|
Fármacos |
Laboratorio |
Comentarios |
|---|---|---|
|
Apatorsen HSPB1 |
Oncogenex |
Apatorsen es un inhibidor de la producción de Hsp27 que se está ensayando para el tratamientondel cáncer de próstata, páncreas, vejiga, pulmón y adenocarcinoma. |
|
Durvalumab |
AstraZeneca |
Durvalumab es un anticuerpo monoclonal anti PD-L1 con actividad sobre varios tipos de tumores. |
|
Lucitanib (E-3810) |
Advenchen Pharmaceuticals |
Lucitanib es un inhibidor dual de los receptores enduteliales vasculares del factor de crecimiento y de los receptores del factor de crecimiento de fibroblastos con capacidad para inhibir la angiogénesis y proliferación celular de tumores. |
|
Selinexor KPT-330) |
Karyopharm |
Selinexor es un inhibidor de XPO, proteína de exportación nuclear cuya inhibición conduce a la acumulación nuclear de proteínas supresoras de tumor (PST). XPO1 se sobreexpresa en muchos tumores, incluido el cáncer de la próstata (PrCa). |
|
Sonidegib (LDE225) |
Novartis |
Sonidegib es un inhibidor específico del receptor SMO con actividad oncolítica. |
Archive
Revista PAM: 380
Número 380, Enero – Febrero 2015
Micofenolato de mofetilo: Riesgo de bronquiectasias e hipogammaglobulinemia
En la Unión Europea, se han revisado los datos de farmacovigilancia del medicamento micofenolato en cuanto al riesgo identificado de bronquiectasias y de hipogammaglobulinemia y se ha concluido que el micofenolato (mofetilo y sódico) administrado en combinación con otros inmunosupresores puede causar hipogammaglobulinemias y bronquiectasias, por lo que deberá realizarse determinación de inmunoglobulinas séricas a todos aquellos pacientes tratados con este fármaco que desarrollen infecciones recurrentes; en caso de hipogammaglobulinemia sostenida clínicamente relevante, se deberá considerar la acción clínica más apropiada y se recomienda llevar a cabo una monitorización lo más precoz posible de aquellos pacientes que desarrollen síntomas pulmonares persistentes como tos y disnea.
El Comité europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) de la EMA (Agencia Europea de Medicamentos) ha llevado a cabo una revisión de los datos recientes sobre nuevos riesgos del micofenolato, en particular casos de bronquiectasias y de hipogammaglobulinemias. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha publicado1 los acuerdos tomados al respecto.
Tanto el micofenolato mofetilo como el micofenolato sódico son profármacos que tras ser administrados se absorben rápida y completamente transformándose en su forma farmacológica activa, el ácido micofenólico, dotado de potentes efectos citostáticos sobre los linfocitos T y B. En combinación con ciclosporina y corticosteroides, el micofenolato sódico está indicado para la profilaxis del rechazo agudo en pacientes adultos sometidos a trasplante renal alogénico, y el micofenolato mofetilo para la profilaxis del rechazo agudo de este mismo trasplante así como del cardíaco y del hepático.
Una revisión reciente, llevada a cabo por el comité europeo PRAC, de los casos notificados y de los estudios publicados hasta el momento, ha puesto de manifiesto que el micofenolato mofetilo administrado en combinación con otros inmunosupresores, puede causar hipogammaglobulinemia y bronquiectasias. A lo largo de la misma revisión, se puso de manifiesto que para el micofenolato sódico también deben ser considerados los citados riesgos.
Es conocido que el descenso de las inmunoglobulinas incrementa la probabilidad de desarrollar infecciones recurrentes, al tiempo que se asocia con una menor esperanza de vida y un mayor riesgo de sufrir rechazo agudo del trasplante. La acción inhibitoria que el micofenolato mofetilo ejerce sobre los linfocitos, se ha postulado como el mecanismo causal de la hipogammaglobulinemia de estos pacientes.
El riesgo de bronquiectasias parece estar relacionado con la propia hipogammaglobulinemia o con un efecto farmacológico directo sobre el pulmón. Cabe mencionar que también se han producido casos aislados de enfermedad pulmonar intersticial y fibrosis pulmonar, algunos de los cuales fueron mortales.
En España se encuentran comercializados con micofenolato mofetilo: Cellcept®, Myfenax® y numerosos genéricos (Micofenolato de Mofetilo Kern Pharma®, Micofenolato de Mofetilo Accord®, Micofenolato de Mofetilo Stada®, Micofenolato Mofetilo Normon®, Micofenolato de Mofetilo Zentiva®, Micofenolato de Mofetilo Sandoz®, Micofenolato de Mofetilo Combix®, Micofenolato Mofetilo Actavis®, Micofenolato de Mofetilo Tecnigen® y Micofenolato de Mofetilo UR®). Con micofenolato sódico únicamente se encuentra comercializado Myfortic®.
Recomendaciones
Dado lo anteriormente expuesto, la AEMPS establece las siguientes recomendaciones dirigidas a los profesionales sanitarios:
- Deberá realizarse determinación de inmunoglobulinas séricas a todos aquellos pacientes en tratamiento con micofenolato (mofetilo o sódico) que desarrollen infecciones recurrentes.
- En caso de hipogammaglobulinemia sostenida clínicamente relevante, se deberá considerar la acción clínica más apropiada. En algunos de los casos notificados, la sustitución del micofenolato (mofetilo o sódico) por otro inmunosupresor, dio lugar a la normalización de los niveles de IgG en suero.
- Se recomienda llevar a cabo una monitorización lo más precoz posible de aquellos pacientes que desarrollen síntomas pulmonares persistentes como tos y disnea. En algunos de los casos confirmados de bronquiectasias, la sustitución del micofenolato (mofetilo o sódico) por otro inmunosupresor, condujo a una mejora de los síntomas respiratorios de los pacientes.
Referencias
- Agencia Española de Medicamentos y Productos Sanitarios. Micofenolato mofetilo y sódico: riesgo de bronquiectasias e hipogammaglobulinemia. Nota informativa MUH (FV) 19/2014, de 12 de diciembre de 2014. http://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2014/NI-MUH_FV_19-micofenolato.htm (consultado 30 enero 2015).
IMPORTANTE
El Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (SEFV-H) se basa en el programa de notificación espontánea de un profesional sanitario (médico, odontólogo, farmacéutico, enfermero, otros) de una sospecha de relación entre un medicamento (incluidos vacunas, sueros, gases medicinales, fórmulas magistrales, plantas medicinales) y un síntoma o signo adverso (reacción adversa, RAM) que manifieste el paciente (programa de tarjeta amarilla). El Real Decreto 577/2013 de Farmacovigilancia de medicamentos de uso humano (BOE núm. 179, de 27 de julio de 2013) entró en vigor el 28 de julio de 2013. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) coordina el SEFV-H.
¿Qué notificar?
Se deben notificar las sospechas de RAM:
– con medicamentos autorizados, incluidas las de aquellos que se hayan utilizado en condiciones diferentes a las autorizadas o con medicamentos extranjeros importados con autorización de la AEMPS,
– principalmente las RAM ‘graves’ (mortales, o que amenacen la vida, prolonguen o provoquen una hospitalización, causen incapacidad o sean médicamente importantes y las trasmisiones de un agente infeccioso a través de un medicamento) o RAM ‘inesperadas’ de cualquier medicamento,
– con medicamentos de ‘seguimiento adicional’ (durante sus primeros 5 años desde la autorización, identificados con un triángulo negro invertido (▼) a la izquierda del nombre del medicamento en el material informativo, en el prospecto y en la ficha técnica); ver la lista mensual de los medicamentos con triángulo negro en la web de la AEMPS, en la sección de CIMA con criterio de búsqueda del «Triángulo negro»: http://www.aemps.gob.es/cima/pestanias.do?metodo=accesoAplicacion
– las que sean consecuencia de ‘errores de medicación’, que ocasionen daño en el paciente,
– las originadas por ‘interacciones’ con medicamentos, plantas medicinales, incluso alimentos (zumo de pomelo, ahumados, crucíferas, etc).
¿Cómo notificar?
No olvide notificar cualquier sospecha de RAM a su Centro Autonómico o Regional de Farmacovigilancia mediante las ‘tarjetas amarillas’. Consulte en este directorio su Centro Autonómico de Farmacovigilancia correspondiente.
NUEVO MÉTODO: se puede notificar a través del sitio web https://www.notificaRAM.es/, y el sistema electrónico hace llegar a su centro correspondiente la notificación de sospecha de RAM. Sirve para profesionales sanitarios y para ciudadanos, en formularios diferentes. La nueva legislación europea de farmacovigilancia establece esta posibilidad para facilitar la notificación de las sospechas de RAM por la población en general.
¿Dónde conseguir tarjetas amarillas?
Consultando a su Centro correspondiente del SEFV-H. Podrá encontrar el directorio de Centros en las primeras páginas del «Catálogo de Medicamentos» y en las páginas de Internet http://www.portalfarma.com y http://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/docs/dir_serfv.pdf.
¿Dónde consultar las fichas técnicas y prospectos de los medicamentos?
En la página web de la AEMPS http://www.aemps.gob.es >> seleccionando >> «CIMA: Centro de Información on-line de Medicamentos de la AEMPS, Humanos», se pueden consultar por nombre comercial o por sus principios activos. También están disponibles en la base de datos BOT Plus.
NOTA: la mención de marcas comerciales en el texto solo tiene fines de identificación, y en absoluto se les debe asignar directamente lo descrito en el texto.
Daclatasvir DAKLINZA® (Bristol-Myers Squibb)
Resumen
El daclatasvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5A, una proteína con diversas funciones relacionadas con la replicación del VHC y el ensamblaje de los viriones. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos. Presenta un mecanismo de acción diferente de los fármacos hasta ahora utilizados y su potencia antiviral es, sin duda, muy elevada, actuando – al menos in vitro – sobre todos los genotipos de VHC. No obstante, el daclatasvir tiene una baja barrera de resistencia a las mutaciones para algunos genotipos, particularmente para 1a, 2a, 2b y 3a. Aunque actualmente está siendo objeto de varios ensayos clínicos, los datos clínicos disponibles hasta ahora para el daclatasvir no son equiparables en cantidad ni en diversidad a los de simeprevir y, especialmente, de sofosbuvir; en este sentido, es particularmente relevante la muy escasa experiencia clínica actual en pacientes con cirrosis o enfermedad hepática avanzada.
HEPATITIS C
El virus de la hepatitis C (VHC) se clasifica dentro de la familia Flaviviridae por ser un virus envuelto, de aproximadamente 50 nm de diámetro, con una sola cadena de ARN en sentido positivo (5’-3’), formando parte del género Hepacivirus. Su prevalencia global se estima entre 150 y 180 millones (2-3 % de la población general), con una incidencia anual de 3-4 millones de nuevos casos. En España, la prevalencia de la infección por VHC C se sitúa en torno al 1,2-1,7% de la población general, lo que supone en torno a 800.000 personas infectadas, la mayoría aún sin diagnosticar; más del 50% de los pacientes que han necesitado un trasplante de hígado son pacientes con hepatitis C que han evolucionado a una enfermedad hepática terminal.
Existen varios genotipos del VHC. En Europa y EEUU, el más frecuente es el 1 (70% de los casos), seguido por el 2 y 3 (25%). El 2 (a y b) y 4 (b, c, e y m) son más frecuentes en África del Norte y Central, y el 6p y 2i en Asia. En concreto, en Europa predomina el genotipo 1b, seguido de 2a, 2b, 2c y 3a; por su parte, en Norte América predomina el genotipo 1a, seguido de 1b, 2a, 2b y 3a.
Una vez en el interior del hepatocito, el virus de la hepatitis C (VHC) utiliza la maquinaria celular para replicarse. La expresión del ARN viral conduce a la síntesis de una única proteína (poliproteína) de gran tamaño (3.011 aminoácidos), que requiere de la acción de varias proteasas para dar lugar a las formas activas de las proteínas virales, que incluyen a tres proteínas estructurales (E) y a siete no estructurales (NS). Esta poliproteína (C(F)-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B) genera al menos diez proteínas maduras al ser procesada por la señal peptidasa del retículo endoplásmico, liberando las proteínas C, E1, E2 y p7 que forman la cápside o núcleo viral (C), la envoltura de la partícula (E1 y E2) y p7 que se parece intervenir en la liberación del genoma al formar canales iónicos en la envoltura viral; la región de las proteínas no estructurales (NS2-NS5B) que participan en la replicación son procesadas por dos enzimas virales: NS2 y NS3 que generan un corte autocatalítico en las regiones NS2-NS3 y NS3-NS4A respectivamente y permiten la formación del complejo proteasa de serina NS3/4A que procesa el resto de la poliproteína viral, provocando la maduración de las proteínas NS4B, NS5A y NS5B con función de polimerasa de ARN dependiente de ARN (NS5B) y de fosfoproteína (NS5A); por el momento, la función de la proteína NS4B no está plenamente comprendida.
Las proteínas NS toman el ARN viral formando un complejo de replicación – sistema replicón – que es asociado a membranas citoplasmáticas modificadas. La capacidad de replicación del VHC es enorme, calculándose en un billón (1012) el número de nuevos virus que son capaces de ser producidos durante un día en una persona infectada.
La proteína NS3 del VHC es una enzima multifuncional ya que presenta en el primer tercio de su estructura una actividad de proteasa de serina y en el resto una función de ARN helicasa DexH/D que revierte el enrollamiento de cadenas dobles de ARN formadas durante la replicación viral. El extremo NS4A es una proteína anfipática de 54 aminoácidos que se asocia no covalentemente con las cadenas A0 y A1 de NS3 y provoca una reorganización en su estructura, optimizando así la actividad de proteasa de NS3; además NS4A promueve la localización del complejo NS3/4A a la membrana del retículo endoplásmico donde es procesada la poliproteína viral.
Por su parte la proteína NS5A es una proteína intensamente fosforilada que, una vez escindida de la poliproteína viral, localiza a las membranas donde se une a la fracción 3’-terminal del ARN viral recién sintetizado y participa en la replicación del genoma viral, en parte a través de interacciones con la ARN polimerasa dependiente del ARN viral (NS5B). Finalmente, la NSB5 es la ARN polimerasa dependiente del ARN viral que desarrolla un papel esencial en la replicación del ARN viral, utilizando la cadena de este última como molde para nuevas cadenas y catalizando el proceso de polimerización de los ribonucleótido-trifosfato (rNTP) durante este proceso de replicación, siendo responsable de producir la hebra de ARN complementaria – ARN(-) – que servirá de molde para fabricar las auténticas hebras de ARN viral – ARN(+) –, que a su vez podrán ser de nuevo replicados y traducidas, o bien empaquetados en las proteínas estructurales para formar nuevas partículas virales de VHC, que son liberadas mediante un proceso de exocitosis.
En el año 2011 se produjo un cambio notable en el panorama terapéutico de la hepatitis C, al comercializarse el boceprevir y el telaprevir – la primera generación de inhibidores selectivos y reversibles de la proteasa NS3 – para el tratamiento de pacientes infectados por VHC de tipo 1, tanto no tratados previamente (naïve) como tratados, en combinación con peginterferón alfa y ribavirina (terapia triple). En estas circunstancias, las tasas de respuesta tanto en pacientes naïve como en pretratados llegaban a doblarse prácticamente (de un 40% a un 70%, en término medio), permitiendo acortar la duración del tratamiento en muchos de los pacientes de 48 a 24 semanas. Sin embargo, boceprevir y telaprevir presentan un perfile toxicológico importante que obliga a suspender el tratamiento en un porcentaje de pacientes netamente superior a los tratados solo con peginterferón alfa y ribavirina, amén de un amplio abanico de interacciones farmacológicas, lo cual, asociado con la propia complejidad del tratamiento, dejaba un amplio margen para la mejora.
{kind=link}
Tras esta primera generación de inhibidores de la proteasa del VHC ha llegado una nueva oleada de agentes con propiedades farmacodinámicas, farmacocinéticas y toxicológicas más satisfactorias, que está en la fase final del proceso de autorización o ya la han recibido. Se trata del asunaprevir, inhibidor de la NS3); simeprevir, faldaprevir y vaniprevir, inhibidores duales de NS3 y NS4A; daclatasvir y ledipasvir, de la NS5A; y sofosbuvir y deleobuvir, de la NS5B (Cuéllar, 2014).
ACCIÓN Y MECANISMO
El daclatasvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5A, una proteína con diversas funciones relacionadas con la replicación del VHC y el ensamblaje de los viriones. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos.
Daclatasvir es el primero de la clase de los inhibidores de NS5A y el inhibidor de VHC más potente hasta ahora conocido. El fármaco se une selectivamente en concentraciones picomolares a la proteína NS5A, alterando su localización subcelular, sus procesos de hiperfosforilación e inhibiendo la síntesis de ARN viral. La proteína NS5A es una proteína intensamente fosforilada que, una vez escindida de la poliproteína viral, localiza a las membranas donde se une a la fracción 3’-terminal del ARN viral recién sintetizado y participa en la replicación del genoma viral, en parte a través de interacciones con la ARN polimerasa dependiente del ARN viral (NS5B).
A pesar de su extraordinaria actividad frente al VHC, que se manifiesta en prácticamente los genotipos virales más comunes (1a, 1b, 2a, 2b, 3a, 4a, 5a y 6a), el daclatasvir tiene una baja barrera de resistencia a las mutaciones para algunos genotipos, particularmente para 1a, 2a, 2b y 3a, mientras que dicha barrera es bastante más elevada para los genotipos 1b, y 4a. El grado de susceptibilidad (expresado como EC501) de las cepas salvajes (no mutadas) oscila entre 0,003 nM (1b) y 0,26 (3a), lo que implica una diferencia de casi 100 veces en potencia antiviral.
La mayoría las mutaciones más resistentes en la NS5A del VHC de genotipo 1a se deben a la sustitución de un solo aminoácido, siendo una de las más peculiares la Q30E (sustitución de glutamina por ácido glutámico en la posición 30), lo que eleva la EC50 a 111 nM, 18.500 veces mayor que en el tipo salvaje (0,006 nM). Otras mutaciones únicas que confieren resistencia a daclatasvir en genotipo 1a son M28T, Q30E, Q30H, Q30R, Q30S, L31M, L31V, Y93C y Y93H, con valores de EC50 entre 1 y 111 nM; algunas mutaciones dobles, como Q30R-L31M y Q30H-Y93H también producen altos niveles de resistencia a al genotipo 1a (EC50 410-868 nM); por el contrario, estas mutaciones – salvo la Q30H-Y93H – no afectan sustancialmente a la sensibilidad al daclatasvir por los VHC de genotipo 1b. La mutación L31M reduce la susceptibilidad al daclatasvir de los genotipos 2a y 2b en 440 y 2.600 veces, respectivamente. La frecuencia de cepas mutadas encontradas en pacientes no tratados previamente oscila entre el 1% y el 7% para el genotipo 1a, llegando al 60% en los genotipos 2.
ASPECTOS MOLECULARES
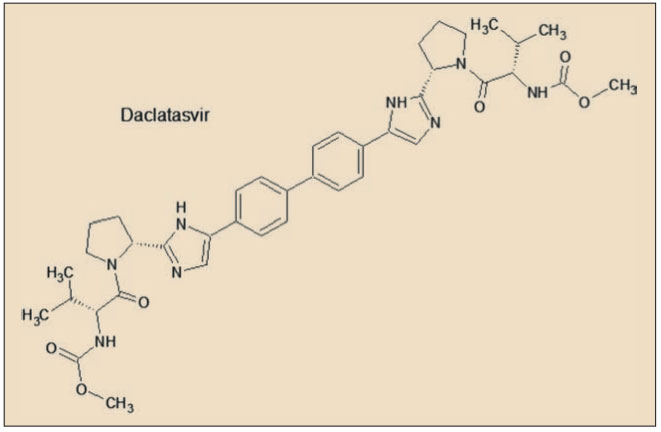
Daclatasvir presenta una característica estructura simétrica, en la que destacan básicamente dos aspectos fundamentales. En primer lugar, el núcleo central de la molécula está formado por un grupo bifenilo ligado por sus dos extremos a sendos anillos imidazólicos, formando los cuatro anillos aromáticos un amplio sistema resonante, de alta densidad electrónica, el cual facilita la unión a la proteína NS5A, estabilizando su estructura y bloqueando sus funciones. En segundo lugar, se aprecian sendas estructuras en los extremos de la molécula que emulan secuencias peptídicas (son, en realidad, carbamatos), que facilitan la inserción del fármaco en los huecos (bolsillos) de la proteína.
A diferencia de las proteínas NS3 (inhibidas por simeprevir, telaprevir y boceprevir) y NS5B (sofosbuvir), no se ha identificado ninguna función enzimática específica hasta el momento para la proteína NS5A, aunque es crucial en la producción de virus y se ha demostrado que está implicada en la modulación de la respuesta inmune del huésped y en la patogenicidad y la replicación del VHC. La proteína NS5A del VHC tiene 447 aminoácidos y su forma activa es como un homodímero que se organiza en tres ámbitos diferentes, de los cuales el dominio I es el más conservado y con una estructura más compleja. La NS5A puede existir en estado fosforilado o hiperfosforilado.
Daclatasvir tiene un mecanismo único, con un sitio de unión simétrico. Los anillos de bifenilo descansan en la parte superior de los dos grupos metilo del aminoácido Thr(T)95 en los dos monómeros de la NS5A. Los anillos de los dos residuos Tyr(Y)93 extienden el canal hidrófobo para acomodar el resto del grupo bifenilo y uno de los anillos laterales del daclatasvir. Las cadenas de Lys(K)26, Ile(I)27, y Ile(I)28 de las dos α-hélices y Pro(P)97 de cada monómero completan las esquinas hidrófobas, permitiendo que el fármaco se ajuste fácilmente en la ranura hidrófoba formada en la esquina de los bolsillos de la proteína NS5A. Los restos Arg(R)30 y Gln(Q)54 forman cuatro enlaces de hidrógeno simétricos con daclatasvir (Barakat, 2014).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad del daclatasvir en la indicación autorizada han sido contrastadas mediante dos ensayos clínicos principales de fase III, tanto en pacientes naïve (no tratados previamente) como en aquellos pretratados. En todos los casos, la variable primaria de eficacia utilizada fue la tasa de pacientes con respuesta virológica sostenida, es decir, aquellos con niveles séricos de menos de 25 copias/ml de ARN del VHC, medidos 12 semanas después de haber finalizado el tratamiento completo antiviral (RVS12). La dosis empleadas fueron de 60 mg/24 h de daclatasvir, 400 mg/24 h de sofosbuvir, 500 o 600 mg/12 h (según peso) de ribabvirina, en todos los casos por vía oral, y de la peginterferón alfa-2a fue de 180 µg/semana, por vía subcutánea.
El estudio principal (AI444040; Sulkowski, 2014) es un ensayo clínico abierto y multicéntrico realizado sobre 211 pacientes en total, en el que se incluyeron pacientes con infección por VHC de genotipo 1a/1b no tratados previamente (naïve, 126 pacientes) o pretratados y con fallo virológico a boceprevir/telaprevir (41 pacientes), así como pacientes naïve con VHC de genotipo 2/3 (44 pacientes).
Los pacientes naïve con VHC de genotipo 1a/1b fueron aleatoriamente asignados a un tratamiento de 12 semanas con daclatasvir y sofosbuvir, con o sin ribavirina (DAC/SOF/RBV12, DAC/SOF12) o de 24 semanas (DAC/SOF/RBV24, DAC/SOF24); los pacientes pretratados con boceprevir/telaprevir y con fallo virológico y los pacientes naïve con VHC de genotipo 2/3 fueron tratados durante 24 semanas (DAC/SOF/RBV24, DAC/SOF24).
En los pacientes naïve con VHC de genotipo 1a/1b (122), la mediana de edad era de 54-56 años, un 51% eran varones y un 80% eran de raza blanca; la mediana de la carga viral era de 6,1-6,8 log10 (1,3-6,3 millones de copias de ARN por ml). Un 79% presentaban VHC de genotipo 1a y el 21% restante 1b. La tasa global de respuesta (RVS12) fue del 98,2% con DAC/SOF/RBV (con ribavirina) y del 100% con DAC/SOF (sin ribavirina). Las tasas fueron del 100% en ambos grupos de 24 semanas de duración (DAC/SOF/RBV24 y DAC/SOF24), del 100% con DAC/SOF12 y del 95,1% con DAC/SOF/RBV12.
En los pacientes con VHC de genotipo 1a/1b pretratados y con fallo virológico al boceprevir/telaprevir (41) la mediana de edad era de 57-59 años, un 61% eran varones y un 90% eran de raza blanca; la mediana de la carga viral era de 6,3-6,4 log10 (2,0-2,5 millones de copias de ARN por ml). Un 80% presentaban VHC de genotipo 1a y el 20% restante 1b. La tasa global de respuesta fue del 98% tanto con DAC/SOF/RBV24 (con ribavirina) como DAC/SOF24 (sin ribavirina).
Finalmente, en los pacientes con VHC de genotipo 2/3 naïve (44) la mediana de edad era de 50-52 años, un 50% eran varones y un 86% eran de raza blanca; la mediana de la carga viral era de 6,7-6,9 log10 (5,0-7,9 millones de copias de ARN por ml). Un 59% presentaban VHC de genotipo 2 y el 41% restante 3. La tasa global de respuesta fue del 93,2%, un 92,9% con DAC/SOF/RBV24 (con ribavirina) y un 93,3% con DAC/SOF24 (sin ribavirina). Considerando específicamente los genotipos virales, la tasa de respuesta viral sostenida fue del 96,2% para el genotipo 2 y del 88,9% para el 3.
El estudio AI444042 es un ensayo clínico abierto y multicéntrico realizado sobre 125 pacientes naïve con infección por VHC de genotipo 4, asignados aleatoriamente a recibir peginterferón alfa y ribavirina (IFN/RBV), junto con daclatasvir (60 mg/24 h) o placebo. En aquellos pacientes que mostraron niveles indetectables de ARN viral, se completó el tratamiento hasta la semana 24, mientras que los restantes siguieron recibiendo la combinación IFN/RBV hasta la semana 48. La mediana de edad de los pacientes era de 48 años, un 73% eran varones y un 77% eran de raza blanca; la mediana de la carga viral era de 5,7 log10 (0,5 millones de copias de ARN por ml). La la tasa de respuesta viral sostenida fue del 81,7% con daclatasvir vs. 42,9% con placebo.
