glutamatérgicos por la tianeptina – a través de mecanismos aún no bien conocidos – podría justificar su actividad frente a la influencia negativa de la neurogénesis en el hipocampo en estado de estrés crónico, de la proliferación celular y del remodelado dendrítico, todos los cuales se encuentran profundamente alterados en los estados depresivos. De acuerdo con ello, la tieneptina podría ayudar a normalizar la función sináptica glutamatérgica, determinante para un correcto funcionamiento afectivo (McEwen, 2010).
En este sentido, conviene recordar que el glutamato es el neurotransmisor excitatorio principal en el cerebro, donde tiene una distribución amplia y ubicua. En el cerebro humano, las neuronas glutamatérgicas proyectan sus axones desde la corteza a regiones subcorticales, tales como el locus coeruleus, núcleos del rafe y sustancia nigra, donde modulan vías monoaminérgicas, lo que permite al sistema glutamatérgico participar en una amplia gama de funciones fisiológicas, que incluyen la memoria y la cognición, así como acciones neurotróficas y neurotóxicas y la inducción de la plasticidad neuronal (Álamo, 2012).
ASPECTOS MOLECULARES
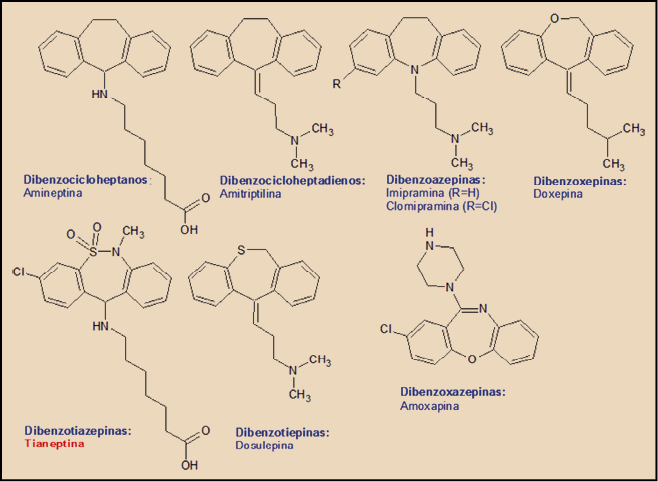
En términos estructurales, la tiamineptina se encuadra dentro del grupo genéricamente conocido como antidepresivos tricíclicos (ADT), término quealude al elemento estructural común de todos ellos, un triple ciclo condensado linealmente, formado por dos anillos bencénicos en los extremos y uno central con siete eslabones (-epina), uno o dos de los cuales – eventualmente – son heteroátomos, tales como el azufre (S; tia-), el nitrógeno (N; aza-) o el oxígeno (O; oxa-). De acuerdo con ello, encontramos derivados del dibenzocicloheptano (amineptina), del dibencicloheptadieno (amitriptilina), de la dibenzoazepina (imipramina, clomipramina), de la dibenzoxepina (doxepina), de la dibenzoxazepina (amoxapina), de la dibenzotiepina (dosulepina) y, como es el caso de la tianeptina, del núcleo de dibenzotiazepina.
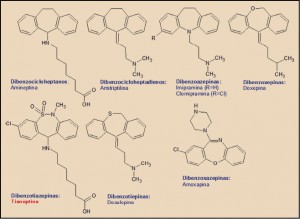
Además del núcleo central, determinante de la actividad antidepresiva, estos fármacos presentan restos laterales que modulan dicha actividad, potenciando o reduciendo la actividad – generalmente inhibidora – sobre otros múltiples receptores celulares (histamina, acetilcolina, etc.). En el caso de la tianeptina, presenta un peculiar resto de ácido hepatanoico que solo es compartido con la amineptina y que se asocia con algunas peculiaridades farmacológicas, especialmente con la modulación de la transmisión glutamatérgica, mecanismo al que se atribuye un papel relevante en los efectos de la tianeptina sobre la neurogénesis y el remodelado de las dendritas neuronales.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de la tianeptina han sido adecuadamente contrastadas mediante varios ensayos clínicos aleatorizados, doblemente ciegos y controlados con comparadores activos (fluoxetina, paroxetina, sertralina, escitalopram). Como criterios de eficacia se han utilizado fundamentalmente las tasas de variación de la depresión (y las correspondientes tasas de respuesta), determinada mediante escalas clínicas validadas, principalmente HDRS y MADRS, realizadas mediante entrevista a los pacientes por psiquiatras especializado, antes, durante y tras finalizar el tratamiento.