Los datos de seguridad clínica del daclatasvir muestran un perfil toxicológico benigno, sin efectos adversos específicos, que motiva la suspensión del tratamiento solo en el 1-2% de los casos. En el ensayo clínico principal ninguno de los pacientes experimentó ningún evento adverso de grado 3/4 relacionado con el tratamiento, a pesar de recibir también sofosbuvir y, en algunos pacientes, ribavirina. Los eventos adversos más comunes fueron fatiga (31,7% para DAC/SOF/RBV12 vs. 30,6% para DAC/SOF/RBV24, y 31,7% para DAC/SOF12 vs. 25,0% para DAC/SOF24), cefalea (14,6% vs. 26,5% y 14,6% vs. 18,8%) y náusea (12,2% vs. 20,4% y 17,1% vs. 10,0%).
ASPECTOS INNOVADORES
El daclatasvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5A, una proteína con diversas funciones relacionadas con la replicación del VHC y el ensamblaje de los viriones. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos.
La eficacia de la combinación con sofosbuvir ha sido claramente demostrada en pacientes con genotipo viral 1, 2, 3 y 4, con tasas de respuesta viral sostenida por encima del 90%. En el caso de los genotipos 1a y 1b también se ha demostrado esta misma elevada tasa de respuesta en pacientes que habían experimentado un fallo viral previo a boceprevir o telaprevir. Presenta un perfil de seguridad muy benigno, con efectos adversos inespecíficos (fatiga, cefalea, náusea) y poco intensos.
Se trata de un nuevo agente activo frente al VHC que presenta un mecanismo de acción diferente de los fármacos hasta ahora utilizados y su potencia antiviral es, sin duda, muy elevada, actuando – al menos in vitro – sobre todos los genotipos de VHC. No obstante, el daclatasvir tiene una baja barrera de resistencia a las mutaciones para algunos genotipos, particularmente para 1a, 2a, 2b y 3a. Aunque actualmente está siendo objeto de varios ensayos clínicos (16 en la Unión Europea; EMA, 2015), los datos clínicos disponibles hasta ahora para el daclatasvir no son equiparables en cantidad ni en diversidad a los de simeprevir y, especialmente, de sofosbuvir; en este sentido, es particularmente relevante la muy escasa experiencia clínica actual en pacientes con cirrosis o enfermedad hepática avanzada.
La última versión de la Guía de Práctica Clínica de la Asociación Europea para el Estudio del Hígado (EASL, 2014) establece como opciones preferentes de tratamiento de la hepatitis C crónica producida por VHC de genotipo 1a y 1b las combinaciones de sofosbuvir o de simeprevir con peginterferon alfa + ribavirina. Como alternativas recomienda daclatasvir junto con peginterferon + ribavirina, sofosbuvir + simeprevir durante 12 semanas o sofosbuvir + daclatasvir durante 12 o 24 semanas, según las condiciones de los pacientes. Para el genotipo 2 se recomienda una combinación de ribavirina y sofosbuvir durante 12 semanas (16-20 en casos de pacientes cirróticos). Para el genotipo 3, la combinación es de sofosbuvir con peginterferon alfa-2a y ribavirina durante 12 semanas, y como alternativas ribavirina y sofosbuvir durante 12 semanas (no aconsejable en pacientes cirróticos) o sofosbuvir y daclastavir durante 12 (naïve) o 24 semanas (pretratados). Para los cuadros asociados al genotipo 4, se aconseja el simeprevir durante 12 semanas asociados a peginterferón y ribavirina durante 24 o 48 semanas (naïve o pretratados), citándose como alternativas peginterferón más ribavirina y daclastavir durante 24 semanas o sofobuvir y ribavirina durante 24 semanas.
Tras la primera oleada de antivirales específicos frente al VHC, con boceprevir y telaprevir, que ciertamente modificaron en 2011 el pronóstico y las expectativas terapéuticas en este campo, el daclatasvir forma parte de una segunda oleada – junto con simeprevir y sofosbuvir, incorporados en 2014 – que completa el perfil de mecanismos farmacológicos relacionados con el bloqueo de la proteasa del VHC. Estos tres fármacos, con el sofosbuvir a la cabeza como referente, presentan elevadas tasas de curación en diversos subtipos de pacientes, a través de regímenes orales sencillos y bien tolerados, con la particularidad de que permiten eliminar en algunos casos la otrora imprescindible presencia del interferón alfa e incluso de la ribavirina en el tratamiento, origen de importantes y frecuentes efectos adversos, amén de acortar sustancialmente la duración del tratamiento y con ello facilitar la adherencia al mismo y su tolerabilidad.
A esta tríada de nuevos inhibidores de la proteasa del VHC habrá que añadir próximamente otros fármacos, como ledipasvir o asunaprevir, pero ya no aportarán nuevos mecanismos y es poco probable que se mejoren sustancialmente la tasa de respuesta viral (ya muy elevada); también es inminente la autorización de combinaciones (combos) de varios antivirales en formulaciones únicas, al estilo de las existentes para el tratamiento de la infección por VIH.
|
VALORACIÓN |
|
|---|---|
|
Daclatasvir
► DAKLINZA®
(Bristol-Myers Squibb) |
|
|
Grupo Terapéutico (ATC): J05AE. ANTIINFECCIOSOS SISTÉMICOS. Antivirales de acción directa: inhibidores de la proteasa. |
|
|
Indicaciones autorizadas: Tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar. |
♣ ♣ ♣ |
|
Novedad clínica: Alta tasa de eficacia, posibilidad de asociar con otros tratamientos actualmente en vigor y utilidad en cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
⇑ |
|
Novedad molecular: Mecanismo de acción innovador |
⇑ |
|
Novedad toxicológica: Mejora el perfil toxicológico con relación a la terapia farmacológica estándar. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Boceprevir |
Victrelis |
Merck Sharp Dohme |
2011 |
|
Telaprevir |
Incivo |
Janssen Cilag |
2011 |
|
Simeprevir |
Olysio |
Janssen Cilag |
2014 |
|
Sofosbuvir |
Sovaldi |
Gilead |
2014 |
|
Daclatasvir |
Daklinza |
Bristol-Myers Squibb |
2015 |
BIBLIOGRAFÍA
Fumarato de dimetilo TECFIDERA® (Biogen Idec)
Resumen
El fumarato de dimetilo es un agente antiinflamatorio, inmunomodulador y neuroprotector, que ha sido autorizado para el tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. Desde el punto de vista de la seguridad, no parece que la toxicidad sea un problema grave, aunque la frecuencia de eventos adversos puede ser relativamente alta. Los más relevantes son sofocos, diarrea, dolor abdominal y exantema, con una incidencia ligeramente mayor que la observada con glatirámero. Se trata de una nueva opción terapéutica a considerar en el tratamiento de la forma remitente-recurrente de la esclerosis múltiple. Su mecanismo de acción es complejo y todavía no bien comprendido, aunque los datos clínicos resultan contundentes y la mejora clínica ha sido objetivamente contrastada, aunque está por ver en qué medida podría afectar a la progresión de la enfermedad.
ACCIÓN Y MECANISMO
El fumarato de dimetilo es un agente antiinflamatorio, inmunomodulador y neuroprotector, que ha sido autorizado para el tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. En realidad, se trata de un profármaco que es hidrolizado en el interior celular, dando lugar al monometiléster (fumarato ácido de metilo) y al ácido fumárico libre, aunque se desconoce en qué grado contribuye cada uno de estas sustancias a la respuesta farmacológica.
En general, el fumarato de dimetilo activa el sistema del factor nuclear eritroide-2 Nrf2, que representa una de las principales vías intracelulares protectoras frente al daño oxidativo. Las especies reactivas de oxígeno (reactive oxygen species, ROS) son radicales libres y peróxidos que presentan electrones no apareados y, por ello, son químicamente muy reactivos, siendo capaces de oxidar a numeras sustancias, tanto orgánicas como inorgánicas. Las ROS se forman espontáneamente como subproductos normales del metabolismo oxidativo y ejercen papeles decisivos en la señalización bioquímica celular. Sin embargo, cuando se generan en exceso o no son neutralizadas adecuadamente por los sistemas antioxidantes del propio organismo, pueden provocar daños sustanciales a las estructuras celulares en lo que se conoce como estrés oxidativo. De hecho, las ROS parecen estar íntimamente implicadas en la fisiopatología de la esclerosis múltiple, actuando como promotores de la migración de los leucocitos a través del endotelio vascular, responsables últimos del daño axonal y de la mielina de las vías nerviosas en la esclerosis múltiple. En este sentido, el fumarato de dimetilo ha demostrado tener un efecto protector neto sobre las neuronas y oligodendrocitos en la esclerosis múltiple (di Nuzzo, 2014).
El factor nuclear eritroide-2 o Nfr2 regula la expresión inducible de numerosos genes de enzimas destoxificantes y antioxidantes, mediante su unión a una secuencia específica del ADN (ARE, Antioxidant Response Element), que puede ser activada por diversos compuestos oxidantes y/o electofílicos. La actividad del factor Nrf2 se encuentra constitutivamente reprimida debido a su unión con un dímero de la proteína citoplásmica Keap1 (Kelch-like ECH-associated protein 1) y al citoesqueleto. Dicha unión fomenta la permanente degradación de Nrf2 por el sistema ubiquitina-proteosoma, por lo que el control primario de su función radica principalmente en su distribución subcelular, más que en la síntesis de novo. La activación de Nrf2 conduce a la inducción de genes que codifican enzimas que destruyen las ROS o que aumentan los niveles de glutatión, tales como la NAD(P)H:quinona oxidorreductasa, la tioredoxina y la hemo oxigenasa 1, todas las cuales resultan fundamentales para los mecanismos preventivos del daño oxidativo.
El fumarato de dimetilo activa la vía de Nrf2 mediante una depleción inicial de los depósitos fisiológicos de glutatión, pero sin llegar a alcanzar el límite que activaría los mecanismos celulares de apoptosis (muerte celular programada). Este mecanismo, conocido como hormesis, es similar a otros implicados en sistemas defensivos como el de tolerancia a la isquemia, a través del cual el cerebro se protege de los efectos negativos de la isquemia cerebral mediante mínimos efecto isquémicos, que permiten acondicionar el cerebro y prevenir un mayor daño neuronal.
Los astrocitos y la microglía, igualmente implicados en la cascada neuroinflamatoria de la esclerosis múltiple, son también objetivo del fumarato de dimetilo a través de la activación de la vía Nrf2 y de la inhibición del Factor Nuclear potenciador de las cadenas ligeras Kappa de las células B activadas (NF-kB). Esto último es asociado con la inducción del hemo oxigenasa-1, la activación de la síntesis de glutatión y la inhibición de la producción de citocinas proinflamatorias.
El fumarato de dimetilo inhibe la maduración de las células dendríticas y la diferenciación de los linfocitos T en las formas autorreactivas TH1 y TH17, implicadas en la respuesta inmune adaptativa que participa de forma relevante en la patogénesis de la esclerosis múltiple. En su lugar, el fármaco facilita la formación de células dendríticas de tipo II, que producen interleucina 10 (IL-10) en lugar de IL-12 e IL-23, y linfocitos TH2 (productores de IL-4) en lugar de los TH1 y TH17.
ASPECTOS MOLECULARES
{kind=link}
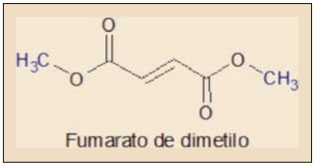
La acción farmacológica del fumarato de dimetilo (trans-1,2-etilendicarboxilato de metilo) se debe fundamentalmente a sus propiedades químicas de tipo electrofílico, siendo responsable de la activación del sistema Nfr2. En efecto, el ácido fumárico y sus ésteres constituyen un sistema α,β-insaturado – es decir, conjugado – con la reactividad característica de estas sustancias, entre las que cabe destacar las reacciones de tipo Diels-Alder, susceptibles de producir modificaciones moleculares múltiples1.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del fumarato de dimetilo en la indicación autorizada han sido contrastadas mediantes dos ensayos clínicos principales de fase III, multicéntricos, doblemente ciegos, aleatorizados y controlados con placebo (DEFINE) o con placebo y acetato de glatirámero (CONFIRM), realizados en pacientes con esclerosis múltiple remitente recurrente, utilizando dosis orales de fumarato de dimetilo de 240 mg/12 h o 240 mg/8 h, vs. placebo o acetato de glatirámero (20 mg/24 h, SC), a lo largo de 96 semanas (2 años).
El primero de estos estudios (DEFINE; Gold, 2012) se llevó a cabo sobre un conjunto de 1.234 pacientes mayores de edad (18-55 años) con diagnóstico confirmado de esclerosis múltiple remitente recurrente con un nivel basal de discapacidad de ≤5 puntos en la escala EDSS (Expanded Disability Status Scale), con al menos una recaída en los últimos doce meses y diagnóstico por imagen (RMN) positivo mostrando lesiones cerebrales congruentes con la esclerosis múltiple en los últimos seis meses. Los pacientes tenían una mediana de edad de 39 años (52% <40 años), un 74% eran mujeres y un 79% eran de raza blanca, con una mediana de peso de 68 kg. La mediana de tiempo desde los primeros síntomas de esclerosis era de 7,0 años y desde el diagnóstico de 4,0, con una mediana de 2 recaídas en los tres últimos años (al menos una de ellas en el último) y con una puntuación mediana de 2,0 en la escala EDSS. Un 55% de los pacientes habían recibido anteriormente tratamiento (interferón beta 1A, 27%; interferón beta 1B, 14%; glatirámero, 15%; natalizumab, 3%; corticosteroides, 24%; etc.).
La variable primaria de eficacia utilizada fue la proporción de pacientes que tuvieron una recaída confirmada por un comité independiente en los dos años de estudio. Como variables secundarias se determinaron las tasas anualizadas de recaídas, el tiempo medio hasta progresión confirmada de la discapacidad (al menos 1 punto en la escala EDSS durante al menos tres meses) y las variaciones experimentadas en las imágenes de RMN, particularmente de nuevas lesiones T2 hipertensas o un agrandamiento de las anteriores.
Los resultados mostraron que el porcentaje de pacientes que habían experimentado al menos un recaída en los dos años de estudios fue del 24% con 240 mg dos veces al día (cada 12 h) de fumarato de dimetilo (FDM2), 23% con FDM3 y 42% con placebo, lo que implica una reducción del riesgo del 49% con FDM2 (hazard risk, HR=0,51; IC95% 0,40 a 0,66; p<0,0001) y del 50% con FDM3 (HR=0,50; IC95% 0,39 a 0,65; p<0,0001), con respecto al placebo. Las tasas anualizadas de recaídas fueron de 0,172 (IC95% 0,138 a 0,214) con FDM2, de 0,189 (IC95% 0,153 a 0,231) con FDM3 y de 0,364 (IC95% 0,303 a 0,436) con placebo, lo que implica unas reducciones del 53% (HR=0,47; IC95% 0,37 a 0,61; p<0,0001) con FDM2 y del 48% (HR=0,52; IC95% 0,40 a 0,67; p<0,0001) con FDM3, en relación al placebo.
Asimismo, la proporción de pacientes que experimentaron progresión de la discapacidad en los dos años fueron del 16,4% con FDM2, del 17,7% con FDM3 y del 27,1% con placebo, lo que implica una reducción del riesgo de progresión del 38% (HR=0,62; IC95% 0,44 a 0,87; p=0,005) con FDM2 y del 34% (HR=0,66; IC95% 0,48 a 0,92; p=0,0128) con FDM3, con respecto al placebo. Los valores medios de nuevas lesiones (o aumento del tamaño de antiguas) determinadas por RMN fue de 2,6 con FDM2, 4,4 con FDM3 y 17,0 con placebo, lo que supone unas reducciones del 85% (HR=0,15; IC95% 0,10 a 0,23; p<0,0001) con FDM2 y del 74% (HR=0,26; IC95% 0,17 a 0,38; p<0,0001) con FDM3, en relación al placebo.
En general, los efectos favorables del tratamiento en relación al placebo fueron demostrados en cada uno los subgrupos de pacientes analizados (edad, sexo, tamaño y número de las lesiones cerebrales, etc.) comprobándose que las reducciones del riesgo de recaída oscilaron entre un 68% y un 26% con FDM2 y del 66% al 25% con FDM3 (Bar-Or, 2013). Asimismo, se registró una mejora significativa en las puntuaciones de calidad de vida, utilizando el cuestionario breve SF-36 (Kappos, 2014).
El segundo estudio (CONFIRM; Fox, 2012) comparó el fumarato de dimetilo en dos difentes psoologías (FDM2 y FDM3) con placebo y con glatirámero, aunque el estudio no fue diseñado para ensayar la superioridad o no inferioridad del fumarato de dimetilo sobre el glatirámero. Fue desarrollado en 1.417 pacientes adultos con una mediana de edad de 37 años (59% <40 años), un 70% eran mujeres y un 84% eran de raza blanca, con una mediana de peso de 69 kg. La mediana de tiempo desde los primeros síntomas de esclerosis era de 7,1 años y desde el diagnóstico de 3,0, con una mediana de 2 recaídas en los tres últimos años (al menos una de ellas en el último) y con una puntuación mediana de 2,5 en la escala EDSS. Un 55% de los pacientes habían recibido anteriormente tratamiento (interferón beta 1A, 21%; interferón beta 1B, 11%; natalizumab, 1%; corticosteroides, 15%; etc.), pero ninguna glatirámero.
La variable primaria de eficacia utilizada fue la tasa anualizadas de recaídas y como variables secundarias se determinaron las variaciones experimentadas en las imágenes de RMN, particularmente de nuevas lesiones T2 hipertensas o un agrandamiento de las anteriores, la proporción de pacientes que tuvieron una recaída confirmada en los dos años de estudio y la progresión de la discapacidad.
Los resultados mostraron que las tasas anualizadas de recaídas fueron de 0,224 (IC95% 0,179 a 0,282) con FDM2, de 0,198 (IC95% 0,156 a 0,252) con FDM3, de 0,286 (IC95% 0,232 a 0,353) con glatirámero (GLA) y de 0,401 (IC95% 0,329 a 0,488) con placebo, lo que implica unas reducciones del 44,0% (HR=0,560; IC95% 0,423 a 0,740; p<0,0001) con FDM2, del 50,5% (HR=0,495; IC95% 0,369 a 0,662; p<0,0001) con FDM3 y del 28,6% (HR=0,714; IC95% 0,548 a 0,931; p=0,0128) con GLA, en relación al placebo.
El porcentaje de pacientes que habían experimentado al menos un recaída en los dos años de estudios fue del 26% con FDM2, del 22% con FDM3, del 30% con GLA y del 39% con placebo, lo que implica una reducción del riesgo del 34,0% con FDM2 (HR=0,66; IC95% 0,51 a 0,86; p=0,002), del 44,6% con FDM3 (HR=0,55; IC95% 0,41 a 0,73; p<0,0001) y del 28,6% con GLA (HR=0,71; IC95% 0,55 a 0,92; p=0,0097), con respecto al placebo.
Por otro lado, la proporción de pacientes que experimentaron progresión de la discapacidad en los dos años fueron del 12,8% con FDM2, del 13,0% con FDM3, del 15,6% con GLA y del 16,9% con placebo, lo que implica una reducción del riesgo de progresión del 21,4% (HR=0,79; IC95% 0,52 a 1,19; p=0,2536) con FDM2, del 23,8% (HR=0,76; IC95% 0,50 a 1,16; p=0,2041) con FDM2 y del 7,3% (HR=0,93; IC95% 0,63 a 1,37; p=0,7036) con GLA, con respecto al placebo.
Los valores medios de nuevas lesiones (o aumento del tamaño de antiguas) determinadas por RMN fue de 5,7 con FDM2, 5,1 con FDM3, 5,1 con GLA y 19,9 con placebo, lo que supone unas reducciones del 71% (HR=0,29; IC95% 0,21 a 0,41; p<0,0001) con FDM2, del 73% (HR=0,27; IC95% 0,20 a 0,38; p<0,0001) con FDM3 y del 54% (HR=0,46; IC95% 0,33 a 0,63; p<0,0001) con GLA, en relación al placebo.
Como en el estudio anterior, los efectos favorables del tratamiento en relación al placebo fueron demostrados en cada uno los subgrupos de pacientes analizados (edad, sexo, tamaño y número de las lesiones cerebrales, etc.) comprobándose que las reducciones del riesgo de recaída oscilaron entre un 53% y un 34% con FDM2 y del 67% al 13% con FDM3 (Hutchinson, 2013). También se registró una mejora significativa en las puntuaciones de calidad de vida, utilizando el cuestionario breve SF-36 (Kita, 2014).
Tal como se indicó, este estudio no fue diseñado para determinar la posible superioridad o no inferioridad del fumarato de dimetilo (FDM2 y FDM3) frente a glatirámero (GLA), aunque los datos comparativos directos muestran una reducción de la tasa de anualizada de recaídas del 22% favorable a FDM2 (HR=0,78; IC95% 0,59 a 1,05; no significativa) y del 31% favorable a FDM3 (HR=0,69; IC95% 0,51 a 0,94), una reducción del número nuevas lesiones o agrandamiento de estas del 36% favorable a FDM2 (HR=0,64; IC95% 0,46 a 0,88) y del 41% favorable a FDM3 (HR=0,59; IC95% 0,43 a 0,82).
La reducción en la proporción de pacientes que recayeron fue de 8% favorable a FDM2 (HR=0,92; IC95% 0,70 a 1,22) y del 22% favorable a FDM3 (HR=0,78; IC95% 0,58 a 1,04), mientras que la reducción de la progresión de la discapacidad fue del 15% favorable a FDM2 (HR=0,85; IC95% 0,56 a 1,29) y del 18% favorable a FDM3 (HR=0,82; IC95% 0,54 a 1,26). Las diferencias entre fumarato de dimetilo y glatirámero en estas dos variables no fueron estadísticamente significativas.
Desde el punto de vista de la seguridad clínica, el perfil toxicológico del fumarato de dimetilo es amplio, aunque no parece revestir una especial gravedad en relación al placebo y al glatirámero. Los eventos adversos más frecuentemente descritos en los dos ensayos clínicos anteriores, con una diferencia de al menos 3 puntos porcentuales con respecto al placebo son: sofocos (34% FDM2, 29% FDM3, 2% GAL y 5% Placebo), diarrea (14%, 17%, 4% y 10%), náusea (12%, 14%, 5% y 9%), dolor abdominal (9%, 8%, 1% y 4%), exantema (8%, 7%, 3% y 3%) y rubefacción (7%, 7%, 1% y 2%). El porcentaje de pacientes que suspendieron el tratamiento por este motivo fue del 8% con fumarato de dimetilo, 3% con glatirámero y 4% con placebo.
ASPECTOS INNOVADORES
El fumarato de dimetilo es un agente antiinflamatorio, inmunomodulador y neuroprotector, que ha sido autorizado para el tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. Su eficacia y la seguridad clínicas han sido contrastadas mediantes dos amplios ensayos clínicos principales de fase III, multicéntricos, doblemente ciegos, aleatorizados y controlados con placebo y glatirámero, utilizando dosis orales de fumarato de dimetilo de 240 mg/12 h o 240 mg/8 h, a lo largo de dos años.
Los resultados de dichos ensayos clínicos son robustos y concluyentes, encontrándose unas tasas de reducción con respecto al placebo del 44-50% en el porcentaje de pacientes que experimentaron alguna recaída, en el 21-38% de aquellos que experimentaron progresión en la discapacidad neurológicas asociada a la esclerosis y del 71-85% en el número de nuevas lesiones neurológicas o agrandamiento de las antiguas. Esta diferencia se mantiene en todos los subgrupos analizados (sexo, edad, tamaño y número de las lesiones neurológicas, discapacidad basal, etc.).
Los datos comparativos con glatirámero muestran una tendencia a la superioridad favorable para el fumarato de dimetilo, pero las diferencias no llegaron a ser significativas estadísticamente. En cualquier caso, el estudio comparativo no fue específicamente diseñado para mostrar superioridad o no inferioridad del fumarato de dimetilo frente al glatirámero, sino frente al placebo.
Desde el punto de vista de la seguridad, no parece que la toxicidad sea un problema grave para el fumarato de dimetilo, aunque la frecuencia de eventos adversos puede ser relativamente alta. Los más relevantes son sofocos, diarrea, dolor abdominal y exantema, con una incidencia ligeramente mayor que la observada con glatirámero.
En definitiva, una nueva opción terapéutica a considerar en el tratamiento de la forma remitente-recurrente de la esclerosis múltiple (la más común). Su mecanismo de acción es complejo y todavía no bien comprendido, aunque los datos clínicos resultan contundentes y la mejora clínica ha sido objetivamente contrastada, está por ver en qué medida podría afectar a la progresión de la enfermedad. Actualmente, hay diversas opciones que dan lugar a respuestas terapéuticas significativas y que tienen una amplia experiencia clínica; por ello, se ha sugerido que en los pacientes recientemente diagnosticados los tratamientos inyectables como el interferón beta (1a y 1b) o el acetato de glatirámero irán siendo progresivamente reemplazados por los nuevos tratamientos orales, como el fumarato de dimetilo o la teriflunomida, más cómodos de manejar. No obstante, no parece que haya justificación para cambiar el tratamiento en aquellos pacientes que venían siendo tratados con los fármacos inyectables con una buena tasa de respuesta y un perfil aceptable de efectos adversos (Sorensen, 2014).
|
VALORACIÓN |
|
|---|---|
|
Fumarato de dimetilo ► TECFIDERA® (Biogen Idec) |
|
|
Grupo Terapéutico (ATC): N07XX. SISTEMA NERVIOSO. Otros fármacos. |
|
|
Indicaciones autorizadas: Tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |
♣ ♣ |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma indicación terapéutica. |
⇑ |
|
Novedad físico-química: Vía de administración más cómoda para el paciente. |
⇑ |
BIBLIOGRAFÍA
Teriflunomida AUBAGIO® (Sanofi Aventis)
Resumen
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente. Se trata del estereoisómero Z del principal metabolito activo de la leflunomida, utilizada para el tratamiento de la artritis reumatoide y de la artritis psoriásica. Los datos clínicos disponibles muestran un nivel de eficacia modesto aunque clínicamente relevante, con diferencias con respecto al placebo similares a las observadas con otros agentes (glatirámero, interferón beta, etc.), con una reducción en torno al 30% del riesgo de progresión. Por otro lado, la mayor parte de los pacientes que participaron en los ensayos clínicos presentan un bajo nivel de afectación (EDSS 2,0-2,5; sobre un nivel máximo de 10). Desde el punto de vista de la seguridad clínica, la toxicidad no parece ser un problema importante, aunque son comunes los trastornos de naturaleza hepática y digestiva. El mecanismo inmunológico es ciertamente poco específico, pero eso mismo puede achacarse a la mayoría de sus teóricos competidores. Como una potencial – y modesta – ventaja frente a estos, podría mencionarse su cómoda administración oral, una vez al día; pero nada más.
ESCLEROSIS MÚLTIPLE
La esclerosis múltiple es una enfermedad inflamatoria crónica autoinmune del sistema nervioso central, habitualmente de carácter lentamente progresivo, caracterizada por la presencia de múltiples placas diseminadas de desmielinización, distribuidas a lo largo del cerebro y la médula espinal. La enfermedad se caracteriza por múltiples y variados síntomas y signos de disfunción del sistema nervioso central, con remisiones y exacerbaciones recurrentes. En general, los que suelen manifestarse más precozmente son parestesias en una o más extremidades, el tronco o un lado de la cara; debilidad o torpeza de un miembro inferior o la mano; o trastornos visuales; debilidad muscular o fatiga inusual de un miembro, alteraciones leves de la marcha, dificultad en el control vesical, vértigo y trastornos emocionales leves. El curso habitual de la enfermedad se caracteriza por la presencia de remisiones y recaídas, con alguna discapacidad acumulativa, y aunque dicha discapacidad física puede aparecer desde el inicio de la enfermedad, lo más frecuente que comience a manifestarse tras varios años de evolución.
También es denominada como esclerosis en placas o diseminada. Las placas son en realidad lesiones formadas por infiltrados de células, con desmielinización y gliosis (destrucción de células gliales, que constituyen el elemento fundamental del tejido neurológico de sostén). La enfermedad afecta especialmente a adultos jóvenes, en los que produce manifestaciones clínicas muy diversas. Además, tiene un curso variable, observándose frecuentemente brotes y periodos de remisión. Es considerada como la causa de tipo no traumático más frecuente de invalidez o discapacidad neurológica del adulto joven en el mundo occidental.
El comienzo de la enfermedad sucede habitualmente entre los 16 y 50 años de edad, particularmente entre los 20 y los 40, y es dos veces más común en las mujeres, en las cuales suele comenzar más temprano. Es muy rara su aparición antes de los 10 años o después de los 70 años de edad. Se estima que afecta a unos 2,5 millones de personas en todo el mundo, aunque distribuidos de forma irregular; en este sentido, la región de menor prevalencia (<5 casos por 100.000 habitantes) es Asia central, mientras que en la mayoría de los países occidentales, especialmente los más desarrollados (Unión Europea, Estados Unidos, Canadá, Australia, etc.), la prevalencia supera ampliamente los 30 casos por 100.000 habitantes. En la Unión Europea hay aproximadamente 500.000 pacientes, siendo la enfermedad más frecuente en el norte de Europa y menos común en el área mediterránea. En España hay aproximadamente unos 40.000 pacientes1.
La esclerosis múltiple constituye la causa más frecuente de enfermedad por alteración de la mielina en el sistema nervioso central. Se estima que la susceptibilidad de padecer esclerosis múltiple depende de la interrelación con factores ambientales en determinadas áreas geográficas y que parecen operar antes de la pubertad, con una predisposición heredada de tipo multigénico. En este sentido, se ha cuantificado el grado de incremento del riesgo de padecer la enfermedad en función del parentesco familiar. Así, tener un primo afectado supone un 1%; padre o hijo, un 2%; hermano gemelo dicigótico, un 7%; padre y hermano, un 13%; padre y madre, un 20%; padre, madre y un hermano, un 23%; y un hermano gemelo monocigótico, un 30%.
Parece tratarse de una enfermedad de tipo autoinmune, es decir, producida por la reacción inmunológica contra elementos constitutivos del propio organismo, que son detectados – anómalamente – como antígenos. Se ha propuesto que un auténtico antígeno, todavía desconocido pero probablemente de origen externo, actuaría mimetizando proteínas de la mielina de las neuronas. Ese antígeno se presentaría sobre la superficie de los macrófagos en combinación con moléculas de clase 2 del Complejo Mayor de Histocompatibilidad (CMH). La resultante estimulación de los linfocitos T facilitadores (helper, Th1) provocaría la expresión de LFA-1 (lymphocyte function-associated antigen-1)2 y de VLA-4 (Integrin alpha4beta1; Very Late Antigen-4), facilitando la unión de dichos linfocitos T a moléculas de adhesión, como la ICAM-1 ICAM-1 (Intercellular Adhesion Molecule 1, también conocido como CD54 o Cluster of Differentiation 54) y la VCAM-1 (Vascular cell adhesion molecule 1 o CD106, cluster of differentiation), la principal molécula responsable del incremento de la adhesión leucocitaria al endotelio; actuando todos ellos sobre las células endoteliales de los vasos sanguíneos, facilitando su migración a través del endotelio y su penetración en el sistema nervioso central, donde atacarían a las células nerviosas y destruyendo específicamente su capa de mielina.
La destrucción de la mielina se debe a tres mecanismos complementarios:
- liberación de Factor de Necrosis Tumoral alfa (TNFα) por los linfocitos Th1
- liberación de TNFα y de radicales libres de oxígeno, óxido nítrico y proteasas por parte de macrófagos activados
- activación de la cascada del complemento mediante anticuerpos
Una de las causas propuestas es la infección por un virus latente (posiblemente herpesvirus o retrovirus humanos, como el HTLV-1, que causa la paraparesia espástica tropical) en la cual la activación vírica y su expresión desencadenan una respuesta inmune secundaria. Se considera, no obstante, que es más probable que infecciones comunes (tales como las producidas por virus del sarampión u otros similares) a una determinada edad desencadenen en personas susceptibles mecanismos de mimetismo molecular por los que presentarían una sensibilización cruzada contra la mielina. Por otro lado, la incidencia familiar y la asociación con ciertos alotipos HLA justifican la relativa susceptibilidad genética observada.
La esclerosis múltiple es, por consiguiente, una enfermedad básicamente desmielinizante, que conduce a la muerte neuronal desde las etapas iniciales de la enfermedad. Probablemente, esta pérdida neuronal es lo que contribuye decisivamente a la creciente discapacidad a la que se enfrentan los pacientes con esclerosis múltiple. Se caracteriza por la presencia de placas de desmielinización con destrucción de la oligodendroglía e inflamación perivenular diseminadas en el sistema nervioso central, especialmente en la sustancia blanca, con cierta especificidad por las columnas laterales y posteriores de la médula espinal, los nervios ópticos y las áreas periventriculares. También se encuentran afectadas las vías del mesencéfalo, la protuberancia y el cerebelo, al igual que la sustancia gris cerebral y medular.
El término de esclerosis se debe a la sustitución de la mielina normal por una proliferación de astrocitos. El infiltrado rodea a los pequeños vasos venosos – perivenular – y está formado por linfocitos T CD8+, CD4+, células fagocitarias, linfocitos B y células plasmáticas (estas últimas en menor número), y puede haber edema. A medida que las vainas de mielina van degenerando, se hace más apreciable la proliferación de células fagocitarias (macrófagos/microglia) y de astrocitos. Los cuerpos neuronales y los axones no suelen estar afectados, sobre todo en las lesiones recientes, pero posteriormente puede haber destrucción axonal, principalmente en las vías largas, y gliosis fibrosa que produce esclerosis de los haces de fibras, lo que justifica la irreversibilidad observada de algunos síntomas.
La principal consecuencia de la destrucción de las vainas mielínicas es la alteración de la conducción de los impulsos nerviosos transmitidos por las fibras desmielinizadas. La velocidad de transmisión se hace más lenta y los estímulos no se transmiten correctamente o incluso no lo hacen en modo alguno. El grado de anormalidad de la conducción puede variar dependiendo de circunstancias como la temperatura corporal, el ejercicio o la composición iónica del espacio extracelular. La sintomatología de la enfermedad depende de este fenómeno. En el caso concreto de la temperatura tiene consecuencias clínicas importantes, ya que la mayoría de los pacientes experimentan un empeoramiento de sus síntomas al aumentar la temperatura corporal. Se distingue actualmente cuatro variedades o formas clínicas de esclerosis múltiple:
- Remitente-recurrente (RR). Es el tipo más frecuente y afecta a más del 80% de las personas con esclerosis múltiple. En las fases iniciales puede no haber síntomas, a veces incluso durante varios años; sin embargo, a pesar de la ausencia de síntomas, sí se van produciendo lesiones inflamatorias en el sistema nervioso central. Los brotes son imprevisibles y pueden aparecer síntomas en cualquier momento – nuevos o ya conocidos – que pueden durar desde algunos días a varias semanas, desapareciendo posteriormente. Entre las recidivas no parece haber progresión de la enfermedad.
- Progresiva secundaria (SP). El grado de discapacidad persiste o incluso empeora entre brotes. Puede aparecer después de una fase recurrente-remitente y se considera una forma avanzada de la esclerosis múltiple. Entre un 30% y un 50% de los pacientes que sufren inicialmente la forma recurrente-remitente desarrolla la forma secundaria progresiva, habitualmente entre los 35 y los 45 años. Se caracteriza por una progresión continua con o sin recidivas ocasionales, remisiones poco importantes y fases de estabilidad.
- Progresiva primaria (PP). Afecta al 10% de todos los pacientes. Se caracteriza por la ausencia de brotes definidos, pero hay un comienzo lento y un empeoramiento constante de los síntomas sin un periodo intermedio de remisión. No hay episodios de recidiva ni periodos de remisión, sólo fases de estabilidad ocasionales y mejorías pasajeras poco importantes.
- Progresiva recidivante (PR). Es la forma más atípica, con progresión desde el comienzo, aunque a diferencia de aquellos con la forma progresiva primaria (PP), hay brotes agudos claros, con o sin recuperación completa. Los períodos entre brotes se caracterizan por una progresión continua.
Existe una gran controversia sobre la existencia real de una quinta forma de esclerosis múltiple, la benigna (B), que se caracterizaría por tener tan solo una recidiva inicial y, posiblemente, solo un brote adicional y una recuperación completa entre estos episodios, pudiendo transcurrir hasta 20 años hasta que se produzca una segunda recidiva, por lo que el proceso únicamente progresa de forma limitada. Para algunos especialistas, la forma benigna sería en realidad un cuadro recurrente-remitente (RR) sintomáticamente muy leve y con discapacidad mínima. No obstante, estos pacientes acaban progresando en su mayoría y experimentan deterioro cognitivo. Se estima que constituyen aproximadamente el 15% de todos los casos diagnosticados de esclerosis múltiple.
La forma sintomática de la enfermedad incluye una amplia variedad de síntomas y signos, entre los que resultan más frecuentes los mentales (apatía, alteración del juicio o inatención, etc.), los de pares craneales (especialmente oculares y acústicos), los motores (incremento de los reflejos tendinosos profundos, la combinación de espasticidad y ataxia cerebelosa puede llegar a ser totalmente incapacitante; además, las lesiones hemisféricas pueden producir hemiplejia), los sensitivos (parestesias, entumecimiento y embotamiento de la sensibilidad) y los autónomos (urgencia urinaria, dificultad para la micción, retención urinaria parcial o incontinencia leve, estreñimiento, etc.).
En general, a los 5 años de la aparición de los primeros síntomas, algo más del 50% de los pacientes tiene algún tipo de afectación leve, en otro 40 % hay afectación moderada y en menos de un 10 % es grave; un 70% de los pacientes están en condiciones de trabajar habitualmente. No obstante, a los 15 años solo el 25-30% de los pacientes continúa con una afectación leve y un 50% requieren ayuda para caminar. A los 20 años, un 35% continúa en condiciones de trabajar y un 20% ha muerto como consecuencia de las complicaciones.
Más del 60% de los pacientes con esclerosis múltiple evidencia un deterioro de la movilidad, que aparece en todos los tipos de esclerosis, incluso en etapas tempranas; en este sentido, en España, un 42% de los pacientes con una antigüedad de diagnóstico de ≤5 años reportan dificultades en la marcha y un 53% pérdida de equilibrio. De hecho, la mayoría de los pacientes con valores ≥4 en la escala EDSS3 tienen problemas para caminar y dos de cada tres consideran que su vida familiar se ve significativamente afectada por sus problemas de movilidad (Arroyo, 2013).
La esclerosis múltiple produce globalmente una reducción media de unos 9 años sobre la duración de vida en los varones y hasta de 14 en las mujeres. La esperanza de vida es de unos 25 años tras el comienzo de la enfermedad, aunque con notables variaciones interindividuales. En este sentido, la supervivencia depende sobre todo del grado de incapacidad existen en los paciente: sólo el 7 % de los enfermos que caminan han fallecido a los 10 años, mientras que asciende al 49 % para los que apenas se mantienen en pie y al 84 % para los encamados de forma permanente.
El pronóstico depende fundamentalmente del número de ataques, siendo un signo de mal pronóstico la existencia de una elevada frecuencia de recaídas durante los primeros años de enfermedad (la frecuencia media de ataques en los primeros años es de uno anual). Igualmente, el tipo de ataques es relevante para el pronóstico, ya que los síntomas primarios de tipo motriz, ataxia o problemas bulbares se asocian con peores pronósticos, mientras que si son de tipo visual, el pronóstico es más favorable.
Por el momento, no existe ningún tratamiento curativo de la enfermedad y sus objetivos consisten en reducir la gravedad y la frecuencia de las recaídas, limitar la discapacidad persistente, aliviar los síntomas y promover la reparación tisular. El tratamiento de elección para las recaídas agudas son los corticosteroides, atendiendo al carácter inflamatorio e inmunológico de la esclerosis múltiple. Reducen la intensidad y la duración de la recaída, probablemente reduciendo el edema, pero no afecta a la progresión de la discapacidad. El régimen preferible consiste en el empleo de metilprednisolona IV en dosis elevadas (1 g durante tres días), aunque si se opta por la vía oral, la pauta recomendada es una dosis inicial de 60 mg de prednisona por día, durante una semana, reduciéndola posteriormente a lo largo de tres semanas. No obstante, en la forma recidivante-remitente la tendencia es que la eficacia de los corticosteroides disminuya con el tiempo. Por otro lado, en los cuadros agudos graves resistentes a corticosteroides, se opta por un cambio de plasma en días alternos, medida que produce excelentes resultados en más del 40% de los pacientes afectados.
La terapia modificadora de la enfermedad se emplea únicamente en las formas recidivante-remitente y en la secundariamente progresiva, pero no en la progresiva primaria. En la forma recidivante-remitente, este tratamiento busca reducir la frecuencia e intensidad de los ataques y prevenir la acumulación de discapacidad asociada con la transición a la forma secundariamente progresiva. Hasta el momento las terapias inmunomoduladoras indicadas en la esclerosis múltiple son los interferones beta, el acetato de glatirámero, el fingolimod, el natalizumab y los agentes citotóxicos, como azatioprina o ciclofosfamida.
El interferón beta 1b consiste en una leve variación molecular del interferón beta humano, difiriendo en un aminoácido y no está glucosilado. En pacientes con forma recidivante-remitente, la administración de dosis subcutáneas de 8 MU cada dos días reduce en un 34% la tasa de recaídas, pero no parece reducir de forma significativa la acumulación de discapacidad. Por su parte, en pacientes con esclerosis múltiple secundariamente progresiva el tratamiento de dos años ha demostrado un significativo incremento del tiempo hasta la progresión de la enfermedad, así como otros importantes beneficios, tales como el retardar la necesidad de utilizar silla de ruegas, reducir el consumo de corticosteroides y el número de hospitalizaciones. Por su parte, el interferón beta 1a es estructuralmente idéntico a la citocina humana, tanto en la secuencia de aminoácidos como en los restos glucídicos. Reduce en un tercio la tasa de recaídas, prolongando una media de cinco meses el tiempo transcurrido hasta la primera recaída. También incrementa de forma significativa el periodo transcurrido hasta la progresión sostenida de la enfermedad.
El glatirámero es una mezcla de péptidos sintéticos formados por copolímeros de ácido L-glutámico, L-alanina, L-tirosina y L-lisina, parcialmente acetilados. No se conoce su mecanismo de acción, aunque se ha sugerido que podría actuar como un péptido que mimetiza a la proteína base de la mielina, provocando un efecto inductor de los linfocitos T supresores, deficitarios en la esclerosis múltiple, e inhibiendo el efecto de los antígenos anti-mielina del sistema nervioso central, al inhibir e efecto de los linfocitos T autorreactivos. El acetato de glatirámero actúa sobre las células dendríticas, que tienen una intensa capacidad presentadora de antígenos, orquestando las respuestas Th1 y Th2. Es capaz de reducir en un 30% el número de recaídas, y la discapacidad resultante, en los pacientes con esclerosis múltiple de tipo remitente-recidivante. Sin embargo, no hay evidencia de que este tratamiento tenga efectos beneficiosos sobre la duración o gravedad de la recaída. Tampoco hay datos clínicos significativos en pacientes afectados con formas progresivas de la enfermedad.
El fingolimod es, previa transformación en el fosfato correspondiente, un modulador del receptor 1 de la esfingosina-1-fosfato (S1P), localizado en la superficie de los linfocitos y al que se une con alta afinidad, actuando como un antagonista funcional al inducir su desacoplamiento o internalización. Este proceso hace a los linfocitos insensibles al S1P, bloqueando así la señal bioquímica que induce la salida de los linfocitos desde los órganos linfoides y, en consecuencia, provoca una redistribución linfocitaria4. Como consecuencia de ésta, se reduce la infiltración de los linfocitos al sistema nervioso central, y con ello reduce el riesgo de provocar inflamación y lesiones en el tejido nervioso en los pacientes con esclerosis múltiple. El fingolimod ha sido autorizado en monoterapia para el tratamiento modificador del curso de la enfermedad en la esclerosis múltiple remitente recurrente muy activa para pacientes tratados con interferón beta o aquellos con una evolución rápida. El tratamiento crónico con fingolimod da lugar a una reducción del recuento de linfocitos, especialmente de linfocitos T y B que circulan a través de los órganos linfoides. Sin embargo, los linfocitos que circulan en sangre periférica – implicados principalmente en la defensa inmunológica periférica – no son afectados significativamente por el fármaco. En este sentido, el fingolimod reduce levemente (20%) los niveles de neutrófilos y prácticamente no afecta a los de monocitos.
El natalizumab es un anticuerpo monoclonal que inhibe selectivamente las moléculas de adhesión, uniéndose a la subunidad a4 de las integrinas humanas, evitando la penetración de los leucocitos al sistema nervioso central inflamado, facilitando con ello la reducción de la inflamación y de las lesiones neurológicas asociadas a la esclerosis múltiple. En este sentido, es capaz de reducir el número recaídas en pacientes con esclerosis múltiple recidivante-remitente, así como el riesgo de progresión de la discapacidad. Se emplea en monoterapia en la forma remitente recidivante muy activa que no responda adecuadamente al interferón beta o en esclerosis múltiple remitente recidivante grave de evolución rápida. Presenta una pauta posológica cómoda, consistente una única administración al mes. Sin embargo, se ha mencionado la persistencia de anticuerpos inactivadores en el 6% de los pacientes, que anulan su actividad e inducen cuadros de hipersensibilidad. Aún más importante es que su uso ha sido asociado con la aparición – aunque excepcionalmente infrecuente: 1 caso por cada 2.000 pacientes tratados – de leucoencefalopatía multifocal (LMP), una enfermedad subaguda progresiva del SNC causada por la reactivación del virus JC, predominantemente en pacientes inmunodeprimidos y que suele provocar una discapacidad grave o la muerte. La sintomatología de la LMP es muy similar a un brote de esclerosis múltiple y el riesgo de desarrollar la enfermedad parece aumentar a partir de los dos años de tratamiento.
Entre los tratamientos inmunosupresores, los fármacos más utilizados son la ciclofosfamida y la azatioprina. El fundamento de su aplicación la disminución de las células en rápida proliferación, entre ellas las linfoides responsables de la destrucción de la mielina del SNC. Sin embargo, el uso de fármacos inmunosupresores en las formas progresivas más graves no ha mostrado un beneficio uniforme y tienen notables riesgos tóxicos. La ciclofosfamida puede ser beneficiosa en pacientes de menos de 40 años de edad, pero produce efectos tóxicos graves y su uso parece contar cada vez con menos partidarios. La azatioprina, por su parte, se ha utilizado mucho más que la anterior, porque sus efectos tóxicos son menos acusados y el manejo clínico es más sencillo; administrada sola o junto con corticoides orales en dosis bajas, ha demostrado una eficacia modesta en algunos aspectos clínicos como la rapidez de progresión o el número de recaídas, pero no en la discapacidad. Este leve beneficio es el principal motivo por justifica que continúe siendo un fármaco usado en pacientes con múltiples brotes o en rápida progresión. Otros fármacos inmunosupresores ensayados y con resultados más o menos decepcionantes (por su escasa eficacia o por su inaceptable toxicidad) han sido la ciclosporina, la mioxantrona, el clorambucilo, la cladribina y el metotrexato.
Desde el punto de vista de los tratamientos sintomáticos, el fármaco más relevante es la fampridina, un bloqueante de canales iónicos de potasio (K+) dependientes del voltaje que limita la fuga de iones potasio a través de dichos canales en los axones desmielinizados de los pacientes con esclerosis múltiple, prolongando la repolarización e intensificando el potencial de acción en las neuronas afectadas y, con ello, mejorando algunas de las funciones neurológicas perturbadas en estos pacientes, particularmente la marcha en pacientes adultos. No obstante, sus efectos clínicos son modestos y solo son observados en un tercio de la población susceptible de su uso; por otro lado, presenta un perfil toxicológico nada desdeñable, todo lo cual limita su potencial terapéutico (Cuéllar, 2013).
ACCIÓN Y MECANISMO
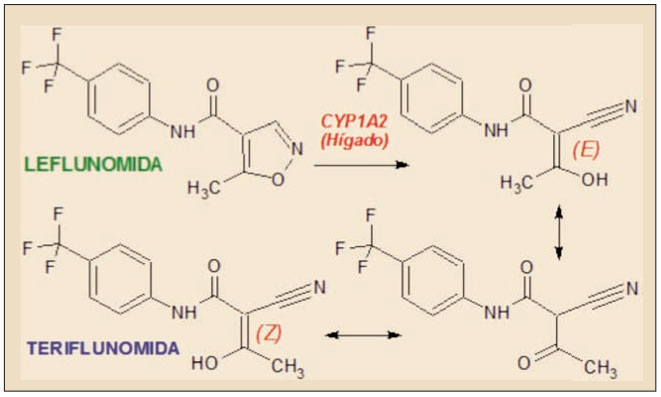
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente.
La etapa limitante en la biosíntesis de bases nucleicas de tipo pirimidínico es la formación del N-carbamilaspartato, partiendo del carbamilfosfato y del aminoácido aspartato y gracias al enzima aspartato-transcarbamilasa. El núcleo pirimidínico se genera a partir del N-carbamilfosfato, formándose el dihidroorotato, el cual, mediante la DHODH, da lugar a orotato y, tras incorporar ribosa-fosfato, forma orotidilato, un precursor del uridilato (UMP), el cual a su vez puede ser convertido en los nucleótidos citidilato (en el ARN) y timidilato (en el ADN).
Los requerimientos de nucleósidos y de sus correspondientes bases nucleicas para la síntesis de ADN y ARN en los procesos de proliferación celular pueden ser cumplimentados mediante la administración exógena (fundamentalmente, con la dieta) o por la síntesis de novo a partir de precursores bioquímicos. Sin embargo, la capacidad para reutilizar o reconvertir nucleótidos a partir de restos de ácidos nucleicos propios, permite que el organismo humano pueda prescindir prácticamente de la obtención de bases purínicas y pirimidínicas a partir de la dieta. De hecho, la vía de la reutilización constituye la principal fuente de nucleótidos para la síntesis de ADN, ARN y cofactores enzimáticos (AMP, ATP, GMP, etc). Esto es así para la mayoría de las células humanas, pero no tanto para las células con alta capacidad de división (en función de las demandas orgánicas), como ocurre con los linfocitos B y T activados.
Por este motivo, el bloqueo de la síntesis de novo de pirimidinas en los linfocitos T y B es capaz de interferir con la síntesis proteica y de ARN en estas células, activando las moléculas sensoras que bloquean la progresión del ciclo celular en la fase G1 (crecimiento). Esto se manifiesta como un efecto fundamentalmente citostático. Este efecto es capaz de bloquear la acción combinada de los linfocitos T y B activados.
La esclerosis múltiple es considerada como una enfermedad de tipo autoinmune, posiblemente relacionado con la existencia de un antígeno – aún sin identificar – que se presentaría sobre la superficie de los macrófagos en combinación con moléculas de clase 2 del Complejo Mayor de Histocompatibilidad (CMH). La resultante estimulación de los linfocitos T facilitadores (Th1) provocaría la expresión de LFA-1 y de VLA-4, facilitando la unión de dichos linfocitos T a moléculas de adhesión, como la ICAM-1 y la VCAM-1, sobre las células endoteliales de los vasos sanguíneos, facilitando su migración a través del endotelio y su penetración en el sistema nervioso central, donde atacarían a las células nerviosas y destruyendo específicamente su capa de mielina. Esta destrucción de la mielina se debe a tres mecanismos complementarios: liberación de Factor de Necrosis Tumoral alfa (TNFa) por los linfocitos Th1, así como del propio TNFa y de radicales libres de oxígeno y óxido nítrico, y proteasas por parte de macrófagos activados y la activación de la cascada del complemento mediante anticuerpos.
{kind=link}
En este sentido, además de la inhibición de la proliferación de linfocitos B y T activados, y de la interferencia de la adhesión de estos a las células endoteliales, la teriflunomida disminuye la síntesis de citocinas inmunosupresoras, como el Factor de Crecimiento Transformante beta (TGF–β). Asimismo, inhibe diversas isoenzimas tirosina cinasa, el Factor Nuclear potenciador de las cadenas ligeras Kappa de las células B activadas (NF-kB) y metaloproteasas (MMP). También parece desarrollar efectos antiinflamatorios específicos, al reducir la liberación de histamina e inhibir la inducción de la ciclooxigenasa 2 (COX 2).