La escala de puntuación de la depresión de Hamilton (Hamilton Depression Rating Scale; HDRS) es posiblemente la escala más utilizada en la práctica clínica y en la investigación para medir la gravedad de la depresión y monitorizar la evolución de los síntomas. Aunque la versión original constaba originalmente de 21 ítems o preguntas (HDRS21), la forma más habitualmente utilizada actualmente es una versión reducida de 17 ítems (HDRS17), entre los que se incluyen ánimo depresivo, sentimientos de culpa, suicidio, insomnio precoz, medio y tardío, trabajo y actividades, inhibición, agitación, ansiedad psíquica y ansiedad somática, síntomas somáticos gastrointestinales, síntomas somáticos generales, síntomas sexuales (disfunción sexual y alteraciones de la menstruación), hipocondría, pérdida de peso y capacidad de entendimiento. La puntuación global se obtiene sumando las puntuaciones de cada ítem, con un rango de puntuación que en la versión española es de 0 (ausencia completa de ningún síntoma depresivo) a 54 (gravedad máxima). Habitualmente, se considera que no hay depresión con valores de 0 a 7 puntos, es ligera o menor entre 8 y 13, moderada entre 14 y 18, grave de 19 a 22 y muy grave por encima de 22.
Por su parte, la escala de puntuación de la depresión de Montgomery-Asberg (Montgomery-Asberg Depression Rating Scale, MADRS) ha sido específicamente diseñada para evaluar la intensidad de los síntomas depresivos en adultos, así como los efectos del tratamiento antidepresivo. Es un instrumento breve, formado por 10 ítems relativos a 10 síntomas depresivos distintos. Cada ítem es evaluado mediante una subescala con 7 grados de gravedad (de 0: ausencia del síntoma, a 6: máximo nivel de gravedad del síntoma). Sumando las puntuaciones parciales de cada ítem se obtiene la puntuación global de la escala que oscila entre 0 puntos (ausencia de depresión) y 60 (máximo nivel de depresión). Se considera que una puntuación inferior a 10 puntos indica ausencia de trastorno depresivo.
Es habitual en los ensayos clínicos con antidepresivos utilizar como variables de eficacia la tasa de respuesta (porcentaje de pacientes con una disminución del 50% o más de los valores iniciales de HDRS o MADRS) y la tasa de remisión (porcentaje de pacientes con unos valores finales de HDRS menores a 7 o de MADRS menores de 9). Adicionalmente, a la HDRS y la MADRS, también se utiliza la escala clínica global de impresión de la gravedad de la enfermedad (Clinical Global Impressions-Severity of Illness, CGI-S).
En un meta-análisis (Kasper, 2002) se comparó la eficacia de la tianeptina frente a varios antidepresivos inhibidores de la recaptación de serotonina en el tratamiento a corto plazo de la depresión. El estudio incluyó a cinco ensayos clínicos, dos frente a fluoxetina, otros dos frente a paroxetina y uno frente a sertralina, totalizando 1.348 pacientes. Considerando a todos los pacientes y específicamente aquellos con una puntuación MADRS >28, ninguno de las variables de eficacia (tasa de respondedores y de remisiones) mostró diferencia significativa alguna entre los brazos comparados de tratamiento. Asimismo, el análisis basado en la impresión clínica global (CGI) tampoco encontró diferencias, excepto para el ítem 3 de esta escala (índice terapéutico), para el que se apreció una tendencia estadística (p=0,06-0,07) favorable a la tianeptina. En definitiva, el meta-análisis concluyó que la tianeptina es al menos tan eficaz como los inhibidores de la recaptación de serotonina y con tendencia a una mejor tolerabilidad del tratamiento por parte de los pacientes deprimidos.
Otro estudio multicéntrico y aleatorizado (Jeon, 2014) comparó durante 12 semanas los efectos neurocognitivos de la tianeptina (12,5 mg/8 h) y el escitalopram (10 mg/24 h) en 167 pacientes con depresión mayor. Al final del periodo de tratamiento, el grupo tratado con tianeptina mostró una significativa (p=0,002) mejoría en la comisión de errores, en la memoria verbal inmediata (p=0,001), la mini prueba de estado mental (Mini-Mental State Examination, p<0,0001), memoria retardada (p<0,0001) y capacidad de razonamiento (p=0,001); por su parte, el escitalopram mejoró la memoria retardada y la capacidad de razonamiento, pero ningún otro parámetro neurocognitivo. La comparación de ambos grupos mostró que el grupo de pacientes tratados con tianeptina experimentó una significativa mejoría en la puntuación de comisión de errores (F=6,64; p=0,011) y memoria verbal inmediata (F=4,39; p=0,038), en comparación con el escitalopram y tras ajustar los datos por edad, sexo, años de educación, puntuaciones basales y cambios en el nivel de gravedad de la depresión. Ambos fármacos mejoraron el deterioro cognitivo subjetivo de la memoria y la concentración.
En otro ensayo clínico abierto y multicéntrico (Vukovic, 2009) se estudió la eficacia, seguridad y tolerabilidad de la tianeptina durante 8 semanas en 77 pacientes “frágiles” (mayores de 55 años o con adicción al alcohol) con depresión leve a moderada. Al final de las ochos semanas de tratamiento, las tasas de remisión (≤12 puntos MADRS) fueron del 51% entre los mayores de 55 años y del 84% entre los adictos al alcohol, observándose una significativa reducción de la puntuación MADRS a partir de la primera semana de tratamiento. Los pacientes mayores de 55 años toleraron peor el tratamiento (4,5% tuvieron náusea, 4,4% fatiga en las piernas, 2,2% irritabilidad), mientras que solo el 3,1 de los pacientes deprimidos adictos al alcohol mostraron vértigos.