ASPECTOS MOLECULARES
La teriflunomida es el estereoisómero Z del principal metabolito activo de la leflunomida, un agente inmunomodulador autorizado para el tratamiento de la artritis reumatoide y de la artritis psoriásica. La leflunomida es transformada en el hígado humano mayoritariamente (70%) en el metabolito M1 principalmente a través del citocromo P450 (CYP), particularmente por el isoenzima CYP1A2, mediante la apertura del anillo isoxazólico y dando lugar a una mezcla interconvertible de estereoisómeros enólicos (E y Z) y de una cetona, de los que el estereoisómero enólico Z es el más estable y, por tanto, el predominante.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínica de la teriflunomida han sido adecuadamente contrastadas en las indicaciones autorizadas mediante tres ensayos clínicos principales de fase 3, multinacionales, aleatorizados, doblemente ciegos, con diseño de grupos paralelos y controlados con placebo o con comparador activo (interferón beta 1a). Dichos ensayos clínicos fueron llevados a cabo sobre pacientes con esclerosis múltiple mayoritariamente (>91%) remitente-recurrente y moderadamente afectados, con una puntuación no superior a 5,5 (sobre un máximo de 10) en la escala de discapacidad de Kurtzke o EDSS5 (Expanded Disability Status Scale, Escala Expandida del Estado de la Discapacidad), que mide el estado funcional de los pacientes con esclerosis múltiple, aunque la mediana fue de 2,0 a 2,5; los pacientes experimentaron los primeros síntomas entre 4 y 7 años antes y habían sido diagnosticados hace entre uno y cuatro años. Asimismo, los pacientes debían haber experimentado al menos un recaída en el último año o dos recaídas en los dos últimos años.
En todos los estudios se empleó teriflunomida por vía oral, tanto en dosis de 7 como de 14 mg/24 h, durante un periodo no inferior a 48 semanas (en caso contrario, se valoró como suspensión prematura del tratamiento) y, en su caso, el correspondiente placebo. En el estudio comparativo con interferón beta 1a, éste se empleó con dosis de 44 µg tres veces por semana6, por vía subcutánea. Como variable primaria de eficacia se midió en los estudios controlados con placebo la tasa anualizada de recaídas confirmadas, mientras que entre las variables secundarias se determinó la mediana de tiempo trascurrido hasta progresión de la discapacidad (aumento persistente durante al menos 12 semanas de al menos un punto en la escala EDSS). En el estudio comparativo con interferón beta, se determinó la mediana de tiempo hasta fallo terapéutico (definido como la aparición de la primera recaída confirmada o la suspensión del tratamiento por cualquier causa, lo que sucediese en primer lugar). Otros parámetros determinados fueron la variación experimentada en la escala de impacto de fatiga (Fatigue Impact Scale, FIS) y el cuestionario de satisfacción del tratamiento (Treatment Satisfaction Questionnaire for Medication, TSQM).
Dos de los estudios fueron controlados con placebo y tuvieron un diseño similar. El primero de ellos (TEMSO; O’Connor, 2011) se llevó a cabo sobre 1.088 pacientes con una mediana de edad de 38 años, 72% mujeres, 98% de raza caucásica, mediana de 2,5 en la escala EDSS (un 23% tenía >3,5) y un 27% habían recibido tratamiento previamente. El tratamiento se prolongó durante 108 semanas.
Los resultados mostraron una tasa anualizada de recaídas de 0,37 (IC95% 0,32 a 0,43) con teriflunomida 7 mg, de 0,37 (IC95% 0,31 a 0,44) con teriflunomida 14 mg y de 0,54 (IC95% 0,47 a 0,62) con placebo; esto supone una reducción del riesgo de recaída con respecto al placebo del 31%, tanto con la dosis de 7 mg (HR= 0,69; IC95% 0,56 a 0,84; p= 0,0002) como con la de 14 mg (HR= 0,69; IC95% 0,55 a 0,85; p= 0,0005). Por su parte, el porcentaje de pacientes con progresión de la enfermedad fue del 18,6% con teriflunomida 7 mg, del 17,3% con teriflunomida 14 mg y del 23,7% con placebo, lo que implica una reducción del riesgo de progresión de la enfermedad del 24% con la dosis de 7 mg (HR= 0,76; IC95% 0,56 a 1,05; p= 0,0835, no significativa) y del 30% con la de 14 mg (HR= 0,70; IC95% 0,51 a 0,97; p= 0,0279).
En un análisis secundario (O’Connor, 2013), los pacientes tratatados con las dosis de 7 y de mg de teriflunomida mostraron una reducción de la tasa anualizada de recaídas con secuelas neurológicas del 32% y del 36% respectivamente (ambas, estadísticamente significativas). También se apreció una reducción estadísticamente significativa en la tasa de recaídas que finalizaron en la hospitalización del paciente (36% y 59%, respectivamente para las dosis de 7 y 14 mg), así como en la de pacientes que requirieron la administración IV de corticosteroides para tratar las recaídas (29% y 34%).
El segundo estudio pivotal (TOWER; Confavreux, 2014) fue realizado en 1.169 pacientes con una mediana de 38 años, 71% mujeres, 82% de raza caucásica, mediana de 2,5 en la escala EDSS (un 24% tenía >3,5) y un 33% habían recibido tratamiento previamente (16% con interferón beta y 12% con glatirámero).
Se registró una tasa anualizada de recaídas de 0,39 (IC95% 0,33 a 0,46) con teriflunomida 7 mg, de 0,32 (IC95% 0,27 a 0,38) con teriflunomida 14 mg y de 0,50 (IC95% 0,43 a 0,58) con placebo; todo lo cual implica una reducción del riesgo de recaída con respecto al placebo del 22% con la dosis de 7 mg (HR= 0,78; IC95% 0,63 a 0,96; p= 0,0183) y del 36% con la de 14 mg (HR= 0,64; IC95% 0,51 a 0,79; p= 0,0001). Los correspondientes porcentajes de pacientes con progresión de la enfermedad fueron del 16,0% con teriflunomida 7 mg, del 11,9% con teriflunomida 14 mg y del 16,8% con placebo, lo que implica una reducción del riesgo de progresión de la enfermedad del 4% con la dosis de 7 mg (HR= 0,96; IC95% 0,68 a 1,35; p= 0,7620, no significativa) y del 31% con la de 14 mg (HR= 0,69; IC95% 0,47 a 1,00; p= 0,0442).
El estudio comparativo con interferón beta 1a (TENERE; Vermersch, 2014) incluyó a 324 pacientes con una mediana de 35 años, 68% mujeres, 100% de raza caucásica, mediana de 2,0 en la escala EDSS (un 12% tenía >3,5) y un 19% habían recibido tratamiento previamente. Los datos registrados indicaron que el porcentaje de pacientes que habían experimentado fallo terapéutico fue del 48,6% con 7 mg de teriflunomida, del 37,8% con 14 mg y del 42,3% con interferón beta, siendo la probabilidad de fallo a las 96 semanas del 59% (HR= 0,59; IC95% 0,46 a 0,71) con teriflunomida 7 mg, del 41% (HR= 0,41; IC95% 0,31 a 0,51) con teriflunomida 14 mg y del 44% (HR= 0,44; IC95% 0,34 a 0,54) con interferón beta; en este sentido, no encontrándose superioridad de ninguna de las dosis de teriflunomida vs. interferón beta 1a (OR= 1,12; IC95% 0,75 a 1,67; p= 0,5190, no significativa, para la dosis de 7 mg; OR= 0,86; IC95% 0,56 a 1,31; p= 0,5953, no significativa, para la de 14 mg).Las puntuaciones en el índice de fatiga (FIS) indicaron una fatiga más frecuente con interferón beta, aunque las diferencias fueron solo estadísticamente significativas con teriflunomida 7 mg. También las puntuaciones en el cuestionario de satisfacción del tratamiento (TSQM) fueron más favorables para la teriflunomida.
Desde el punto de vista de la seguridad clínica, los eventos adversos más frecuentemente asociados al tratamiento con teriflunomida son de carácter hepático, digestivo e inmunológico: alteraciones de las pruebas funcionales hepáticas (17,8% con la dosis de 14 mg de teriflunomida; 17,0% con la de 7 mg y 10,5% con placebo), diarrea (17,3%; 14,5% y 8,3%), náusea/vómitos (16,6%; 11,7% y 9,0%), alopecia (14,7%; 11,4% y 4,3%), parestesia/disestesia (14,7%; 12,1% y 10,2%), neutropenia (4,6%; 2,3% y 0,7%), infecciones virales (6,5%; 4,4% y 1,9%), hipertensión (4,6%; 3,5% y 1,9%), metrorragia (3,1%; 1,2% y 0,5%), dolor muscular (3,1%; 4,0% y 1,4%). La tasa de eventos adversos graves fue del 16,1% (14 mg), 12,8% (7 mg) y 7,6% (placebo), provocando directamente la suspensión del tratamiento en un 11,8%, 9,1% y 7,6%, respectivamente.
ASPECTOS INNOVADORES
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente. Se trata del estereoisómero Z del principal metabolito activo de la leflunomida, utilizada para el tratamiento de la artritis reumatoide y de la artritis psoriásica.
El meta-análisis de los datos agregados de los dos estudios controlados con placebo muestra una tasa anualizada de recaídas de 0,354 para la teriflunomida 14 mg y de 0,534 para placebo (P< 0,0001), siendo los respectivos porcentajes de pacientes con progresión sostenida del 17,9% vs. 25,2%, lo que supone una reducción del riesgo del 30,5%. Por lo que respecta a la comparación con interferón beta 1a, el ensayo pivotal correspondiente no ha sido capaz de mostrar superioridad con ninguna de las dos dosis de teriflunomida ensayadas. No se dispone de ningún otro estudio directamente comparativo con otros medicamentos utilizados en la esclerosis múltiple.
Desde el punto de vista de la seguridad clínica, la toxicidad no parece ser un problema importante, aunque son comunes los trastornos de naturaleza hepática y digestiva. Hasta un 12% de los pacientes tratados con la dosis de 14 mg suspendieron el tratamiento por eventos adversos, frente a un 9% con placebo. El perfil toxicológico no parece presentar diferencias con por su precursor farmacológico, la leflunomida, por lo que no son previsibles grandes sorpresas en este campo.
Los datos clínicos registrados solo corresponden a la forma recidivante-recurrente – por otro lado, la más común – y muestran un nivel de eficacia modesto aunque clínicamente relevante, con diferencias con respecto al placebo similares a las observadas con otros agentes (glatirámero, interferón beta, etc.), con una reducción en torno al 30% del riesgo de progresión. Por otro lado, la mayor parte de los pacientes que participaron en los ensayos clínicos presentan un bajo nivel de afectación (EDSS 2,0-2,5; sobre un nivel máximo de 10).
El mecanismo inmunológico es ciertamente poco específico, pero eso mismo puede achacarse a la mayoría de sus teóricos competidores. Como una potencial – y modesta – ventaja frente a estos, podría mencionarse su cómoda administración oral, una vez al día; pero nada más.
|
VALORACIÓN |
|
|---|---|
|
Teriflunomida ► AUBAGIO® (Sanofi Aventis) |
|
|
Grupo Terapéutico (ATC): L04AX. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores selectivos. |
|
|
Indicaciones autorizadas: Tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad físico-química: Vía de administración más cómoda para el paciente. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Leflunomida |
Arava |
Sanofi Aventis |
2000 |
|
Teriflunomida |
Aubagio |
Sanofi Aventis |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
BIBLIOGRAFÍA
Ruxolitinib JAKAVI® (Novartis)
Resumen
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. Atendiendo a la condición de enfermedad rara, el medicamento fue designado como huérfano. El nuevo fármaco ha demostrado ser capaz de reducir de forma notable la esplenomegalia en el 29-42% de los pacientes, frente a menos del 0-1% con placebo o con el mejor tratamiento alternativo disponible. Desde el punto de vista de la seguridad, el ruxolitinib se comporta básicamente como un agente mielotóxico, dando lugar a trombocitopenia, anemia, hemorragias y neutropenia, como consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. Aunque se trata de un efecto mielotóxico importante, es previsible y manejable mediante transfusiones y ajustes de la posología del fármaco. En definitiva, se trata de un paso adelante en el tratamiento de una enfermedad rara, aunque su eficacia es modesta y su efecto real sobre la supervivencia global está todavía sujeto a estudio.
MIELOFIBROSIS
Las enfermedades mieloproliferativas crónicas comparten la característica de ser clonales; es decir, se originan en una célula madre de la médula ósea con capacidad de diferenciación pluri- y multipotente. En dichas patologías se produce una proliferación, diferenciación y maduración efectiva de las células en la médula ósea, generando una médula hipercelular y un número incrementado de células maduras en sangre periférica; asimismo, es frecuente encontrar hepatoesplenomegalia debida a hematopoyesis extramedular y/o a secuestro de las células sanguíneas (Novelli, 2011).
Según la clasificación de la Organización Mundial de la Salud, los síndromes mieloproliferativos crónicos incluyen a la leucemia mieloide crónica Filadelfia+ BCR/ABL+, la leucemia neutrofílica crónica, la leucemia eosinofílica crónica/síndrome hipereosinofílico, la policitemia vera, la trombocitemia esencial y la mielofibrosis crónica idiopática, junto con otras enfermedades mieloproliferativas inclasificables.
En concreto, la mielofibrosis crónica idiopática (metaplasia mieloiode, mieloesclerosis) se caracteriza por una intensa fibrosis de la médula ósea, esplenomegalia, hematopoyesis extramedular y leucoeritroblastosis (granulocitos y eritrocitos inmaduros) en sangre periférica y glóbulos rojos en forma de lágrimas. En su fase temprana, se caracteriza por un número elevado de células CD34+ en la médula, mientras que las fases posteriores de fibrosis medular muestran una disminución de células CD34+ en la médula y un aumento de congestión esplénica y hepática con células CD34+. Además, hay formas secundarias de mielofibrosis, que derivan secundariamente de otros trastornos mieloproliferativos, especialmente la policitemia vera y la trombocitemia esencial1.
La mielofibrosis se origina por una mutación somática en una célula madre hematopoyética pluripotente, lo que le confiere a ésta ciertas ventajas proliferativas respecto a los progenitores normales. Esa población celular anormal libera diversas citocinas y factores de crecimiento (PDGF, TGFβ, VEGF, bFGF y calmodulina) en la médula, dando lugar a la producción de fibrosis colágena, osteoesclerosis y angiogénesis, como fenómeno secundario y la colonización de órganos y sitios extramedulares, como el hígado y el bazo, a los que produce un sobrecrecimiento (hepato y esplenomegalia).
Presenta una incidencia de 4-14 casos por millón de habitantes/año, con una mediana de la edad de presentación de 65 años, aunque aproximadamente la tercera parte de los pacientes están asintomáticos al diagnóstico. Se suele diagnosticar entre los 50 y 80 años (menos del 10% son menores de 45 años), pero puede ocurrir a cualquier edad, y afecta tanto a hombres como a mujeres.
Aunque en muchos casos se ignora la etiología de esta enfermedad, en un 50% de los casos está presente una mutación del gen JAK2 y en un 10% una mutación del gen MLP2. La expresión de los genes JAK conduce a la síntesis de la familia de tirosina cinasas Janus (Janus tirosine kinases, JAK; de los que se conocen cuatro tipos, de la JAK1 a la JAK4), un miembro de la superfamilia de los receptores de citocinas. Las JAK parecen ejercer un papel esencial en los procesos de transducción de señales bioquímicas de diversas citocinas; de hecho, la eritropoyetina, la trombopoyetina y el factor de estimulante de colonias de granulocitos y macrófagos (GM-CSF) actúan exclusivamente sobre receptores que utilizan homodímeros de JAK2 en sus procesos de señalización intracelular.
En respuesta a la activación del receptor de citocinas, estas tirosina cinasas (que no forman parte propiamente de dicho receptor) son rápidamente fosforiladas dando lugar a una activación celular en cascada y supone la activación de varias vías de transducción de señales, que determinan la aparición de cambios en la expresión genética que promueven la progresión y maduración celular.
En realidad, la mutación de JAK2 no parece ser la causa primaria de la enfermedad sino más bien una mutación somática secundaria. Aunque la mutación JAK2V617F3 parece ser la más común (aproximadamente, el 65%) de las asociadas a la mielofibrosis primaria, se han identificado otras mutaciones que activan anormalmente al JAK2, como la MPLW515L/Kin del receptor de MPL. Sea como fuere, los altos niveles de citocinas encontradcos en los pacientes con mielofibrosis, particularmente de interleucina 6 (IL-6) parece ser los responsables del estado hipercatabólico y de los síntomas constitucionales de la enfermedad (pérdida de peso, fatiga, etc.). Muchas de estas citoxinas utilizan tirosina cinasas de tipo JAK1 en su mecanismo señalizador celular.
Las principales causas de muerte en los pacientes con mielofibrosis son la insuficiencia medular evolutiva, la transformación a leucemia no linfoblástica aguda, las infecciones oportunistas, las complicaciones trombohemorágicas, la insuficiencia cardíaca y la hipertensión portal. Los factores pronósticos más relevantes son:
- Edad de 65 años o más.
- Anemia (hemoglobina <10 g/dl).
- Síntomas inespecíficos: fiebre, sudores nocturnos o pérdida de peso.
- Leucocitosis (>25 × 109/l).
- ≥1% de blastocitos circulantes.
Los pacientes sin ninguna característica adversa, excepto la edad, tienen una mediana de supervivencia de más de 10 a 15 años (mediana de 135 meses: 11,25 años), pero la presencia de uno de los mencionados factores adversos la reduce a 95 meses (7,9 años), dos factores a 48 meses (4 años) y tres o más a 27 meses (2,25 años). Las anomalías cariotípicas también pueden afectar el pronóstico. En este sentido, algunas series retrospectivas han permitido observar que las deleciones 13q y 20q y la trisomía 9 se han correlacionado con una mayor supervivencia y ninguna transformación leucémica, en comparación con el peor pronóstico de la trisomía 8, un cariotipo complejo, –7/7q-, i(17q), inv(3), -5/5q-, 12p– o el reordenamiento 11q23.
Los pacientes asintomáticos de riesgo bajo solo requieren un seguimiento clínico, sin ninguna intervención terapéutica, que solo estaría justificada en caso de aparición de anemia sintomática, leucocitosis marcada, sudores nocturnos abundantes, pérdida de peso, fiebre o esplenomegalia sintomática.
El trasplante alogénico de células madre hematopoyéticas es el único tratamiento curativo de la mielofibrosis, pero su aplicabilidad está limitada por la avanzada edad de la mayoría de los pacientes y por la existencia de diversas condiciones comórbidas (Odenike, 2013)
Hasta ahora, el único tratamiento mielosupresor específicamente indicado en España (y algunos otros países de la Unión Europea, como Francia, Italia o Suecia) en la mielofibrosis. El ruxolitinib, atendiendo a su condición de inhibidor de JAK1 y JAK2, ha sido autorizado para reducir la esplenomegalia y los síntomas debilitantes de la pérdida de peso, la fatiga y los sudores nocturnos de los pacientes de mielofibrosis primaria, mielofibrosis posterior a trombocitopenia esencial o mielofibrosis posterior a policitemia vera, tanto positivos como negativos para JAK2.
ACCIÓN Y MECANISMO
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia.
Las JAK parecen ejercer un papel esencial en los procesos de transducción de señales bioquímicas de citocinas hematopoyéticas como la eritropoyetina, la trombopoyetina y el factor de estimulante de colonias de granulocitos y macrófagos (GM-CSF). En respuesta a la activación del receptor de citocinas, estas tirosina cinasas (que no forman parte propiamente de dicho receptor) son rápidamente fosforiladas dando lugar a una activación celular en cascada y supone la activación de varias vías de transducción de señales, que determinan la aparición de cambios en la expresión genética que promueven la progresión y maduración celular. El ruxolitinib impide dichos procesos de fosforilación al competir directamente con el ATP, impidiendo de esta manera los efectos proliferativos de dichas citocinas.
ASPECTOS MOLECULARES
El ruxolitinib está estrechamente relacionado farmacológicamente con otros miembros de la serie de inhibidores de tirosina cinasas. Se han desarrollado modelos moleculares de relación estructura-actividad, en los que las interacciones estéricas y electrostáticas han demostrado ser las más determinantes para el grado de inhibición y el tipo de tirosina cinasa susceptible.
{kind=link}
En concreto, la estructura del ruxolitinib emula la del ATP (trifosfato de adenosina); de hecho, se trata de un análogo isostérico de la adenosina. En tal calidad, el ruxolitinib actúa como un antagonista competitivo del ATP específicamente en la JAK1 y JAK2, lo que conduce a un bloqueo del proceso de la fosforilación en la cadena de señalización bioquímica intracelular de determinadas citocinas hematopoyéticas.
La mutación genética más comúnmente registrada en pacientes con mielofibrosis afecta a la estructura de la JAK2 (JAK2V617F), pero lo hace en la región externa al bolsillo de unión al ATP del enzima, por lo que los inhibidores de la cinasa JAK2 competitivos del ATP – como el ruxolinitib – no son capaces de distinguir entre las enzimas JAK2 normales y las mutadas. Por ello, este tipo de fármacos producen una mielosupresión no selectiva que, si bien facilita el control la hiperproliferación de las células hematopoyéticas en mielofibrosis y en otras patologías mieloproliferativas (pero también es responsable de efectos adversos mielosupresores), pueden no ser capaces en eliminar los clones mutantes.
EFICACIA Y SEGURIDAD CLÍNICAS
Le eficacia y la seguridad clínica del ruxolitinib en la indicación autorizada han sido contrastadas mediante dos ensayos clínicos principales aleatorizados de fase III; el primero de ellos (COMFORT-I; 309 pacientes) es un estudio doblemente ciego y controlado con placebo, mientras que el segundo (COMFORT-II, 209 pacientes) es un estudio abierto, comparando el tratamiento con ruxolitinib con el mejor tratamiento disponible (seleccionado por el investigador en función de las circunstancias específicas de cada paciente). Las pautas posológicas de ruxolitinib fueron ajustadas en función del recuento plaquetario basal de los pacientes, con dosis orales de partida de 15 mg/12 h (100.000-200.000 plaquetas/µL) o 20 mg/12 h (>200.000 plaquetas/µL), con incrementos de 5 mg/12 h cada 4 semanas de tratamiento, de acuerdo a la evolución del recuento plaquetario y de la respuesta al tratamiento (según el volumen palpable del bazo).
La mediana de edad de los pacientes en ambos estudios es de 66 y 70 años (35-91); entre un 51% y un 58% eran varones. Presentaban mielofibrosis primaria en un 45-55% de los casos, secundaria a policitemia vera en un 27-33% y secundaria a trombocitemia esencial en un 14-23%. Un 40-47% tenían un grado 3 de fibrosis, un 33-41% de grado 2 y un 4-14% de grado 1. Tenían un tamaño mediano de bazo palpable bajo el margen costal de 14-16 cm, mutación de JAK2 en un 67-80% y habían sido tratados previamente con hidroxiurea en un 57-75%.
En el estudio COMFORT-I (Verstovsek, 2012) la duración del tratamiento fue de 24 semanas y la variable primaria de eficacia fue la proporción de pacientes con una reducción del volumen esplénico de al menos un 35% al final de las 24 semanas de tratamiento, determinado por técnicas de imagen (resonancia magnética). Entre las variables secundarias se incluyeron la durabilidad de la respuesta, la variación en las puntuaciones de los síntomas (MFSAF, Formulario de Evaluación de los Síntomas de Mielofibrosis, v2.0) y la supervivencia global de los pacientes.
El 41,9% (IC95% 34,1 a 50,1) de los pacientes tratados con ruxolitinib alcanzaron la variable primaria, en comparación con el 0,7% (IC95% 0 a 3,6) con placebo. Asimismo, el porcentaje de pacientes que experimentaron una reducción de al menos un 50% en la puntuación de síntomas al cabo de 24 semanas fue del 45,9% vs. 5,3% (p<0,0001), con una variación media de -8,6 puntos (desde un nivel basal de 18,0) vs. +3,2 (desde 16,5). La supervivencia global fue del 91,6% vs. 84,3%, con una reducción del riesgo de muerte del 50% (HR= 0,50; IC95% 0,25 a 0,98; p= 0,04). La tasa de descontinuación del tratamiento durante el estudio debido a eventos adversos fue del 11,0% con ruxolitinib vs. 10,6% placebo.
Tras un seguimiento medio de dos años (Verstovsek, 2013) de los pacientes que habían formado parte de la rama de tratamiento con ruxolitinib en el estudio anterior, 100 de los 155 inicialmente asignados al mismo mantenían el tratamiento (65%), mientras que todos aquellos asignados inicialmente a placebo comenzaron a ser tratados con el fármaco o suspendieron definitivamente su participación en el estudio, dentro de los tres meses posteriores al análisis primario. Los resultados mostraron una reducción media del volumen esplénico del 31,&% a la semana 24 y del 34,9% a la semana 96, manteniéndose igualmente los parámetros de calidad de vida de los pacientes. La supervivencia global al final del periodo fue claramente mejor con ruxolitinib, con una reducción del 42% del riesgo de muerte (HR= 0,58; IC95% 0,36 a 0,95; p= 0,03).
Por su parte, en el estudio COMFORT-II la duración del tratamiento fue de 48 seamanas, siendo la variable primaria de eficacia la proporción de pacientes con una reducción del volumen esplénico de al menos un 35% en las primeras 48 semanas de tratamiento, observándose que un 28,5% (IC95% 21,3 a 36,6) de los pacientes tratados con ruxolitinib habían alcanzado la variable primaria, en comparación con el 0% (IC95% 0 a 5,0) con el mejor tratamiento alternativo; a las 24 semanas, los valores correspondientes habían sido del 31,9% vs. 0%.
Un análisis sectorializado de los datos (Guglielmelli, 2014) ha mostrado que las respuesta sobre los síntomas y la esplenomegalia en el estudio COMFORT-II, así como el riesgo de desarrollar anaemia y trombocitopenia por el ruxolitinib fue similar entre los pacientes con diferentes perfiles mutacionales. Además, se apreció una reducción del riesgo de muerte entre aquellos portadores de mutaciones asociadas a peor pronóstico (ASXL1, EZH2, SRSF2, IDH1/2), con una reducción del 43% (HR= 0,57; IC95% 0,30 a 1,08) vs. mejor tratamiento, aunque no se llegó a alcanzar la significación estadística.
Tras un seguimiento medio de tres años (151 semanas) de los pacientes que inicialmente habían sido tratados con ruxolitinib en el estudio COMFORT-II y que habían mantenido el tratamiento posteriormente (45% al final de este periodo de seguimiento), se observó que la reducción se la esplenomegalia se había mantenido durante al menos 144 semanas, con la probabilidad del 50% (IC95% 36 a 63) entre los pacientes que habían obtenido algún grado de respuesta (Cervantes, 2013). La tasa de mortalidad de los pacientes originalmente aleatoriamente asignados a ruxolitiniv fue un 52% inferior a la de aquellos asignados al mejor tratamiento alternativo (HR= 0,48; IC95% 0,28 a 0,85; p= 0,009).
Desde el punto de vista de la seguridad, el perfil toxicológico del ruxolitinib se caracteriza fundamentalmente por la mielotoxicidad, manifestada comúnmente por trombocitopenia, anemia y neutropenia, consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. La incidencia de eventos adversos asociados al tratamiento con ruxolitinib en los ensayos clínicos principales es elevada, particularmente anemia (82%), trombocitopenia (70%), hemoorragias (33%) y neutropenia (16%); fueron también muy frecuentes las alteraciones de los valores de transaminasas (19-27%), hipercolesterolemia (17%), vértigo (15%), cefalea (14%), infecciones del tracto urinario (12%) y aumento de peso (10%). Considerando específicamente aquellos valorados como graves o muy graves (grados 3-4), los más comunes fueron anemia (43%), trombocitopenia (11%), neutropenia (6,6%), hemorragia (4,7%), aumento de niveles de transaminasas (1,3%) e infecciones del tracto urinario (1,0%).
Aproximadamente el 60% de los pacientes tratados con ruxolitinib tuvieron reducciones en su poslogía y cerca del 30% suspendieron el tratamiento, aunque la mayoría lo hizo por motivos diferentes a efectos adversos. Aunque muy comunes y eventualmente graves, las citopenias pudieron ser manejadas adecuadamente mediante ajuste posológico y transfusiones sanguíneas.
ASPECTOS INNOVADORES
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. Atendiendo a la condición de enfermedad rara, el medicamento fue designado como huérfano.
El nuevo fármaco ha demostrado ser capaz de reducir de forma notable la esplenomegalia en el 29-42% de los pacientes, frente a menos del 0-1% con placebo o con el mejor tratamiento alternativo disponible. Una reducción del 35% del volumen del bazo determinado mediante técnicas de imagen (resonancia magnética nuclear o tomografía computarizada) viene a equivaler a una reducción del 50% en el tamaño palpable externamente, lo que es considerado internacionalmente como un criterio objetivo de mejoría clínica, que se asocia con una mejora sustancial de los síntomas abdominales y del riesgo en caso de requerirse una esplenotomía. Por otro lado, es preciso indicar que la mayoría (57-75%) de los pacientes incluidos en los estudios habían sido tratados previamente con hidroxiurea, el único tratamiento actualmente autorizado de la mielofibrosis.
Los pacientes que más se benefician del tratamiento con ruxolitinib son los portadores de la mutación JAK2v617F+, con tasas de eficacia del 33-48% vs. 0-1%; una mutación que está presente, en término medio, en el 65% de los pacientes con mielofibrosis. Sin embargo, también es significativa la respuesta en pacientes no portadores de dicha mutación, con tasas de respuesta que siguen estando significativamente por encima (14-28% vs. 0-1%) de las obtenidas con placebo o con tratamientos alternativos (Becker, 2014).
Los datos procedentes de estudios de seguimiento de los estudios clínicos pivotales muestran que el tratamiento continuado es capaz de mantener la respuesta durante periodos de 1 a 3 años, si bien es cierto que en este tipo de estudios se produce la pérdida del seguimiento, por motivos diversos, de un buen número de los pacientes inicialmente incluidos, lo que limita la robustez de los resultados. No obstante, parece apreciarse un incremento de la supervivencia global de los pacientes tratados, que es preciso confirmar con nuevos estudios de seguimiento que incluyan un mayor número de pacientes y periodos más prolongados de observación.
Desde el punto de vista de la seguridad, el ruxolitinib se comporta básicamente como un agente mielotóxico, dando lugar a trombocitopenia, anemia, hemorragias y neutropenia, como consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. Aunque se trata de un efecto mielotóxico importante, es previsible y manejable mediante transfusiones y ajustes de la posología del fármaco.
El efecto mielotóxico del ruxolitinib no depende del estatus mutacional JAK2V617F, ya que esta mutación afecta a la estructura de la región por fuera del bolsillo de unión al ATP del enzima, por lo que los inhibidores de la cinasa JAK2 competitivos del ATP – como el ruxolitinib – no pueden distinguir entre las enzimas JAK2 normales y las mutadas. Por ello, este tipo de fármacos producen una mielosupresión no selectiva que, si bien es útil para controlar la hiperproliferación de las células hematopoyéticas en mielofibrosis y en otras patologías mieloproliferativas, pueden no ser capaces de eliminar los clones mutantes.
Aunque es evidente que el ruxolitinib forma parte del ya muy amplio grupo de inhibidores de tirosina cinasas (TKI, tyrosine-kinase inhibitors) disponibles, es preciso matizar que se trata de un fármaco muy peculiar, dada su acción altamente selectiva sobre tirosina cinasas de tipo Janus (JAK), las cuales están estrechamente ligadas a los procesos de señalización celular hematopoyética, lo que determina marcadamente tanto su perfil farmacológico como el toxicológico, agrupados por el efecto mielosupresor del fármaco; en este sentido – pero solo en éste – cabría hablar de cabeza de serie. En cualquier caso, se ha convertido en un referente para el desarrollo de nuevos fármacos más selectivos y eficaces en esta indicación, ofreciendo asimismo la posibilidad de combinar varios mecanismos con un objetivo terapéutico común, permitiendo diluir las consecuencias toxicológicas (Rosenthal, 2014).
En definitiva, se trata de un paso adelante en el tratamiento de una enfermedad rara, aunque su eficacia es modesta y su efecto real sobre la supervivencia global está todavía sujeto a estudio.
|
VALORACIÓN |
|
|---|---|
|
Ruxolitinib ► JAKAVI® (Novartis) |
|
|
Grupo Terapéutico (ATC): L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Citostáticos: inhibidores directos de la proteína cinasa. |
|
|
Indicaciones autorizadas: Tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar y puede resultar útil en algunos cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
⇑ |
|
Novedad molecular: Incorpora un mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma indicación terapéutica. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Imatinib |
Glivec |
Novartis |
2002 |
|
Erlotinib |
Tarceva |
Roche |
2006 |
|
Sunitinib |
Sutent |
Pfizer |
2007 |
|
Dasatinib |
Sprycel |
Bristol Myers Squibb |
2007 |
|
Sorafenib |
Nexavar |
Bayer |
2007 |
|
Lapatinib |
Tyverb |
Glaxo |
2008 |
|
Nilotinib |
Tasigna |
Novartis |
2008 |
|
Gefitinib |
Iressa |
AstraZeneca |
2010 |
|
Pazopanib |
Votrient |
Glaxo |
2011 |
|
Crizotinib |
Xalkori |
Pfizer |
2014 |
|
Vemurafenib |
Zelboraf |
Roche |
2014 |
|
Dabrafenib |
Tafinlar |
GlaxoSmithKline |
2014 |
|
Axitinib |
Inlyta |
Pfizer |
2014 |
|
Afatinib |
Giotrif |
Boehringer Ingelheim |
2014 |
|
Ruxolitinib |
Jakavi |
Novartis |
2015 |
|
Vandetanib |
Caprelsa |
AstraZeneca |
2015 |
BIBLIOGRAFÍA
Vandetanib CAPRELSA® (AstraZeneca)
Resumen
El vandetanib es un inhibidor de tirosina cinasas, particularmente VEGFR-2, VEGFR-3, EGFR y RET; ha sido autorizado para el tratamiento del cáncer medular de tiroides agresivo y sintomático en pacientes con enfermedad irresecable localmente avanzada o metastásica. Incrementa de forma significativa la duración de la supervivencia libre de progresión tumoral (mediana de 30,5 vs. 19,3 meses); sin embargo, no se ha podido confirmar hasta ahora un efecto sobre la supervivencia global. El medicamento presenta un perfil toxicológico importante, destacando la diarrea y, en particular, el alargamiento del segmento QT del electrocardiograma, un signo de riesgo de graves arritmias cardiacas (especialmente, torsades de pointes). El IPT recomienda el empleo de vandetanib en pacientes con cáncer medular de tiroides agresivo y sintomático con enfermedad metastásica, en los que exista evidencia radiológica de una franca progresión tumoral, así como una clara sintomatología clínica asociada a la enfermedad; también en aquellos en los que las lesiones no sean evaluables radiológicamente y que presenten un tiempo de duplicación de CTN o CEA menor de 24 meses; igualmente, en los pacientes con enfermedad locorregional no controlada y claramente sintomática, tras cirugía y/o tratamiento con radioterapia (incluso en ausencia de metástasis).
CÁNCER MEDULAR DE TIROIDES
Los tumores malignos de tiroides son los más frecuentes y los que mayor mortalidad ocasionan del sistema endocrino; aun así solo son responsables del 1% de los fallecimientos producidos por cáncer. Tradicionalmente, los tumores malignos1 epiteliales primarios o carcinomas de tiroides se clasifican según el tipo celular en carcinomas de células foliculares, de células parafoliculares C y mixtos. Los carcinomas de células foliculares pueden ser diferenciados (papilar y folicular), poco diferenciados (insular y otros) o indiferenciados (anaplásico), mientras que el único tipo de carcinoma de células parafoliculares (células C) es el cáncer medular. Adicionalmente, hay cánceres tiroideos constituidos por poblaciones celulares tumorales mixtas. Desde el punto de vista epidemiológico, la denominación de cáncer de tiroides incluye a todos los tumores derivados de células tiroideas, quedando por tanto excluidos los linfomas y los sarcomas que asientan en esta glándula. Más del 90% de las neoplasias tiroideas corresponden a tumores bien diferenciados (Bos, 2012).
La incidencia de cáncer de tiroides es considerablemente más elevada en mujeres que en varones. Sin embargo, la distribución por edad es semejante en ambos sexos; a partir de los 10-15 años, la incidencia comienza a aumentar (de forma más pronunciada en mujeres) hasta alcanzar un máximo en torno a los 60 años en mujeres y los 55 en varones, con unas tasas de, aproximadamente, 11 y 5 casos por 100.000 en mujeres y varones, respectivamente. A partir de esa edad la incidencia comienza a descender progresivamente. En España las tasas de incidencia oscilan entre el 3,6% y 4,9% en mujeres y el 0,6 y 1,3% en varones. Su incidencia a lo largo de las últimas décadas ha ido aumentando (Lope, 2005).
La exposición a radiación ionizante es el factor de riesgo mejor evidenciado en el cáncer de tiroides. Dicha glándula es uno de los órganos más radiosensibles del organismo, debido a su localización superficial, a su alto grado de oxigenación y a su alta tasa de división celular. El carcinoma papilar es el principal tipo celular inducido por radiación. Las principales fuentes de exposición son la irradiación terapéutica y la contaminación ambiental por accidentes industriales o detonaciones nucleares.
El bocio multinodular endémico asociado a un contenido inadecuado de iodo en la dieta que se presenta en ciertas áreas geográficas con suelos pobres en iodo (normalmente alejados de zonas costeras) parecen estar asociado con un alto riesgo de carcinoma folicular y posiblemente también de anaplásico, mientras que las áreas ricas en iodo tienden a presentar un aumento del riesgo de carcinoma papilar.
Hay abundantes datos que demuestran la marcada asociación entre la presencia de nódulos tiroideos benignos (adenomas) y bocio con el cáncer de tiroides. Esta asociación podría reflejar la existencia de diversas situaciones como una posible relación causal, una lesión precursora, el efecto de un tratamiento, o bien ser factores de riesgo independientes. Sin embargo, existe una discordancia importante en lo concerniente a la asociación entre tiroiditis de Hashimoto y el cáncer de tioides; igualmente, el papel de la tirotoxicosis en la etiología de este tipo de tumor permanece incierto. En relación con otras enfermedades no tiroideas, en algunos estudios se ha observado un exceso de carcinomas tiroideos entre pacientes con cáncer de mama, y viceversa; también, aunque en menor medida, se ha encontrado una asociación con otras neoplasias, hiperparatiroidismo, anemia aplásica, acromegalia y ataxia telangiectásica.
El hecho de que la incidencia del cáncer de tiroides sea de 2 a 3 veces mayor en mujeres que en varones, fundamentalmente durante su edad reproductiva, unido al cambio de tamaño y de actividad que sufre la glándula durante el ciclo menstrual, sugiere que las hormonas femeninas pueden desempeñar un papel importante en la etiología de este tumor. Concentraciones elevadas de la TSH están asociadas con un mayor riesgo de padecer cáncer de tiroides, y la tasa de secreción de esta hormona se eleva durante la pubertad, el embarazo, el parto, el uso de anticonceptivos orales, la tiroidectomía parcial, el consumo de productos bociógenos y la radiación del cuello. Sin embargo, no parece que exista una asociación clara entre el cáncer de tiroides y los factores menstruales y reproductivos, aunque algunos estudios han sugerido un exceso de riesgo de cáncer papilar asociado con el uso de anticonceptivos orales, principalmente entre usuarias habituales. Se han descrito también como posibles factores de riesgo los tratamientos para suprimir la lactancia y los tratamientos fertilizantes, el tratamiento hormonal posmenopáusico y el embarazo a edades tardías. Igualmente se ha comunicado un aumento de riesgo en los primeros años tras el parto, pero no se ha encontrado asociación con historia previa de abortos, infertilidad ni paridad.
El cáncer papilar de tiroides supone más del 70% de los tumores tiroideos y es el de mejor pronóstico (90% supervivencia a los 10 años). Es más frecuente en mujeres (3:1), en la cuarta década de la vida. Se suele presentar como un nódulo único, indoloro, con adenopatías también indoloras y de lento crecimiento. Metastatiza vía linfática y de forma precoz en la cadena ganglionar cervical pero sólo se aprecian metástasis a distancia si está muy avanzado puesto que su diseminación hematógena es rara. Histológicamente es un tumor multicéntrico (20%), bien diferenciado, que deriva de células foliculares, no encapsulado y con calcificaciones que forman los típicos cuerpos de psamoma.
Por su parte, el cáncer folicular supone el 10-15% de los tumores tiroideos. Es también más frecuente en mujeres (3:1), en la quinta década de la vida y en zonas de bocio endémico, debido a escaso aporte nutricional de iodo. Se suele presentar como un nódulo único, indoloro sobre un tiroides con bocio o ya en forma de metástasis. Metastatiza vía sanguínea afectando a pulmón (más típico en gente de edad avanzada y sin síntomas) y hueso (más común en la infancia, produciendo dolor, inflamación y fracturas osteolíticas). Histológicamente es un tumor prácticamente idéntico al tejido tiroideo normal que deriva de células foliculares y su pronóstico de supervivencia se halla entre 63-75 % los 10 años.
El cáncer anaplásico es el menos común y representa en torno al 2% de los tumores tiroideos. También es más frecuente en mujeres, pero solo por encima de los 65 años. Se presenta como un nódulo único, doloroso, de consistencia pétrea, de rápido crecimiento y adherido al tejido circundante, con invasión precoz hacia laringe produciendo afonía y hacia esófago produciendo disfagia. Su diseminación es ganglionar, metastatizando de manera precoz a distancia por lo que el pronóstico es malo y su supervivencia es muy baja, apenas unos meses. Deriva también de células foliculares del tiroides, pero al ser muy agresivo las células están muy distorsionadas e infiltra con rapidez. Algunos tumores papilares pueden degenerar en anaplásicos.
El cáncer medular de tiroides es también infrecuente, constituyendo en torno al 2,5-10% de los tumores tiroideos y, a diferencia de los anteriores, la frecuencia entre hombres y mujeres es casi idéntica (1:1,3). Los casos esporádicos son diagnosticados durante la sexta o séptima década de la vida, pero los ligados a factores genéticos se diagnostica habitualmente en pacientes jóvenes (generalmente, antes de los 20 años). Se presenta como uno o varios nódulos no dolorosos o como metástasis a distancia en el momento del diagnóstico. Es muy típico el aumento de calcitonina en sangre ya que histológicamente deriva de las células parafoliculares del tiroides, productoras de calcitonina (de ahí el término de células C). En más del 10% se observan episodios intensos de diarrea sin que se conozca su etiología. Se disemina rápidamente por vías linfática y hematógena, y su pronóstico es mejor que el anaplásico, pero peor que papilar y folicular.
El componente genético es particularmente importante en el cáncer medular de tiroides, ya que una cuarta parte de los casos aparecen ligados a síndromes hereditarios (síndromes de neoplasia endocrina múltiple, MEN) con herencia autosómica dominante. Las principales vías oncogénicas de inicio y progreso de la carcinogénesis tiroidea son la MAPK (RAS/RAF/mitogen-activated protein kinase) y la PI3/Akt (phophatidylinositide 3-kinase), debido a la importancia que tienen en la supervivencia, proliferación, diferenciación y motilidad, y considerando que la malignificación celular implica la progresiva acumulación cinasas activadas o de genes supresores inactivados. Entre los condicionantes genéticos relacionados con el cáncer medular de tiroides, los más relevantes son los polimorfismos del protooncogén RET (Rearranged during transfection), presentes en el 95% de todos los casos familiares de cáncer medular de tiroides y en el 40-50% de los esporádicos. Hasta un 10% de los casos esporádicos también se relacionan con mutaciones RAF (KRAS/HRAS/NRAS) (Alonso-Gordoa, 2015).
El tratamiento del cáncer medular sin metástasis a distancia es la cirugía que, si se practica convenientemente a tiempo, permite con frecuencia considerar curado al paciente. La intervención suele consistir en la extirpación del tiroides junto con los ganglios cervicales, lo que implica un tratamiento con tiroxina de por vida. El principal biomarcador para el seguimiento de la evolución tumoral es el nivel de calcitonina en sangre: Si después de la cirugía persisten altas las concentraciones de calcitonina, esto sugiere que el tumor ha metastasizado. En caso de recidiva tumoral, siempre que no haya metástasis distales, puede reintentarse la extirpación del tumor o, en los casos donde no proceda la cirugía, radioterapia local o regional.
En los cuadros con metástasis distales, el enfoque suele ser conservador dado que se trata de un tumor que crece lentamente y que, con un tratamiento adecuado de los síntomas, puede mantenerse asintomático durante largo tiempo. La quimioterapia tiene una eficacia muy limitada en estos casos (con tasas de respuesta que no superan el 20%), aunque los protocolos con dacarbacina, doxorubicina y fluorouracilo pueden producir una estabilización prolongada del tumor.
La supervivencia a los cinco años para los pacientes con cáncer medular de tiroides de diseminación regional es cercana al 80%, tasa que desciende a la mitad en aquellos con metástasis distales, con una supervivencia media de 2-3 años. Los pacientes con la forma hereditaria de cáncer medular tienen un mejor pronóstico que aquellos con la forma esporádica, aunque posiblemente esto pueda deberse – al menos en parte – a un diagnóstico más precoz y, por tanto, a un tratamiento más eficaz.
ACCIÓN Y MECANISMO
El vandetanib es un inhibidor de diferentes tipos de tirosina cinasas, pero particularmente del receptor 2 del factor de crecimiento endotelial vascular (VEGFR-2), del receptor del factor de crecimiento epidérmico (EGFR, también denominado HER-1, erbB1 o c-erbB) y del protooncogén RET (Rearranged during transfection), así como – en menor medida – del receptor 3 del factor de crecimiento endotelial vascular (VEGFR-3). El medicamento ha sido autorizado para el tratamiento del cáncer medular de tiroides agresivo y sintomático en pacientes con enfermedad no resecable localmente avanzada o metastásica.
El vandetanib es un potente inhibidor de las tirosina cinasas mencionadas, compitiendo con el trifosfato de adenosina (ATP) en la zona catalítica de dichos enzimas, lo que da lugar a un bloqueo de la cascada de fosforilación y, por tanto, de la señalización bioquímica ligada al estímulo de los receptores mencionados por sus respectivos ligandos naturales.
La consecuencia farmacológica principal de la inhibición de las tirosina cinasas mencionadas es una inhibición de los procesos de angiogénesis tumoral; en concreto, la inhibición de las tirosina cinasas asociadas al VEGFR inhibe la formación de neovasos en los tumores. Por su parte, el efecto inhibidor del vandetanib sobre el EGFR impide la estimulación de las células tumorales y endoteliales. Las anomalías en el EGFR están involucradas en el complicado proceso de carcinogénesis de varios tipos de tumores sólidos, incluyendo la proliferación celular incontrolada, la migración celular, invasión del estroma, angiogénesis y resistencia a la apoptosis. Los ligandos específicos del EGFR son el propio EGF (Factor de Crecimiento Epidérmico) y otros péptidos relacionados con éste, entre los que cabe incluir el Factor de Crecimiento Transformante alfa (TGF-a) y la amfiregulina, entre otros. Tanto el EGF como el TGF-a son capaces de desencadenar, tras su acción sobre el EGFR, una serie de eventos citoquímicos necesarios para continuar con el ciclo de división celular.
Adicionalmente, los polimorfismos del protooncogén RET están presentes en el 95% de todos los casos familiares de cáncer medular de tiroides y en el 40-50% de los esporádicos, donde el vandetanib – activo tanto sobre las formas naturales o salvajes como sobre la mayoría de las formas mutadas de este gen – inhibe específicamente la proliferación de las líneas celulares del cáncer medular de tiroides.
ASPECTOS MOLECULARES
El vandetanib está estrechamente relacionado farmacológicamente con otros miembros de la serie de inhibidores de tirosina cinasas. Presenta una evidente relación estructural con el afatanib, un potente inhibidor de las tirosina cinasas asociadas al EGFR.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del vandetanib en la indicación autorizada han sido adecuadamente contrastadas mediante un ensayos clínico principal de fase III (Wells, 2012), multicéntrico, multinacional, aleatorizado, doblemente ciego y controlado con placebo, llevado a cabo sobre 331 pacientes con cáncer medular de tiroides no resecable localmente avanzado o metastásico, confirmado histológicamente y con una esperanza de vida de al menos 12 semanas, en el que los pacientes recibieron por vía oral un tratamiento a base de vandetanib (300 mg/24 h) o placebo hasta la confirmación de la progresión de la enfermedad. La mediana de edad de los pacientes era de 51 años, (18% <40 años), un 57% eran varones, 95% caucásicos, con una mediana de peso de 70 kg. Un 90% de los casos eran esporádicos y en un 95% había metástasis. Un 90% había sido sometido anteriormente a tiroidectomía, un 81% había recibido radioterapia y un 21% quimioterapia. La variable primaria de eficacia fue la supervivencia libre de progresión tumoral2, valorada de forma ciega y centralizada de acuerdo a criterios RECIST3. Como variables secundarias se determinaron la tasa global de respuesta y la supervivencia global, así como otros parámetros de tipo bioquímico.
Tras un seguimiento medio de 24 meses, el porcentaje de pacientes con progresión tumoral o muerte fue del 31,6% con vandetanib vs. 51,0% con placebo, lo que reduce el riesgo en un 54% (HR=0,46; IC95% 0,31 a 0,69; p>0,001). La mediana de tiempo fue de 19,3 meses con placebo pero no pudo calcularse directamente para el vandetanib, debido al insuficiente número de eventos requeridos estadísticamente; no obstante, utilizando una aproximación estadística (modelo de Weibull) la mediana fue de 30,5 meses con vandetanib y de 19,3 meses con placebo. El efecto sobre la supervivencia libre de progresión tumoral fue homogéneo en todos los subgrupos analizados en función del estatus tumoral.
La tasa de respuesta objetiva global fue del 45% vs. 13,0% (HR=5,48; IC95% 2,99 a 10,79; p>0,0001), aunque ninguno de los pacientes manifestó una respuesta completa (todas las respuestas fueron parciales). En cuanto a la supervivencia global, no pudo apreciarse una diferencia estadísticamente significativa debido a que en el momento de la valoración se habían producido menos del 50% (14% de fallacimientos con vandetanib y 16% con placebo) de los eventos requeridos para dicha evaluación (HR=0,89; IC95% 0,48 a 1,65). El tiempo hasta el empeoramiento del dolor4 fue de 7,9 vs. 3,3 meses. Asimismo, hubo una mejoría en el peso en el 28,6% vs. 11,0% (HR=3,24; IC95% 1,69 a 6,76; p=0,0003).
Considerando el estatus genético de los pacientes, la tasa de progresión tumoral a los 12 meses fue del 34,3% vs. 54,0% en pacientes RET+ y del 27,2% vs. 43,2% con estatus RET desconocido (el número de casos de RET- fue pequeño e insuficiente para validación estadística).
Desde el punto de vista de la seguridad clínica, la incidencia de eventos adversos relacionados con el tratamiento con vandetanib y placebo fue del 96,1% vs. 59,6% (55,4% vs. 24,2%, de grado ≥3). La tasa de mortalidad asociada a eventos adversos relacionados con el tratamiento fue del 2,2% vs. 2,0%. El porcentaje de pacientes que requirieron ajustes posológicos para controlar los efectos adversos fue del 35% vs. 3% y el de aquellos que suspendieron el tratamiento por este motivo fue del 12% vs. 3%.
{kind=link}
Los eventos adversos más frecuentemente descritos con vandetanib y placebo en el ensayo clínico principal fueron diarrea (56,7% vs. 27,3%; 10,8% vs. 2,0% de grado ≥3 o 4), erupciones exantemáticas (88,7% vs. 23,2%; 6,9% vs. 0%), náusea (36,4% vs. 20,2%; 1,7% vs. 0%), hemorragias (15,6% vs. 11,1%; 1,7% vs. 0%), eventos relacionados con un alargamiento del intervalo QT del electrocardiograma (15,6% vs. 4,0%; 8,6% vs. 1,0%), hipertensión (32% vs. 5%) y cefalea (26% vs. 9%).
ASPECTOS INNOVADORES
El vandetanib es un inhibidor de tirosina cinasas, particularmente VEGFR-2, VEGFR-3, EGFR y RET; ha sido autorizado para el tratamiento del cáncer medular de tiroides agresivo y sintomático en pacientes con enfermedad irresecable localmente avanzada o metastásica. En este caso, se entiende como agresivos y sintomáticos aquellos casos en los que se constata un rápido deterioro del estado clínico (determinado por síntomas y por signos bioquímicos y radiológicos); es decir, aquellos que tienen necesidad urgente de tratamiento. En estos casos, parece que debe considerarse como la primera opción (Cooper, 2014).