Finalmente, con el fin de determinar la eficacia y tolerabilidad de la tianeptina en combinación con un inhibidor selectivo de la recaptación de serotonina (ISRS) en respondedores parciales o nulos al tratamiento con ISRS, se realizó un estudio abierto de seis semanas de duración sobre 150 pacientes con depresión mayor utilizando la tianeptina adicionalmente al ISRS (Woo, 2013). Los resultados mostraron cambios significativos desde la primera semana de tratamiento, con tasas de respondedores del 65% (HDRS) y del 69% (MADRS), y tasas de remisión del 34% (HDRS) y del 42% (MADRS) al final del tratamiento. La combinación de tianeptina e ISRS fue bien tolerada en general.
Desde el punto de vista de la seguridad clínica, la tianeptina parece ser razonablemente bien tolerada, asociándose a eventos adversos frecuentes aunque en general leves y transitorios, que no suelen requerir la suspensión del tratamiento. Los más comunes, descritos en el 1-10% de los pacientes son: anorexia, pesadillas, insomnio, somnolencia, mareos, cefalea, vértigo, temblor alteraciones visuales (diplopía), taquicardia, palpitaciones, dolor torácico, sofocos, disnea, sequedad de boca, estreñimiento, dolor abdominal, náusea, vómitos, dispepsia, diarrea, flatulencia, pirosis, dolor de espalda, mialgia, astenia. Está contraindicado su uso conjunto con antidepresivos IMAO, debido al riesgo de interacciones clínicamente muy relevantes (potencialmente mortales).
ASPECTOS INNOVADORES
La tianeptina es un agente antidepresivo, autorizado para el tratamiento de la depresión mayor en adultos. Su mecanismo de acción aún no está completamente dilucidado, aunque no parece estar directamente relacionado con el de otros antidepresivos. Parece un jugar un complejo papel en la interrelación de diversos sistemas de neurotransmisores, facilitando la restauración de la neuroplasticidad del sistema límbico y revirtiendo la alteración de la transmisión glutamatérgica inducida por el estrés bioquímico neurológico, que parecen jugar un papel central en las funciones perturbadas en los estados deprimidos.
No es un fármaco reciente, de hecho data de los años 70 del pasado siglo. Los datos clínicos disponibles indican que su eficacia clínica no es inferior a la de los antidepresivos ISRS de referencia (fluoxetina, sertralina, paroxetina); da lugar a la aparición del efecto antidepresivo de forma relativamente rápida (1-2 semanas) y presenta un perfil toxicológico aceptable, comparable a la de los ISRS. Presenta un mecanismo de acción antidepresivo que resulta sugerente aunque no está completamente establecido; en cualquier caso, se ha contrastado en clínica su capacidad para mejorar el estado neurocognitivo de los pacientes deprimidos. Asimismo, ha demostrado su eficacia, en combinación a un ISRS, en pacientes con respuesta insatisfactoria a estos últimos.
Asimismo, su uso ha sido contrastado en pacientes con edades avanzadas e incluso en pacientes deprimidos con tendencias adictivas al alcohol. Esto último puede tener una particular relevancia, dado que su antecesor inmediato es la amineptina (Survector®), que fue retirada del mercado farmacéutico ante el riesgo de provocar dependencia, sobre todo en pacientes con adicción a otros productos1.
Sin embargo, cabe indicar también que la mayoría de los estudios clínicos que avalan su uso en depresión son de corta duración (6-12 semanas) y que la posología recomendada (12,5 mg/8 horas) puede dificultar la adherencia al tratamiento, particularmente durante la siempre complicada fase de latencia terapéutica al principio del tratamiento (1-2 semanas), durante la cual el paciente no experimenta efectos favorables significativos. En definitiva, una opción más a considerar para el tratamiento de la depresión, que puede resultar de cierto interés en pacientes intolerantes o refractarios a otros tratamientos antidepresivos más establecidos.
1 El 21 de enero de 1999, la antigua Comisión Nacional de Farmacovigilancia (órgano consultivo de la Dirección General de Farmacia y Productos Sanitarios, antecesora en sus funciones de la actual Agencia Española de Medicamentos y Productos Sanitarios, AEMPS), recomendó su retirada del mercado.
|
VALORACIÓN |
|
|
TIANEPTINA
 ZINOSAL® (Juste) |
|
|
Grupo Terapéutico (ATC): N06AX. SISTEMA NERVIOSO. Antidepresivos: otros. |
|
|
Indicaciones autorizadas: Tratamiento de la depresión mayor en adultos. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣♣ |
|
Novedad clínica: Posibilidad de asociar con otros tratamientos actualmente en vigor. Utilidad en cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
⇑ |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma o similar indicación terapéutica. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Amineptina |
Survector1 |
Servier |
1982 |
1 Retirada del mercado farmacéutico español en 1999.
BIBLIOGRAFÍA




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}