La superioridad clínica del vandetanib sobre el placebo ha quedado constatada en estos pacientes, en los que incrementa de forma significativa la duración de la supervivencia libre de progresión tumoral (mediana de 30,5 vs. 19,3 meses), una superioridad que ha sido comprobada en función del estatus tumoral. Sin embargo, no se ha podido confirmar hasta ahora (debido a que no se ha registrado un número suficiente de eventos) un efecto sobre la supervivencia global, es decir, la mortalidad general de los pacientes.
Aunque su efecto parece ser clara en pacientes con un estatus genético de RET+ y en aquellos con estatus desconocido, no puede decirse lo mismo de aquellos RET-, cuyos datos son inconcluyentes. Por este motivo, la Agencia Europea de Medicamentos (EMA), dio un carácter condicional a su autorización de comercialización, condicionando la autorización definitiva a la realización de un ensayo clínico abierto en la Unión Europea5 en los que se comparen los resultados obtenidos en pacientes RET+ y RET- con cáncer medular de tipo esporádico (no familiar). En cualquier caso, la existencia de respuestas favorables en algunos casos de RET- como en pacientes con estatus RET desconocido, sugiere que el vandetanib podría ser útil en cuadros con otras mutaciones asociadas al cáncer medular de tiroides no relacionadas con RET (Alonso-Gordoa, 2015).
El medicamento presenta un perfil toxicológico importante, tanto por frecuencia como por relevancia clínica de los efectos adversos, destacando la diarrea y, en particular, el alargamiento del segmento QT del electrocardiograma, un signo de riesgo de graves arritmias cardiacas (especialmente, torsades de pointes). El vandetanib se asocia con un efecto dependiente de la dosis (que con la dosis recomendada de 300 mg/24 h es de 35 mseg de mediana), especialmente en las mujeres (75% vs. 25% en varones); en este sentido, la elevada semivida de eliminación del vandetanib (alrededor de 19 días) supone un problema, dado que la posología se ajusta fundamentalmente en función de la intensidad de los eventos adversos observados, particularmente con el alargamiento del segmento QT del electrocardiograma (y sus manifestaciones arrítmicas) y el potencial deterioro de la función renal, lo que podría conducir a la persistencia de altas concentraciones del fármaco durante periodos prolongados (incluso meses) después de haber finalizado el tratamiento o haber ajustado la posología. Por este motivo, la EMA ha requerido la remisión de datos adicionales para la selección de la dosis en la optimización del cociente beneficio/riesgo.
Por su parte, el Informe de Posicionamiento Terapéutico (AEMPS, 2013) recomienda el empleo de vandetanib en pacientes con cáncer medular de tiroides agresivo y sintomático con enfermedad metastásica, en los que exista evidencia radiológica de una franca progresión tumoral, así como una clara sintomatología clínica asociada a la enfermedad; también en aquellos en los que las lesiones no sean evaluables radiológicamente y que presenten un tiempo de duplicación de CTN o CEA menor de 24 meses; igualmente, en los pacientes con enfermedad locorregional no controlada y claramente sintomática, tras cirugía y/o tratamiento con radioterapia (incluso en ausencia de metástasis).
|
VALORACIÓN |
|
|---|---|
|
Vandetanib
CAPRELSA® (AstraZeneca) |
|
|
Grupo Terapéutico (ATC): L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Citostáticos: inhibidores directos de la proteína cinasa. |
|
|
Indicaciones autorizadas: Tratamiento del cáncer medular de tiroides agresivo y sintomático en pacientes con enfermedad no resecable localmente avanzada o metastásica. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Imatinib |
Glivec |
Novartis |
2002 |
|
Erlotinib |
Tarceva |
Roche |
2006 |
|
Sunitinib |
Sutent |
Pfizer |
2007 |
|
Dasatinib |
Sprycel |
Bristol Myers Squibb |
2007 |
|
Sorafenib |
Nexavar |
Bayer |
2007 |
|
Lapatinib |
Tyverb |
Glaxo |
2008 |
|
Nilotinib |
Tasigna |
Novartis |
2008 |
|
Gefitinib |
Iressa |
AstraZeneca |
2010 |
|
Pazopanib |
Votrient |
Glaxo |
2011 |
|
Crizotinib |
Xalkori |
Pfizer |
2014 |
|
Vemurafenib |
Zelboraf |
Roche |
2014 |
|
Dabrafenib |
Tafinlar |
GlaxoSmithKline |
2014 |
|
Axitinib |
Inlyta |
Pfizer |
2014 |
|
Afatinib |
Giotrif |
Boehringer Ingelheim |
2014 |
|
Ruxolitinib |
Jakavi |
Novartis |
2015 |
|
Vandetanib |
Caprelsa |
AstraZeneca |
2015 |
BIBLIOGRAFÍA
Medicamentos con nuevos principios activos autorizados por EMA/AEMPS
|
MEDICAMENTOS DE USO HUMANO AUTORIZADOS (EMA/AEMPS) DURANTE LOS ÚLTIMOS DOCE MESES, CON NUEVOS PRINCIPIOS ACTIVOS QUE AÚN NO ESTÁN COMERCIALIZADOS EN ESPAÑA |
||||||
|---|---|---|---|---|---|---|
|
Principio activo |
Medicamento® |
Laboratorio |
Autorización |
GT |
Indicación autorizada1 |
|
|
Ramucirumab |
Cyramza |
Lilly |
2014/12/19 |
L01XC |
Cáncer de estómago (O) |
|
|
Olaparib |
Lynparza |
AstraZeneca |
2014/12/16 |
L01XX |
Cáncer de ovario (O) |
|
|
Dulaglutida |
Trulicity |
Lilly |
2014/11/21 |
A10BX |
Diabetes mellitus de tipo 2 |
|
|
Nintendanib |
Vargatef |
Boehringer Ingelheim |
2014/11/21 |
L01XE |
Cáncer de pulmón |
|
|
Naloxegol |
Moventig |
AstraZeneca |
2014/12/08 |
A06AH |
Estreñimiento |
|
|
Darunavir/Cobicistat |
Rezolsta |
Janssen Cilag |
2014/11/19 |
J05AE |
Infección VIH |
|
|
Aclidinio/Formoterol |
Brinika/Duakir Genuair |
Almirall |
2014/11/19 |
R03AL |
EPOC |
|
|
Sofosbuvir/Ledipasvir |
Harvoni |
Gilead |
2014/11/17 |
J05AX |
Hepatitis C |
|
|
Ibrutinib |
Imbruvica |
Janssen Cilag |
2014/10/21 |
L01XE |
Leucemia linfocítica crónica (O) |
|
|
Idelalisib |
Zydelig |
Gilead |
2014/09/18 |
L01XX |
Leucemia/linfoma |
|
|
Ataluren |
Translarna |
PTC Therapeutics |
2017/07/31 |
– |
Distrofia muscular de Duchenne (O) (C) |
|
|
Obinotuzumab |
Gazyvaro |
Roche |
2014/07/23 |
L01XC |
Leucemia linfocítica crónica (O) |
|
|
Peginterferón beta-1a |
Plegridy |
Biogen |
2014/07/18 |
L03AB |
Esclerosis múltiple |
|
|
Trametinib |
Mekinist |
Glaxo |
2017/06/30 |
L01XE |
Melanoma |
|
|
Vedolizumab |
Entyvio |
Takeda |
2014/05/22 |
L04AA |
Enfermedad Inflamatoria Intestinal |
|
|
Siltuximab |
Sylvant |
Janssen Cilag |
2014/05/22 |
– |
Hiperplasia nódulo linfático (O) |
|
|
Empagliflozina |
Jardiance |
Boehringer Ingelheim |
2014/05/22 |
A10BX |
Diabetes tipo 2 |
|
|
Elosulfasa alfa |
Vimizim |
BioMarin |
2014/04/28 |
A16AB |
Mucopolisacaridosis IV (O) |
|
|
Umeclidinio, bromuro |
Incruse |
Glaxo |
2014/04/28 |
R03BB |
EPOC |
|
|
Delamanid |
Deltyba |
Otsuka |
2014/04/28 |
J04AK |
Tuberculosis multirresistente (O) (C) |
|
|
Riociguat |
Adempas |
Bayer |
2014/03/27 |
C02KX |
Hipertensión pulmonar (O) |
|
|
Albiglutida |
Eperzan |
GlaxoSmithKline |
2014/03/21 |
A10BX |
Diabetes mellitus de tipo 2 |
|
|
Lurasidona |
Latuda |
Takeda |
2014/03/21 |
N05AE |
Esquizofrenia |
|
|
Cabozantinib |
Cometriq |
TMC |
2014/03/21 |
L01XE |
Cáncer de tiroides (O) (C) |
|
|
Bedaquilina |
Sirturo |
Janssen Cilag |
2014/03/05 |
J04AX |
Tuberculosis multirresistente (O) (C) |
|
|
Florbetaben (18F) |
Neuraceq |
Piramidal Imaging |
2014/02/20 |
V09AX |
Diagnóstico Alzheimer |
|
1 O= Medicamento huérfano; C= Autorizado condicionalmente; E= Autorizado en condiciones excepcionales
|
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA EN LOS ÚLTIMOS DOCE MESES |
||||||||||
|
PAM |
Principio activo |
Medicamento |
INNOVACIÓN |
Indicación principal |
Laboratorio |
|||||
|
a |
b |
c |
d |
e |
f |
|||||
|
380 |
Daclatasvir |
Daklinza |
⇑ |
⇑ |
⇑ |
|
|
♣♣♣ |
Hepatitis C |
Bristol-Myers Squib |
|
380 |
Vandetanib |
Caprelsa |
⇑ |
|
|
|
|
♣♣ |
Cáncer medular de tiroides |
AstraZeneca |
|
380 |
Ruxolitinib |
Jakavi |
⇑ |
⇑ |
|
|
|
♣♣ |
Mielofibrosis |
Novartis |
|
380 |
Fumarato de dimetilo |
Tecfidera |
⇑ |
⇑ |
|
|
|
♣♣ |
Esclerosis múltiple |
Biogen Idec |
|
380 |
Teriflunomida |
Aubagio |
|
|
|
⇑ |
|
♣♣ |
Esclerosis múltiple |
Sanofi Aventis |
|
379 |
Enzalutamida |
Xtandi |
⇑ |
|
|
|
|
♣♣ |
Cáncer de próstata |
Astellas |
|
378 |
Vacuna herpes zóster |
Zostavax |
⇑ |
|
|
|
|
♣♣ |
Prevención del herpes zóster |
Sanofi Pasteur MSD |
|
378 |
Lipegfilgrastim |
Lonquex |
|
|
|
|
|
♣ |
Prevención de neutropenia |
Teva |
|
378 |
Nalmefeno |
Selincro |
|
|
|
⇑ |
|
♣♣ |
Alcoholismo |
Lundbeck |
|
377 |
Sofosbuvir |
Sovaldi |
⇑ |
⇑ |
⇑ |
⇑ |
|
♣♣♣ |
Hepatitis C |
Gilead |
|
377 |
Decitabina |
Dacogen |
⇑ |
|
|
|
|
♣♣♣ |
Leucemia mieloide aguda |
Janssen Cilag |
|
377 |
Vilanterol/Fluticasona |
Relvar Ellipta |
|
|
|
⇑ |
|
♣♣ |
Asma y EPOC |
Glaxo SmithKline |
|
377 |
Vacuna meningococo B |
Bexsero |
⇑ |
|
|
|
⇑ |
♣♣♣ |
Prevención meningitis |
Novartis |
|
377 |
Dexmedetomidina |
Dexdor |
|
⇑ |
|
|
|
♣♣ |
Sedoanalgesia |
Orion |
|
377 |
Dolutegravir |
Tivicay |
⇑ |
|
|
⇑ |
|
♣♣ |
Infección por VIH |
Glaxo SmithKline |
|
376 |
Brentuximab vedotina |
Adcetris |
⇑ |
⇑ |
|
|
|
♣♣ |
Linfomas |
Takeda |
|
376 |
Ocriplasmina |
Jetrea |
⇑ |
|
⇑ |
|
|
♣♣ |
Tracción vitreomacular |
Alcon Cusi |
|
376 |
Ofatumumab |
Arzerra |
⇑ |
|
|
|
|
♣♣ |
Leucemia linfocítica crónica |
Glaxo SmithKline |
|
376 |
Pirfenidona |
Esbriet |
⇑ |
⇑ |
|
|
|
♣♣ |
Fibrosis pulmonar idiopática |
InterMune |
|
376 |
Simeprevir |
Olysio |
⇑ |
⇑ |
⇑ |
⇑ |
|
♣♣♣ |
Hepatitis C |
Janssen Cilag |
|
376 |
Tetradecilsulfato sódico |
Veinfibro |
|
|
|
|
|
♣ |
Escleroterapia de venas varicosas |
STD |
|
375 |
Afatinib |
Giotrif |
⇑ |
|
|
|
|
♣♣ |
Cáncer de pulmón |
Boehringer Ingelheim |
|
375 |
Linaclotida |
Constella |
⇑ |
⇑ |
|
|
|
♣♣ |
Síndrome del intestino irritable |
Almirall |
|
375 |
Pertuzumab |
Perjeta |
⇑ |
⇑ |
|
|
|
♣♣♣ |
Cáncer de mama |
Roche |
|
375 |
Pomalidomida |
Imnovid |
⇑ |
|
⇑ |
|
|
♣♣ |
Mieloma múltiple |
Celgene |
|
374 |
Axitinib |
Inlyta |
⇑ |
|
|
|
|
♣♣ |
Cáncer renal |
Pfizer |
|
374 |
Brimonidina2 |
Mirvaso |
⇑ |
|
|
|
|
♣♣ |
Rosácea |
Galderma |
|
374 |
Dabrafenib |
Tafinlar |
⇑ |
|
|
⇑ |
|
♣♣ |
Melanoma |
Pfizer |
|
374 |
Vemurafenib |
Zelboraf |
⇑ |
|
|
⇑ |
|
♣♣ |
Melanoma |
Roche |
|
373 |
Lisdexanfetamina |
Elvanse |
⇑ |
|
|
|
|
♣♣ |
Trastorno por déficit |
Shire |
|
372 |
Avanafilo |
Spedra |
|
|
|
|
|
♣ |
Disfunción eréctil |
Menarini |
|
372 |
Mirabegrón |
Betmiga |
|
⇑ |
⇑ |
|
|
♣♣ |
Incontinencia urinaria |
Astellas |
|
372 |
Regadenosón |
Rapiscan |
|
|
|
⇑ |
|
♣♣ |
Diagnóstico en cardiología |
Mallincrockrodt |
|
371 |
Catridecacog |
Novothirteen |
|
|
|
|
⇑ |
♣♣ |
Déficit congénito Factor XIII |
Novo Nordisk |
|
371 |
Florbetapir [18F] |
Amyvid |
⇑ |
|
|
|
|
♣♣ |
Diagnóstico Alzheimer |
Lilly |
A. Clínica: Mejora la eficacia clínica con relación al tratamiento estándar.
B. Molecular: Incorpora variaciones significativas en la estructura molecular
y/o en el mecanismo de acción.
C. Seguridad: Mejora el perfil toxicológico con relación a la terapia estándar.
D. Farmacocinética: Mejora las características farmacocinéticas, con incidencia potencial o demostrada en las condiciones de uso y en la respuesta del paciente.
E. Técnico-Económica: Mejora algún aspecto farmacoeconómico y/o farmacotécnico.
F. Valoración Global
2 La brimonidina estaba previamente comercializada para el tratamiento tópico oftálmico del glaucoma.
VALORACIÓN DE LA INNOVACIÓN TERAPÉUTICA EN PANORAMA ACTUAL DEL MEDICAMENTO
Es importante indicar que se valora el grado de innovación. Todos los medicamentos no innovadores tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas. Asimismo, debe considerarse que ésta es una evolución que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta ese momento, lo que no prejuzga, en ningún caso, el potencial desarrollo de nuevas indicaciones clínicas ni la posible aparición de aspectos desfavorables (efectos adversos graves previamente desconocidos, interacciones, etc.). Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
- SIN INNOVACIÓN. No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas (♣)
- INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar (♣♣)
- INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar (♣♣♣).
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
- Evidencia clínica: mediante estudios controlados específicamente diseñados y desarrollados para demostrar lo que pretende ser un avance o mejora sobre la terapia estándar.
- Potencialidad: existencia de aspectos en el medicamento que racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostradas mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
|
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA DURANTE LOS ÚLTIMOS DOCE MESES, POR GRUPOS TERAPÉUTICOS |
||||
|---|---|---|---|---|
|
GRUPO TERAPéUTICO |
Principio activo |
Medicamento |
Laboratorio |
PAM |
|
A. Tracto alimentario y metabolismo |
Linaclotida |
Constella |
Almirall |
375 |
|
B. Sangre y órganos hematopoyéticos |
Catridecacog |
Novothirteen |
Novo Nordisk |
371 |
|
C. Aparato cardiovascular |
Regadenosón |
Rapiscan |
Mallinckrodt |
372 |
|
Tetradecilsulfato sódico |
Veinfibro |
STD |
376 |
|
|
D. Terapia dermatológica |
Brimonidina |
Mirvaso |
Galderma |
374 |
|
G. Terapia genitourinaria |
Avanafilo |
Spedra |
Menarini |
372 |
|
Mirabegrón |
Betmiga |
Astellas |
372 |
|
|
J. Antiinfecciosos, uso sistémico |
Daclatasvir |
Daklinza |
Bristol-Myers Squibb |
380 |
|
Dolutegravir |
Tivicay |
Glaxo SmithKline |
377 |
|
|
Simeprevir |
Olysio |
Janssen Cilag |
376 |
|
|
Sofosbuvir |
Sovaldi |
Gilead |
377 |
|
|
Vacuna meningococo B |
Bexsero |
Novartis |
377 |
|
|
Vacuna herpes zóster |
Zostavax |
Sanofi Pasterur MSD |
378 |
|
|
L. Antineoplásicos y terapia inmunomoduladora |
Afatinib |
Giotrif |
Boehringer Ingelheim |
375 |
|
Axitinib |
Inlyte |
Pfizer |
374 |
|
|
Brentuximab vedotina |
Adcetris |
Takeda |
376 |
|
|
Dabrafenib |
Tafinlar |
Pfizer |
374 |
|
|
Decitabina |
Dacogen |
Janssen Cilag |
377 |
|
|
Enzalutamida |
Xtandi |
Astellas |
379 |
|
|
Lipegfilgrastim |
Lonquex |
Teva |
378 |
|
|
Ofatumumab |
Arzerra |
Glaxo SmithKline |
376 |
|
|
Pertuzumab |
Perjeta |
Roche |
375 |
|
|
Pirfenidona |
Esbriet |
InterMune |
376 |
|
|
Pomalidomida |
Immovid |
Celgene |
375 |
|
|
Ruxolitinib |
Jakavi |
Novartis |
380 |
|
|
Teriflunomida |
Aubagio |
Sanofi Aventis |
380 |
|
|
Vandetanib |
Caprelsa |
AstraZeneca |
380 |
|
|
Vemurafenib |
Zelboraf |
Roche |
375 |
|
|
N. Sistema nervioso |
Dexmedetomidina |
Dexdor |
Orion |
377 |
|
Fumarato de dimetilo |
Tecfidera |
Biogen Idec |
380 |
|
|
Lisdexanfetamina |
Elvanse |
Shire |
373 |
|
|
Nalmefeno |
Selincro |
Lundbeck |
378 |
|
|
R. Aparato respiratorio |
Vilanterol/Fluticasona |
Relvar Ellipta |
Glaxo SmithKline |
377 |
|
S. Órganos de los sentidos |
Ocriplasmina |
Jetrea |
Alcon Cusi |
376 |
|
V. Varios |
Florbetapir [18F] |
Amyvid |
Lilly |
371 |
|
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA DURANTE LOS ÚLTIMOS DOCE MESES, POR LABORATORIOS |
|||
|---|---|---|---|
|
Laboratorio |
Principio activo |
Medicamento |
PAM |
|
Alcon Cusi |
Ocriplasmina |
Jetrea |
376 |
|
Almirall |
Linaclotida |
Constella |
375 |
|
Astellas |
Enzalutamida |
Xtandi |
379 |
|
Mirabegrón |
Betmiga |
372 |
|
|
AstraZeneca |
Vandetanib |
Caprelsa |
380 |
|
Biogen Idec |
Fumarato de dimetilo |
Tecfidera |
380 |
|
Boehringer Ingelheim |
Afatinib |
Giotrif |
375 |
|
Bristol-Myers Squibb |
Daclatasvir |
Daklinza |
380 |
|
Celgene |
Pomalodomida |
Imnovid |
375 |
|
Galderma |
Brimonidina |
Mirvaso |
374 |
|
Gilead |
Sofosbuvir |
Sovaldi |
377 |
|
Glaxo SmtihKline |
Dolutegravir |
Tivicay |
377 |
|
Ofatumumab |
Arzerra |
376 |
|
|
Vilanterol/Fluticasona |
Relvar Ellipta |
377 |
|
|
InterMune |
Pirfenidona |
Esbriet |
376 |
|
Janssen Cilag |
Decitabina |
Dacogen |
377 |
|
Simeprevir |
Olysio |
376 |
|
|
Lilly |
Florbetapir [18F] |
Amyvid |
371 |
|
Lundbeck |
Nalmefeno |
Selincro |
378 |
|
Mallinckrodt |
Regadenosón |
Rapiscan |
372 |
|
Menarini |
Avanafilo |
Spedra |
372 |
|
Novartis |
Ruxolitinib |
Jakavi |
380 |
|
Vacuna meningococo B |
Bexsero |
377 |
|
|
Novo Nordisk |
Catridecacog |
Novothirteen |
371 |
|
Orion |
Dexmedetomidina |
Dexdor |
377 |
|
Pfizer |
Axitinib |
Inlyta |
374 |
|
Dabrafenib |
Tafinlar |
374 |
|
|
Roche |
Pertuzumab |
Perjeta |
375 |
|
Vemurafenib |
Zelboraf |
374 |
|
|
Sanofi Aventis |
Teriflunomida |
Aubagio |
380 |
|
Sanofi Pasteur MSD |
Vacuna herpes zóster |
Zostavax |
378 |
|
Shire |
Lisdexanfetamina |
Elvanse |
373 |
|
STD |
Tetradecilsulfato sódico |
Veinfibro |
376 |
|
Takeda |
Brentuximab vedotina |
Adcetris |
376 |
|
Teva |
Lipegfilgrastim |
Lonquex |
378 |
Litiasis renal y crisis renoureteral
Resumen
La litiasis renal es la presencia de cálculos en el tracto urinario superior. La mayoría de las litiasis están compuestas por sales cálcicas. Cuando la litiasis se moviliza, o se fragmenta con migración del fragmento, se puede enclavar en el sistema colector y desencadenar un cólico nefrítico. Clínicamente aparece un dolor lumbar agudo, unilateral, severo, irradiado a región genital o inguinal ipsilateral, habitualmente acompañado de náuseas o vómitos. El diagnóstico se basa en la existencia de sintomatología compatible, aunque puede aparecer hematuria, cristaluria y en ocasiones piuria, si existe una infección acompañante. El diagnostico de la crisis renoureteral aguda(CRU) se completa con la realización de una analítica de sangre, orina y una prueba d eimágen (ecografía, Rx de abdomen simple, TAC helicoidal o UIV). El tratamiento se basa en el uso de AINEs i.m. ó p.o durante una semana. El tratamiento de la litiasis renal asintomática debe ser inicialmente expectante con seguimiento periódico. Las litiasis de 5-10 mm se expulsan de manera espontánea en el 50% de las ocasiones. En el supuesto de causar síntomas se debe valorar la litotricia o la nefrolitotomía percutánea. La prevención mediante dietoterapia se basa en una elevada ingesta de líquidos, tomar cantidades normales de calcio (no dietas hipocalcémicas) hiposódica y pobre en grasas animales
Se conoce como crisis renoureteral o cólico nefrítico al dolor cólico lumbar agudo unilateral severo irradiado a ingles o a genitales. En la mayoría de cólicos nefríticos, no se identifica la causa ni hay enfermedad de base. La causa más frecuente identificable es la litiasis renal.
La litiasis renal es una enfermedad que se caracteriza por la aparición de cálculos en el tracto urinario superior (parénquima renal, cálices, pelvis o uréter). Su forma de presentación más frecuente es el cólico nefrítico, cuadro que aparece cuando un cálculo se desprende o se fragmenta y se deposita en el sistema colector del riñón, lo que aumenta la presión intraluminal activando las terminaciones nerviosas de la mucosa y provocando dolor tipo cólico. La prevalencia de la litiasis renal en España es del aproximadamente 4 %.
Tipos de litiasis
La mayoría de litiasis (60–80%) están compuestas por sales de calcio (oxalato cálcico, fosfato cálcico o ambos). El resto son de estruvita (5–15%), ácido úrico (5–10%), cistina (1%), u otras sustancias (1%). El riesgo de padecer un cólico nefrítico es mayor en los hombres (10–20%) que en las mujeres (3–5%), pero esta diferencia en la actualidad, tiende a desaparecer debido a cambios del estilo de vida y a la obesidad. Cuando no se puede diagnosticar la litiasis renal como causante del cólico nefrítico, se procede a identificar la presencia de posibles factores de riesgo de la litiasis renal.
Factores de riego de la litiasis renal
- Varón, historia familiar de litiasis renal, obesidad, síndrome metabólico.
- Enfermedades metabólicas primarias: hipercalciuria, hiperoricusuria, hipocitraturia, hiperoxaluria, cistinuria.
- Hipercalcemia de cualquier etiología: hiperparatiroidismo, neoplasia, sarcoidosis, inmovilización prolongada.
- Enfermedades intestinales: enfermedad de Crohn, abuso de laxantes, bypass yeyuno-ileal.
- Acidosis tubular renal tipo I.
- Gota.
- Infecciones urinarias recurrentes.
- Depleción crónica de volumen: ingesta inadecuada o climas cálidos.
- Anormalidades anatómicas urológicas que faciliten la estasis de la orina: riñón en herradura, estenosis ureteral, obstrucción de la unión pieloureteral, ureterocele, divertículo calicial, quiste calicial y ectasia tubular (riñón en esponja), riñón único, cirugía renal previa.
- Fármacos: diuréticos del asa, antiácidos, acetazolamida, indinavir, corticoesteroides, teofilina, AAS, alopurinol y vitaminas C y D.
- Factores precipitantes
- Como factores precipitantes se puede hablar de la depleción de volumen y/o aumento de la ingesta de proteínas
Diagnostico cólico nefrítico
- Clínica:
- Dolor cólico lumbar agudo unilateral severo irradiado a ingles o a genitales, es por el paso de la litiasis por el uréter. El dolor abdominal suele ser de inicio agudo y de menos de 12 horas de evolución, no mejora con reposo y se suele acompañar de nauseas y vómitos. Cuando la litiasis entra en el uréter suelen aparecer polaquiuria, disuria, urgencia y tenesmo vesical. En TAC se encuentra una litiasis en tan sólo del 34-73% de estos casos.
- Si la litiasis está en la unión urétero-vesical, puede no haber dolor lumbar.
- Si la litiasis está en la pelvis renal, el dolor puede ser de bajo grado o intermitente.
- Exploración Física:
- Constantes vitales para excluir signos de shock o de infección sistémica.
- Exploración abdominal: localizar la zona de mayor hipersensibilidad lumbar (puño percusión positiva) y diferenciarlo de otras entidades.
- Exploraciones complementarias:
- Tira reactiva y/o urinoanálisis- puede existir hematuria, que apoya la sospecha diagnóstica, pero su presencia o ausencia no es suficientemente sensible o específica para el diagnóstico. Un 14,5% de los cólicos nefríticos no presentan hematuria. La presencia de leucocitos o nitritos indican infección. En algunos casos el urinoanálisis nos puede ayudar a la identificación de cristales, presencia de bacterias y/o piuria.
- Si no se puede realizar tira reactiva de orina, el criterio clínico es sufieciente para iniciar tratamiento específico.
- Urocultivo: No indicado.
- Análisis de sangre: No indicado en fase aguda, pero si se realiza no puede descartar la obstruccion, incluso con una función renal dentro de la normalidad
- Pruebas de imagen: Se recomienda realizar estudios radiológicos pasados los 7 días del episodio agudo, siempre que no haya criterios para derivar a urgencias.
Diagnóstico diferencial del cólico nefrítico
- Pielonefritis: pensar en ella si hay fiebre.
- Salpingitis o embarazo ectópico: pensar en él en mujeres en edad fértil.
- Ruptura de aneurisma abdominal: tenerlo en cuenta en mayores de 60 años.
- Diverticulitis.
- Apendicitis.
- Isquemia miocárdica aguda.
- Isquemia intestinal.
- Obstrucción intestinal.
- Dolor músculo esquelético.
- Cólico biliar.
Exploraciones complementarias ante una litiasis renal
- Estudio básico: tira reactiva de orina y una prueba de imagen.
- Se debe realizar un «estudio básico» a menores 65 años con un único episodio de cólico nefrítico, a los enfermos mayores 65 años cuando presentan una recurrencia de cólico; y a los pacientes con factores predisponentes. Está indicado realizarlo a partir de los 3 meses tras haber presentado un cólico nefrítico, puesto que la obstrucción aguda puede inducir ciertas anormalidades.
Factores predisponentes para presentar un cólico nefrítico:
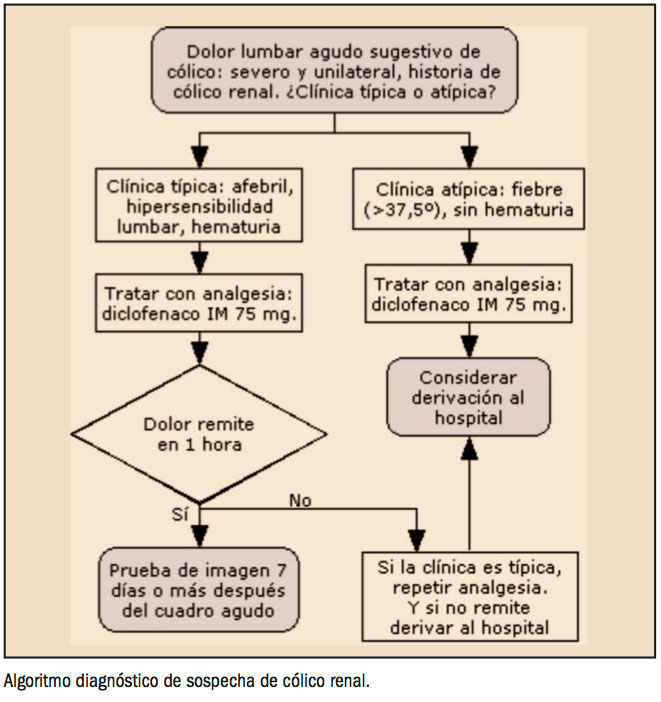{kind=link}
- clínicos: antecedentes familiares de litiasis, enfermedades óseas, gota úrica, hiperparatiroidismo, acidosis tubular renal, infecciones urinarias recidivantes, litiasis infantiles y juveniles (en menores de 20 años), monorreno por litiasis, litiasis recidivantes, enfermedad inflamatoria intestinal, diarrea crónica, malabsorción, historia de cirugía bariátrica, osteoporosis o fracturas patológicas
- radiológicos: litiasis bilateral, litiasis en riñón único, nefrocalcinosis, fragmentos residuales tras litotricia, litiasis coraliforme.
- analíticos: cistinuria, hipercalcemia y/o hipercalciuria, hiperoxalurias, hiperuricemias y/o hiperuricosurias, hipocitraturias.
- Análisis mineralógico del cálculo expulsado: si se obtiene el cálculo, se debería analizar para determinar su composición.
- Análisis completo de la primera orina de la mañana y sangre: los resultados obtenidos no cambian el protocolo de seguimiento urometabólico ni el pronóstico del paciente, ni tampoco las recomendaciones dietéticas a seguir
- Orina:
- urocultivo si hay piuria o bacteriuria
- determinación del volumen urinario de 24 horas y
- cuantificación de la excreción de calcio, ácido úrico, oxalato, citrato, fosfato, urea, creatinina, sodio, potasio y magnesio.
- pH orina- puede sernos útil ya que un valor superior a 7.5 es típico de la litiasis infecciosa e inferior a 5 de la litiasis úrica (algo que ya nos indica la tira reactiva de orina).
- Sangre
- calcio, fosfato, ácido úrico, creatinina, sodio, potasio, cloro, magnesio, fosfatasa alcalina, tiroxina y PTH, pueden orientar el diagnostico hacia diferentes situaciones favorecedores de litiasis renal
Diagnóstico por imagen en litiasis renal
- ecografía reno-vesical después de un primer episodio de cólico nefrítico por accesibilidad y precio
- radiografía simple de abdomen se realizará cuando sospechemos que la litiasis es ureteral
- pielografía intravenosa, considerada clásicamente como la prueba más definitoria, ha sido sustituida por el TAC helicoidal sin contraste; se deberían realizar cuando la ecografía y la radiografía no nos confirman la sospecha diagnóstica.
|
Diagnóstico por imagen de la litiasis renal |
|||
|---|---|---|---|
|
Tipo de prueba |
Sensibilidad Especificidad |
Ventajas |
Desventajas |
|
Ecografía |
19 % 97% |
Accesible. Diagnostica la hidronefrosis y la litiasis renal. |
Dificultad en visualizar litiasis en uréteres. |
|
Radiografía de abdomen |
45 a 59% 71 a 77% |
Accesible. Barato. |
Dificultad en visualizar litiasis ureteral, cálculos radiolúcidos, y cuando existen calcificaciones extraurinarias. |
|
Pielografía intravenosa |
64 a 87% 92 a 94% |
Accesible. Provee de información de la anatomía y funcionamiento de los riñones. |
Usa contraste y requiere preparación previa. Mala visualización en causas no genitourinarias. |
|
TAC helicoidal |
95 a 100% 94 a 96% |
Se visualizan signos indirectos de obstrucción. Provee de información causas no genitourinarias. |
Menos accesible. Caro. No proporciona una medida directa de la función renal. |
Tratamiento del cólico nefrítico agudo
La analgesia parenteral se recomienda usar antiinflamatorios no esteroideos como diclofenaco 75mg IM, que calma el dolor en 20-30 minutos y disminuye admisiones a urgencias, pudiéndose continuar el tratamiento vía oral 7 días más, en medio hospitalario se recurre a ketorolaco 30 mg IM que ha demostrado ser igual de efectivo que el diclofenaco. Otra opción es metamizol IM, que no ha demostrado ser mejor que el diclofenaco. Si la analgesia no ha hecho efecto en 1 hora, y si la clínica es típica, podemos repetir la analgesia. Si no remite, plantear derivación al hospital. No se recomienda usar opiáceos (especialmente la petidina) por la mayor probabilidad de vómitos y mayor necesidad de uso de fármacos de rescate.
La utilización de tamsulosina 0,4 mg al día durante 4 semanas podría ser útil para la expulsión de cálculos ureterales yuxtavesicales además de la disminución de la intensidad y duración del cólico.
Entre las medidas físicas, el calor local seco con una esterilla eléctrica a 42ºC parece ser efectivo en la disminución del dolor y las náuseas.
No puede establecerse ninguna recomendación sobre la efectividad de aumentar la ingesta hídrica. Esta medida tiene como finalidad acelerar el paso de los cálculos y mejorar los síntomas, pero existen escasas evidencias científicas sobre su efectividad. En cualquier caso, debe filtrarse la orina para identificar la expulsión del cálculo.
Criterios de derivación al hospital en cólico nefrítico
- La analgesia con AINES no ha hecho efecto en 1 hora, remitir al hospital por riesgo de afectación de la función renal debido a la obstrucción persistente.
- Náuseas refractarias al tratamiento.
- Fiebre, infección o anuria.
- Comorbilidades debilitantes.
- Mayores de 60 años.
- Embarazadas
- Riñón único funcionante o trasplantados.
- Causas que limitan la analgesia como úlcera duodenal, sangrados, etc., se podría utilizar protección gástrica o un tratamiento alternativo como el metamizol.
Tratamiento de la litiasis renal
A un paciente que se la haya diagnosticado una litiasis ureteral proximal o distal con alta probabilidad de expulsión espontánea y en el que los síntomas estén controlados, se recomienda la observación y evaluación periódica como actuación inicial. Tienen más probabilidad de ser expulsadas espontáneamente las litiasis de <5 mm de diámetro y se recomienda tratamiento conservador y estrecha vigilancia. Se espera que el calculo sea expulsado durante las primeras 4 semanas después del cólico nefrítico. Las litiasis de 5-10 mm se expulsan espontáneamente en el 50% de las veces.
Criterios de derivación al especialista
Cuando la litiasis sea >10 mm ó >5 mm con intolerancia al dolor o múltiples visitas a urgencias. Las opciones de tratamiento serán: litotricia o nefrolitotomía percutánea (útil cuando la litotricia falla o en los que está contraindicada) o la ureteroscopia o la ureterorenoscopia (útil en embarazadas, obesos mórbidos o en coagulopatías). Otro criterio es el dolor persistente por cálculo que no se haya expulsado después de 2-4 semanas de observación.
Litiasis renal recidivante- prevención de las recurrencias
Las dietas habitualmente recomendadas para prevenir las recidivas tienen un impacto muy pobre como para recomendarlas de por vida por lo que tienen que ser individualmente recomendadas según el riesgo alto de recurrencias. La ingesta abundante de líquidos para lograr una diuresis superior o igual a 2 litros al día, parece disminuir las recurrencias
Actualmente no se recomiendan dietas pobres en calcio para prevenir las recurrencias de las litiasis renales porque se ha observado que aumentan la secreción de oxalatos en la orina con el aumento de la formación de complejos de oxalato cálcico. Por el contrario dietas con calcio normal, hiposódica y pobres en proteínas animales, al disminuir la secreción de oxalatos, podrían disminuir la recurrencias de los cálculos renales.
Dicho lo anterior, aunque no existen evidencias claras del beneficio de las dietas ni de la efectividad del tratamiento etiológico, algunos autores recomiendan lo siguiente:
- Litiasis cálcica:
- En pacientes con hipercalciuria:
- Restricción en la ingesta de proteínas, oxalato y sodio.
- No restricción en la ingesta de calcio.
- Tiazidas, citrato potásico, Amiloride.
- En pacientes con hipocitraturia:
- Restricción en la ingesta de proteínas y sodio.
- Suplementos de citrato potásico (citrato sódico si el anterior no es bien tolerado).
- En pacientes con hiperoxaluria: Restricción en la ingesta de oxalatos1.
- En pacientes con hiperuricosuria:
- Restricción en la ingesta de purinas.
- Alopurinol
- Litiasis de ácido úrico: La ingesta de líquidos es poco importante en la prevención de recurrencias.
- En pacientes con pH urinario bajo:
- Restricción en la ingesta de proteínas y sodio.
- Alcalinización de la orina con suplementos de citrato potásico (citrato sódico si el anterior no es bien tolerado).
- En pacientes con hiperuricosuria:
- Restricción en la ingesta de proteínas y sodio.
- Si en pH urinario es bajo, alcalinización de la orina con suplementos de citrato potásico (citrato sódico si el anterior no es bien tolerado).
- En situaciones especiales: Alopurinol.
La ingesta de agua no ha demostrado ser útil para la prevención primaria de la litiasis renal. Existen estudios observacionales prospectivos que recomiendan dietas normocalcémicas y baja en proteínas animales, suplementos de K y Mg y dietas restrictivas de Vitamina C para la prevención primaria de las litiasis, además, un IMC <25 y tener más de 60 años sería un factor protector.
BIBLIOGRAFÍA
Nuevos medicamentos en 2014
Durante 2014 se registraron en España 1.056 presentaciones comerciales o formatos de medicamentos, tal como se recoge en la tabla 1. El dato se alinea con la tendencia mostrada en los periodos analizados (figuras 1a y 1b).
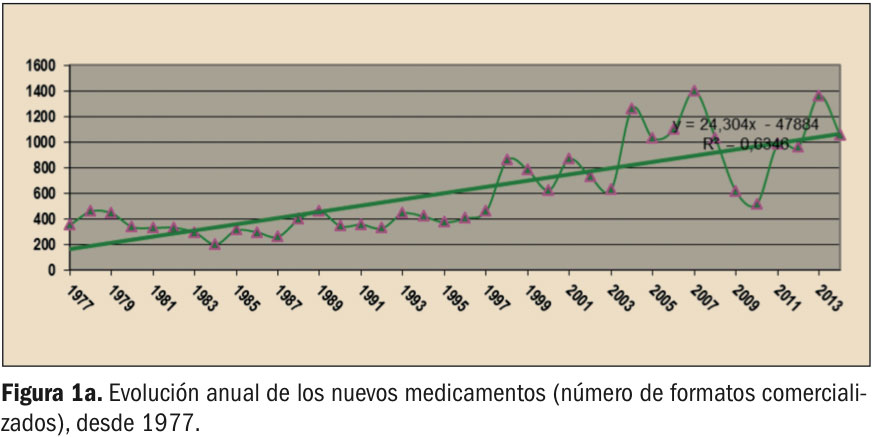{kind=link}
Al 31 de diciembre de 2014, se encontraban en situación de comercialización 16.566 formatos de medicamentos. Por otro lado, durante los últimos diez años se han incorporado al mercado 10.050 presentaciones, lo que supone el 61% del total disponible actualmente; por otro lado desaparecen 8.578, lo que supone un incremento neto de 1.472 formatos. Estas tendencias a la renovación vienen determinadas por el incremento del número de bajas y la paulatina estabilización del de altas (figuras 1b y 1c).
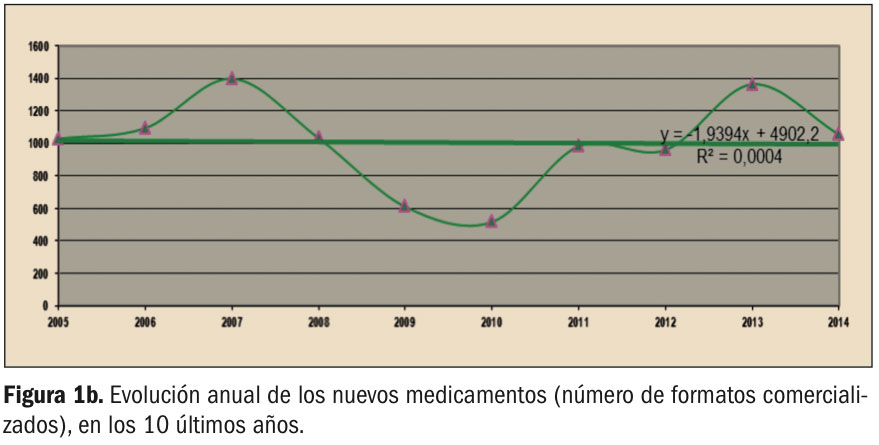{kind=link}
En lo relativo a la composición cuantitativa de los nuevos medicamentos, el 79% de estos en 2014 fueron monocomponente, un 20% contienen dos principios activos y el restante 1% son medicamentos multicomponente (Tabla 2). En este sentido, parece claramente que se mantiene la tendencia hacia los medicamentos monocomponentes, que supone el 87% de los nuevos medicamentos comercializados en los últimos diez años.
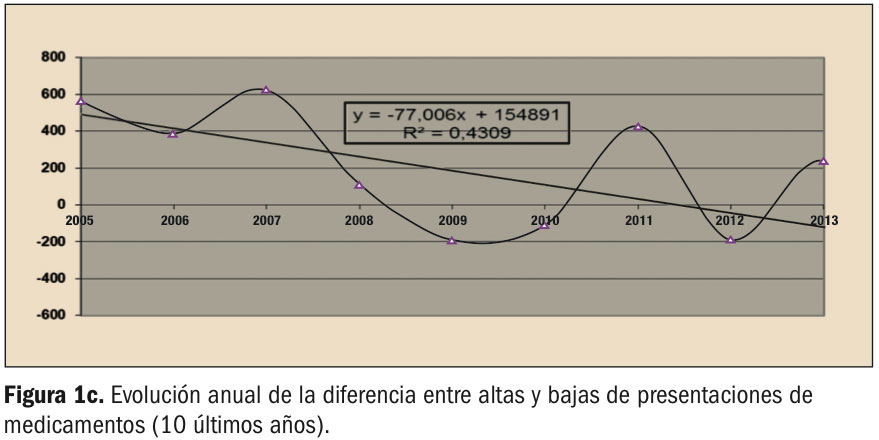{kind=link}
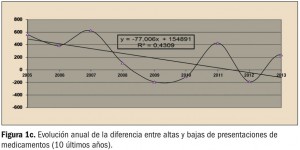
Por lo que se refiere a los nuevos principios activos comercializados en 2014 en nuestro país, han sido un total de 35 (Tabla 3), un año excepcional si se considera que la media de la década 2005-2014 es de 25. Por otro lado, se han comercializado1 un total de 1.056 formatos de medicamentos en 2014, que da un promedio de 30,2 formatos, por debajo de los 38,5 de la década. Aunque globalmente (1977-2014), las tendencias de la incorporación de nuevos principios activos (figura 3a) y de la relación nuevos formatos/nuevos principios activos (figura 3b) son incrementales, en los últimos diez años se ha estabilizado la incorporación de nuevos principios activos (figura 3c).
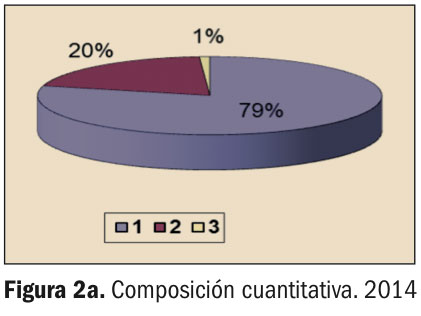{kind=link}
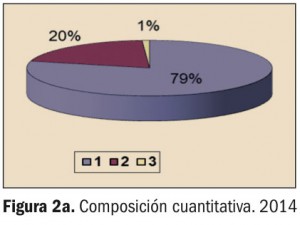
Desde el año 2000 se ha comercializado en España 59 nuevos principios activos como medicamentos huérfanos, lo que supone prácticamente uno de cada seis (16%) de los nuevos principios activos incorporados en ese periodo. La tendencia es ligeramente descendente, a pesar de los 6 nuevos incorporados en 2014, en parte debido a que en los dos últimos años no se incorporó ninguno (figura 3c). En cualquier caso, parece mantenerse una clara proporcionalidad entre el número de nuevos principios activos y el de los incluidos en medicamentos huérfanos, que hace que ambas curvas mantengan un paralelismo ciertamente evidente en la figura 3c.
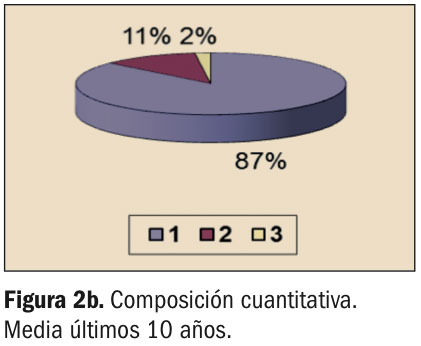{kind=link}
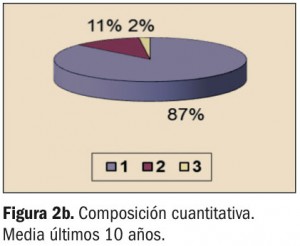
Considerando la clasificación terapéutica de los nuevos principios activos, se han incorporado nuevos principios activos a 11 de los 14 grupos terapéuticos existentes. El grupo con mayor número de nuevos principios activos durante el año ha sido – con gran diferencia sobre el resto – el L (Terapia antineoplásica y agentes inmunomoduladores), nada menos que con 13. Desde el año 1977, en que apareció por vez primera Panorama Actual del Medicamento, se han incorporado un total de 1.053 nuevos principios activos al mercado farmacéutico español.
En cuanto a las tendencias observadas por grupos terapéuticos, el número de nuevos principios activos comercializados en España indica progresión significativa en la última década con relación al promedio global en los grupos L: Terapia antineoplásica y agentes inmunomoduladores (de 3,9 a 6,3/año), V: Varios (1,7 a 2,5); por el contrario, experimentan una tendencia regresiva los grupos C: Aparato cardiovascular (3,2 a 2,3), J: Terapia antiinfecciosa sistémica (3,9 a 3,3), M: Aparato locomotor (1,8 a 0,5), N: Sistema nervioso (3,8 a 3,0), D: Dermatológicos (1,3 a 0,5) y R: Aparato respiratorio (1,2 a 0,8).
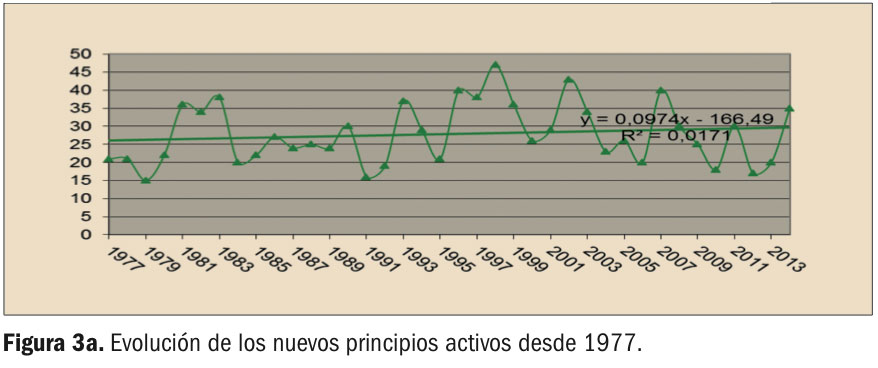{kind=link}
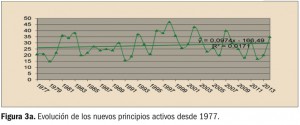
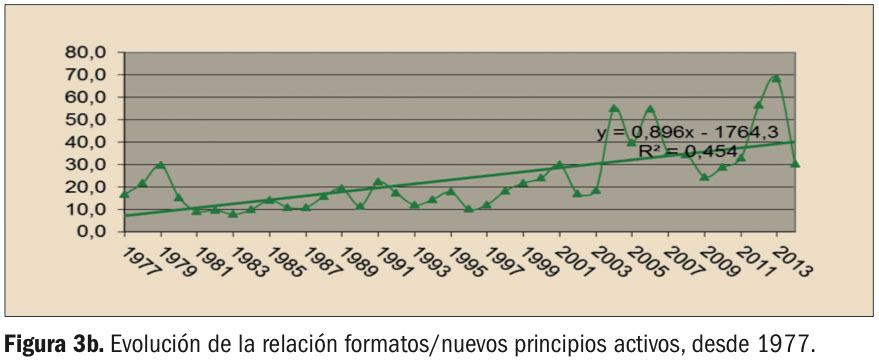{kind=link}
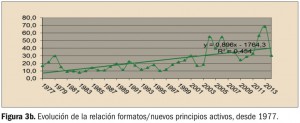
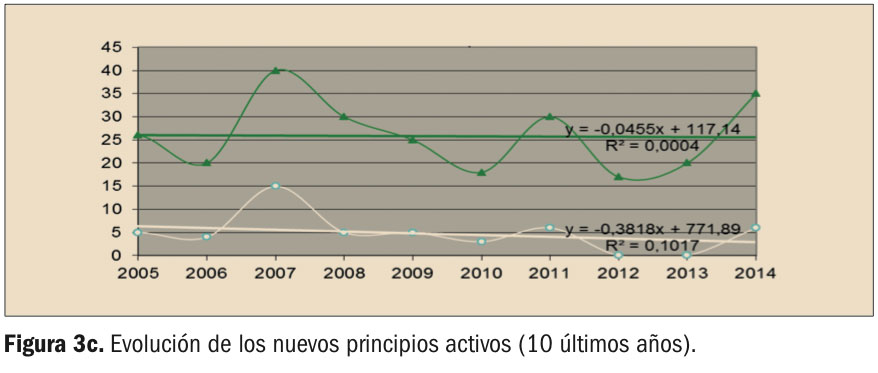{kind=link}
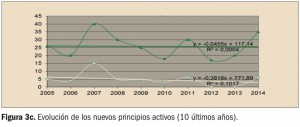
|
Tabla 1. Evolución de los nuevos medicamentos comercializados (formatos comerciales) |
|||
|---|---|---|---|
|
Año |
Altas |
Bajas |
Diferencia |
|
2005 |
1030 |
470 |
560 |
|
2006 |
1095 |
704 |
391 |
|
2007 |
1399 |
774 |
625 |
|
2008 |
1033 |
923 |
110 |
|
2009 |
612 |
801 |
-189 |
|
2010 |
517 |
625 |
-108 |
|
2011 |
984 |
556 |
428 |
|
2012 |
960 |
1147 |
-187 |
|
2013 |
1364 |
1123 |
241 |
|
2014 |
1056 |
1455 |
-399 |
|
Media |
1005 |
858 |
147 |
|
Tabla 2. Evolución de la composición cuantitativa de los nuevos medicamentos (Porcentaje de medicamentos con uno, dos o más principios activos) |
|||
|---|---|---|---|
|
AÑO |
1 PA |
2 PA |
>2 PA |
|
2004 |
91% |
3% |
6% |
|
2005 |
86% |
11% |
3% |
|
2006 |
93% |
6% |
1% |
|
2007 |
91% |
8% |
1% |
|
2008 |
91% |
7% |
2% |
|
2009 |
89% |
9% |
2% |
|
2010 |
81% |
15% |
4% |
|
2011 |
86% |
12% |
2% |
|
2012 |
84% |
15% |
1% |
|
2013 |
86% |
12% |
2% |
|
2014 |
79% |
20% |
1% |
|
Media |
87% |
11% |
2% |
|
Tabla 3. Evolución de los nuevos principios activos comercializados: total (PA) y huérfanos (H) |
|||||
|---|---|---|---|---|---|
|
AÑO |
PA |
Formatos |
F/PA |
Huérfanos |
% H/PA |
|
2005 |
26 |
1030 |
39,6 |
5 |
19% |
|
2006 |
20 |
1095 |
54,8 |
4 |
20% |
|
2007 |
40 |
1399 |
35,0 |
15 |
38% |
|
2008 |
30 |
1033 |
34,4 |
5 |
17% |
|
2009 |
25 |
612 |
24,5 |
5 |
20% |
|
2010 |
18 |
517 |
28,7 |
3 |
17% |
|
2011 |
30 |
984 |
32,8 |
6 |
20% |
|
2012 |
17 |
960 |
56,5 |
0 |
0% |
|
2013 |
20 |
1364 |
68,2 |
0 |
0% |
|
2014 |
35 |
1056 |
30,2 |
6 |
17% |
|
Media |
25 |
1026 |
38,5 |
5 |
19% |
|
Tabla 5. Evolución de los nuevos principios activos, por grupos terapéuticos |
|||
|---|---|---|---|
|
Grupo Terapéutico |
2014 |
Media anual 2005-2014 |
Media anual 1977-2014 |
|
A. Tracto digestivo y metabolismo |
1 |
2,7 |
2,6 |
|
B. Sangre y órganos hematopoyéticos |
1 |
1,7 |
1,6 |
|
C. Aparato cardiovascular |
3 |
2,3 |
3,2 |
|
D. Dermatológicos |
1 |
0,5 |
1,3 |
|
G. Aparato genitourinario |
2 |
1,3 |
1,2 |
|
H. Hormonas no sexuales |
0 |
0,4 |
0,5 |
|
J. Terapia antiinfecciosa, uso sistémico |
6 |
3,3 |
3,9 |
|
L. Terapia antineoplásica y agentes inmunomoduladores |
13 |
6,3 |
3,9 |
|
M. Aparato locomotor |
0 |
0,5 |
1,8 |
|
N. Sistema nervioso |
5 |
3,0 |
3,8 |
|
P. Antiparasitarios |
0 |
0,1 |
0,3 |
|
R. Aparato respiratorio |
1 |
0,8 |
1,2 |
|
S. Órganos de los sentidos |
1 |
0,7 |
0,8 |
|
V. Varios |
1 |
2,5 |
1,7 |
|
TOTALES |
35 |
26,1 |
27,7 |
|
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA DURANTE 2014, POR GRUPOS TERAPÉUTICOS |
||||
|---|---|---|---|---|
|
Grupo terapéutico |
Principio activo |
Medicamento® |
Laboratorio |
PAM |
|
A. Tracto alimentario y metabolismo |
Linaclotida |
Constella |
Almirall |
375 |
|
B. Sangre y órganos hematopoyéticos |
Catridecacog |
Novothirteen |
Novo Nordisk |
371 |
|
C. Aparato cardiovascular |
Regadenosón |
Rapiscan |
Mallinckrodt |
372 |
|
Tetradecilsulfato sódico |
Veinfibro |
STD |
376 |
|
|
Clevidipino |
Cleviprex |
Ferrer |
370 |
|
|
D. Terapia dermatológica |
Brimonidina |
Mirvaso |
Galderma |
374 |
|
G. Terapia genitourinaria |
Mirabegrón |
Betmiga |
Astellas |
372 |
|
Avanafilo |
Spedra |
Menarini |
372 |
|
|
J. Antiinfecciosos, uso sistémico |
Sofosbuvir |
Sovaldi |
Gilead |
377 |
|
Simeprevir |
Olysio |
Janssen Cilag |
376 |
|
|
Elvitegravir (+ Cobicistat/Emtricitabina/Tenofovir) |
Stribild |
Gilead |
370 |
|
|
Dolutegravir |
Tivicay |
Glaxo SmithKline |
377 |
|
|
Vacuna meningococo B |
Bexsero |
Novartis |
377 |
|
|
Vacuna herpes zóster |
Zostavax |
Sanofi Pasterur MSD |
378 |
|
|
L. Antineoplásicos y terapia inmunomoduladora |
Decitabina |
Dacogen |
Janssen Cilag |
377 |
|
Brentuximab vedotina |
Adcetris |
Takeda |
376 |
|
|
Ofatumumab |
Arzerra |
Glaxo SmithKline |
376 |
|
|
Pertuzumab |
Perjeta |
Roche |
375 |
|
|
Afatinib |
Giotrif |
Boehringer Ingelheim |
375 |
|
|
Axitinib |
Inlyta |
Pfizer |
374 |
|
|
Crizotinib |
Xalkori |
Pfizer |
370 |
|
|
Dabrafenib |
Tafinlar |
Pfizer |
374 |
|
|
Vemurafenib |
Zelboraf |
Roche |
375 |
|
|
Enzalutamida |
Xtandi |
Astellas |
379 |
|
|
Lipegfilgrastim |
Lonquex |
Teva |
378 |
|
|
Pirfenidona |
Esbriet |
InterMune |
376 |
|
|
Pomalidomida |
Imnovid |
Celgene |
375 |
|
|
N. Sistema nervioso |
Perampanel |
Fycompa |
Eisai |
370 |
|
Dexmedetomidina |
Dexdor |
Orion |
377 |
|
|
Lisdexanfetamina |
Elvanse |
Shire |
373 |
|
|
Nalmefeno |
Selincro |
Lundbeck |
378 |
|
|
Tafamidis |
Vyndaqel |
Pfizer |
370 |
|
|
R. Aparato respiratorio |
Vilanterol/Fluticasona |
Relvar Ellipta |
Glaxo SmithKline |
377 |
|
S. Órganos de los sentidos |
Ocriplasmina |
Jetrea |
Alcon Cusi |
376 |
|
V. Varios |
Florbetapir [18F] |
Amyvid |
Lilly |
371 |
A. TRACTO ALIMENTARIO Y METABOLISMO
|
LINACLOTIDA |
|---|
|
► CONSTELLA® (Almirall) |
|
A06AX. tracto alimentario y metabolismo Laxantes: otros. |
La linaclotida es un péptido sintético, análogo de la enterotoxina termoestable del Escherichia coli, que actúa uniéndose a los receptores de la guanilato ciclasa C (GC-C) presente en la superficie luminal del epitelio intestinal, dando lugar a un incremento intra y extracelular de guanilato-monofosfato cíclico (GMPc). Este incremento de los niveles de GMPc es capaz de provocar, a nivel extracelular, una redacción de la sensibilidad nociceptiva implicada en el dolor visceral, especialmente en situaciones de hipersensibilidad visceral crónica, mientras que a nivel intracelular provoca un aumento de la secreción celular de cloruro y de bicarbonato a la luz intestinal, un incremento de la cantidad de líquido, una aceleración refleja del tránsito intestinal y una inhibición de la absorción de fluidos en el colon. Todo ello fundamenta que la linaclotida haya sido autorizada para el tratamiento sintomático del síndrome del intestino irritable con estreñimiento entre moderado y grave en adultos.
La eficacia de la linaclotida en esta indicación ha sido confirmada mediante ensayos clínicos, mostrando tasas de respondedores a 12 semanas del 55%, frente a un 40% con placebo; incluso aplicando el criterio de la FDA de Estados Unidos, más exigente en este aspecto, las diferencias entre linaclotida y placebo son marcadas (34% vs. 13-21%). Por otro lado, los datos correspondientes a 26 semanas (6 meses) confirman la persistencia del efecto terapéutico de la linaclotida y su superioridad estadística y clínica frente al placebo. Con todo, aunque las diferencias con el placebo son rigurosamente significativas, la tasa de respondedores (55% según parámetros europeos, 38% según la FDA) están lejos de ser ideales.
LINACLOTIDA: H2N-Cys1-Cys2-Glu3-Tyr4-Cys5-Cys6-Asn7-Pro8-Ala9-Cys10-Thr11-Gly12-Cys13-Tyr14-COOH.
ENTEROTOXINA ST(p) Escherichia coli: H2N-Cys1-Cys2-Glu3-Tyr4-Cys5-Cys6-Glu7–Leu8-Cys9-Cys10-Asn11-Pro12-Ala13-Cys14–Ala15-Gly16-Cys17-Tyr-COOH.
Sin duda, la linaclotida presenta un mecanismo innovador en farmacología, constituyendo un ejemplo paradigmático de cómo transformar un fenómeno adverso, como las temibles diarreas provocadas por enterotoxinas bacterianas, en una herramienta terapéutica útil en condiciones específicas. Por otro lado, actualmente no existe ningún medicamento específicamente autorizado para el síndrome del intestino irritable con estreñimiento y el beneficio terapéutico obtenido con los medicamentos inespecíficos hasta ahora utilizados – mal contrastado, en términos científicos – es harto limitado, tanto en la proporción de pacientes beneficiarios como en el grado de la mejoría clínica obtenida. Frente a eso, la linaclotida ha demostrado de forma consistente una eficacia aceptable y durable, en condiciones de seguridad. Por ello, puede considerarse como primera línea de tratamiento en pacientes que hayan sido diagnosticados siguiendo las recomendaciones del consenso de Roma III
B. SANGRE Y ÓRGANOS HEMAOTOPOYÉTICOS
|
CATRIDECACOG |
|---|
|
► NOVOTHIRTEEN® (Novo Nordisk) |
|
B02BD. SANGRE Y ÓRGANOS Antihemorrágicos: factores de coagulación sanguínea |
El catridecacog es un una forma recombinante de la subunidad A del Factor XIII de la coagulación, que ha sido autorizado para el tratamiento profiláctico a largo plazo de hemorragias en pacientes a partir de 6 años con deficiencia congénita de la subunidad A del factor XIII.
El medicamento ha demostrado prevenir de forma eficaz la aparición de crisis hemorrágicas graves, con una tasa de 0,048 episodios por paciente y año, muy lejos del valor de 2,91 encontrado en los controles históricos de este tipo de pacientes. Evidentemente, la extremada rareza del déficit congénito de Factor XIII (un caso por cada 2-5 millones de habitantes) condiciona su desarrollo clínico, en la medida en que es imposible disponer de un nutrido grupo de pacientes sobre los que investigar diferentes estrategias posológicas y otros aspectos del tratamiento. En cualquier caso, el origen recombinante del catridecacog supone una mejora sobre el Factor XIII de origen extractivo, en tanto que permite soslayar los riesgos inherentes a este origen. Un aspecto práctico relevante es que la conservación incorrecta del catridecacog una vez reconstituido, podría incrementar su nivel de actividad antifibrinolítica y, con ello, el riesgo de trombosis, por lo que es indispensable un riguroso control también en este ámbito. El medicamento fue designado inicialmente como medicamento huérfano en diciembre de 2003 por la EMA, pero en la actualidad ya no está incluido en Registro Comunitario de Medicamentos Huérfanos de la Unión Europea.
c. APARATO CARDIOVASCULAR
|
REGADENOSON |
|---|
|
► RAPISCAN® (Mallinckrodt) |
|
C01EB. APARATO CARDIOVASCULAR Terapia cardiaca: otros |
El regadenosón es un análogo de la adenosina, indicado como agente de la prueba farmacológica de esfuerzo para realizar estudios de imagen de perfusión miocárdica (MPI) con radionúclidos en pacientes adultos que no pueden someterse a una adecuada prueba de esfuerzo con ejercicio. Se trata de un agonista selectivo y de baja afinidad de los receptores A2A de la adenosina; su acción sobre los receptores situados en las membranas de las células musculares lisas de las paredes vasculares de las arterias coronarias da lugar a una vasodilatación momentánea, con el consiguiente incremento, también momentáneo, del flujo sanguíneo coronario en el miocardio.
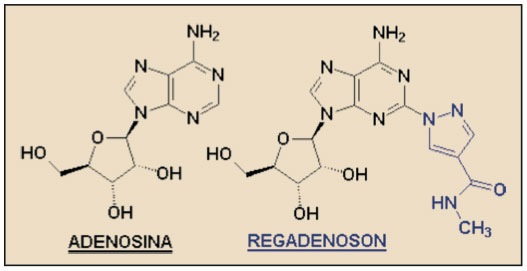{kind=link}
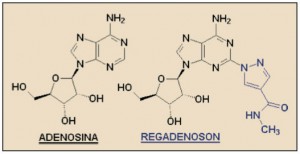
Los datos clínicos comparativos con adenosina muestran niveles de concordancia similares entre adenosina y regadenosón vs. adenosina y adenosina; es decir, si los datos obtenidos en pacientes tratados con regadenosón concuerdan con los obtenidos previamente con adenosina tan frecuentemente como lo hace ésta consigo misma, parece razonable concluir que el regadenosón no es inferior a la adenosina en esta indicación.
Las pruebas de estrés farmacológico usadas son en general seguras y se acompañan de una eficacia diagnóstica similar a la proporcionada por el ejercicio físico. De hecho, el uso de estos agentes ha permitido en los últimos años ampliar sustancialmente el espectro de pacientes capaces de ser estudiados con procedimientos de medicina nuclear. La incorporación de protocolos combinados, las modificaciones de algunos protocolos clásicos y la introducción de nuevos fármacos agonistas específicos hacen posible actualmente la aplicación de pruebas de estrés prácticamente «a la medida» de cada paciente. En concreto, la selectividad del regadenosón sobre los receptores A2A de la adenosina, frente a la ausencia de dicha selectividad en la adenosina y el dipiridamol (indirectamente, inhibiendo la recaptación de la adenosina) hacia el resto de receptores adenosinérgicos, permite un efecto vasodilatador coronario más «limpio», al no afectar significativamente a estos últimos y, en consecuencia, no producir efectos clínicamente menos relevantes sobre el propio corazón o sobre los mastocitos. No obstante, no parece que estas diferencias tengan gran relevancia en términos prácticos. Por el contrario, es mucho más conveniente la administración de un único bolo IV de una dosis fija de 400 µg regadenosón sin necesidad de ajuste posológico, frente a la infusión IV a velocidad controlada y con ajuste dependiente del peso que requieren adenosina y dipiridamol. Por otro lado, los parámetros sensibilidad, especificidad y precisión son muy similares para las EPM realizadas con regadenosón, en comparación con las descritas con adenosina, dipiridamol y dobutamina.
|
TETRADECILSULFATO |
|---|
|
► VEINFIBRO® (STD) |
|
C05BB. APARATO CARDIOVASCULAR Vasoprotectores. Terapia antivaricosa: agentes esclerosantes locales. |
Veinfibro® es un medicamento consistente en una solución de tetradecilsulfato sódico, para administración intravenosa en el tratamiento en adultos de venas varicosas primarias no complicadas, venas varicosas residuales o recurrentes tras cirugía, venas reticulares, vénulas y arañas vasculares de las extremidades inferiores que muestren dilatación. Las concentraciones al 1% y 3% se pueden convertir en espuma previamente a su uso utilizando la técnica Tessari.
El tetradecilsulfato sódico causa un daño endotelial en la vena donde se aplica y, a consecuencia de ello, un vasoespasmo; tras la exposición al agente esclerosante, la superficie interior de la vena se convierte en trombogénica, lo que conduce a la formación de trombos y la oclusión venosa. La vena ocluida acaba siendo sustituida finalmente por tejido conectivo fibroso.
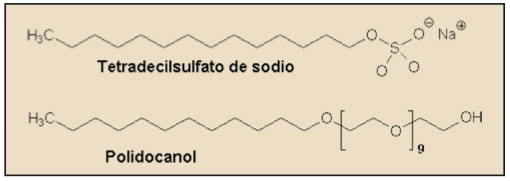{kind=link}
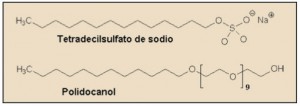
Su eficacia y su seguridad han sido comprobadas, aunque no hay pruebas de que el tetradecilsulfato sódico sea mejor ni más seguro como agente esclerosante que el polidocanol (etoxisclerol®), disponible en España como medicamento desde hace más de 30 años, ni que los pacientes estuvieran más satisfechos. Ambos fármacos están incluidos en las European Guidelines for Sclerotherapy in Chronic Venous Disordersk. La incorporación del tetradecilsulfato sódico viene a completar el arsenal de tensioactivos utilizados en escleroterapia, una técnica con tasas de eficacia en oclusión venas varicosas similar a los métodos térmicos (láser y radiofrecuencia), pero menores que la cirugía, especialmente en los cuadros de varices más profundas y de mayor calibre.
|
CLEVIDIPINO |
|---|
|
► CLEVIPREX® (Ferrer) |
|
C05BB. APARATO CARDIOVASCULAR Bloqueantes de canales de calcio con acción preferente vascular: Dihidropiridinas. |
El clevidipino es un agente antagonista de los canales L de calcio, capaz de reducir rápida e intensamente la presión arterial. Ha sido autorizado para la reducción rápida de la presión arterial en el entorno perioperatorio. Sus efectos antihipertensivos comienzan a manifestarse a los 2-3 minutos de iniciada la administración (infusión IV continua) y la recuperación de la presión arterial previa se obtiene a los 5-15 minutos después de suspender la administración IV.
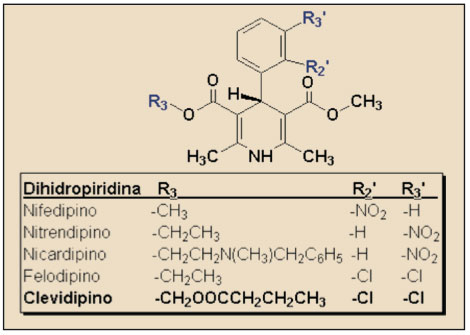{kind=link}
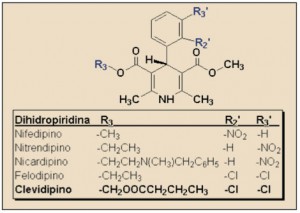
Su eficacia en la indicación autorizada está adecuadamente documentada, así como en cuadros hipertensivos posoperatorios, con tasas de éxito por encima del 90%, alcanzándose los niveles tensionales buscados en 5-6 minutos. En el perioperatorio, su eficacia antihipertensiva parece algo mayor que la de nitroglicerina y nitroprusiato sódico, mientras que en posoperatorio los resultados obtenidos son similares al nicardipino, tanto en términos de eficacia como de seguridad (mortalidad, infarto de miocardio, ictus, insuficiencia renal). Además, su eficacia y su seguridad han sido analizadas tanto en pacientes con insuficiencia cardiaca como con insuficiencia renal, con resultados satisfactorios.
Aunque el clevidipino no presenta particularidades mecanísticas dignas de mención, en cambio su farmacocinética puede resultar más interesante; en efecto, permite una gran inmediatez de efectos y, al mismo tiempo, su acción ultracorta facilita un eficiente ajuste de su posología y su manejabilidad por parte de los anestesistas, a lo que hay que añadir la ausencia de interacciones significativas por la vía del metabolismo hepático a través del citocromo P450 (CYP). Por tanto, una incorporación de cierto interés que amplía las opciones en el complejo y siempre delicado campo perioperatorio.
d. TERAPIA DERMATOLÓGICA
|
BRIMONIDINA |
|---|
|
► MIRVASO® (Galderma) |
|
D11AX. TERAPIA DERMATOLÓGICA Otros preparados dermatológicos. |
La brimonidina es un agente vasoconstrictor que actúa como un agonista selectivo de los receptores α2 adrenérgicos, provocando una reducción del eritema superficial de las zonas donde se aplica localmente. Está estrechamente relacionada química y farmacológicamente con la clonidina y otros análogos de ésta. Ha sido autorizada, en forma tópica dermatológica, para el tratamiento del eritema facial de la rosácea en pacientes adultos. La brimonidina, como principio activo farmacológico, estaba previamente comercializada en España en forma de solución para administración tópica oftálmica para la reducción de la presión intraocular en pacientes con glaucoma de ángulo abierto o hipertensión ocular.
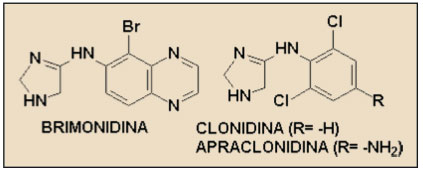{kind=link}
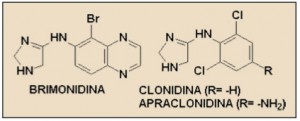
Los datos clínicos indican una significativa superioridad del gel de brimonidina al 5% sobre el placebo, produciendo una mejora de dos grado al final del tratamiento en el 25-30% de los pacientes frente al 9-10% con placebo, a lo largo de todo el día y desde el primer día de tratamiento, manteniéndose durante, al menos, un año de tratamiento, sin que se aprecia taquifilaxia, rebote o agravamiento. Las respuestas más intensas tienden a localizarse entre las 3 y 6 horas posteriores a la aplicación tópica del medicamento, aunque subsiste un efecto clínicamente relevante durante el resto del día con una sola aplicación diaria; en cualquier caso, la respuesta es rápida, ya que a los 30 minutos el efecto clínico es relevante (mejora de al menos un grado CEA) en un 28% con brimonidina vs. 5% con placebo.
El tratamiento tópico con brimonidina solo se dirige a uno de los síntomas de la rosácea, aunque ciertamente es el principal: el eritema facial. Éste tiene una relevancia notable para muchos pacientes, a los que, además de las molestias propias, provoca un aislamiento personal con consecuencias sociales y laborales en absoluto desdeñables. Bien es cierto que la respuesta al fármaco tampoco resulta espectacular (menos de la tercera parte de los pacientes mejoran en dos grados en la escala CEA), pero incluso una pequeña mejora puede tener una repercusión sustancial para determinados pacientes. Parece razonable, por tanto, considerar al nuevo medicamento como una cierta innovación en un campo de la terapéutica que actualmente tiene un carácter fundamentalmente empírico y no muy satisfactorio.
g. TERAPIA GENITOURINARIA
|
MIRABEGRÓN |
|---|
|
► BETMIGA® (Astellas) |
|
G04BD. TERAPIA GENITOURINARIA Antiespasmódicos |
El mirabegrón es un agente agonista selectivo de los receptores β3-adrenérgicos, que ha sido autorizado para el tratamiento sintomático de la urgencia, aumento de la frecuencia de micción y/o incontinencia de urgencia que puede producirse en pacientes adultos con síndrome de vejiga hiperactiva.
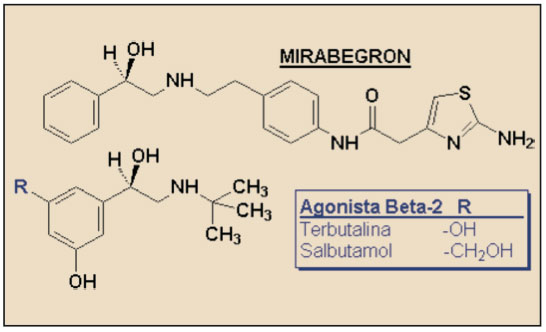{kind=link}
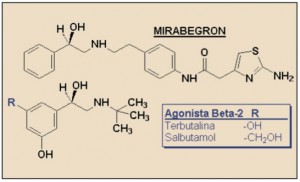
El fármaco produce una moderada aunque significativa reducción del número de micciones y de episodios de incontinencia urinaria, así como mejorías en otras variables clínicas consideradas. La dosis de 50 mg/24 h de mirabegrón se asocia con una reducción de 1,75 micciones diarias, 0,55 más que con placebo. Asimismo, entre aquellos que previamente había experimentado algún episodio de incontinencia previo, este tratamiento redujo significativamente 0,4 episodios diarios más que el placebo; considerando la tasa de respondedores (que experimentaron una reducción de al menos la mitad de los episodios de incontinencia), llega a cerca del 70%, si bien la correspondiente al placebo fue del 60% lo que deja la mejora del mirabegrón en un 10% adicional sobre el placebo.
El mirabegrón aporta un nuevo mecanismo farmacológico; sin embargo, su eficacia clínica es modesta y está en línea con los agentes anticolinérgicos habitualmente utilizados en esta indicación (fesoterodina, tolterodina, solifenacina, etc.); también su nivel de seguridad puede equipararse a estos últimos, aunque sin la siempre molesta sequedad de boca de los agentes anticolinérgicos que hace que algunos pacientes abandonen el tratamiento. Por todo ello, el mirabegrón representa una opción adicional para tratar a pacientes intolerantes o que hayan tenido una respuesta insatisfactoria a los tratamientos previos, pero sin que deban esperarse resultados netamente superiores.
|
AVANAFILO |
|---|
|
► SPEDRA® (Menarini) |
|
G04BE. TERAPIA GENITOURINARIA Preparados urológicos: fármacos usados en disfunción eréctil. |
El avanfilo es un inhibidor reversible y selectivo de la fosfodiesterasa de tipo 5 (PDE-5) específica del GMPc que impide la degradación de éste. Ha sido autorizado para el tratamiento de la disfunción eréctil en hombres. Está química y farmacológicamente relacionado con otros inhibidores de la PDE-5 previamente comercializados, como el sildenafilo, el vardenafilo y el tadalafilo.
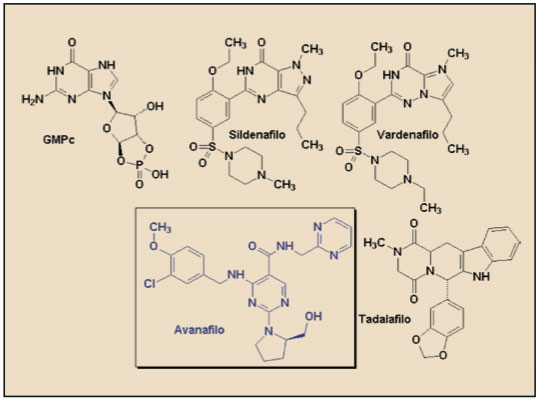{kind=link}
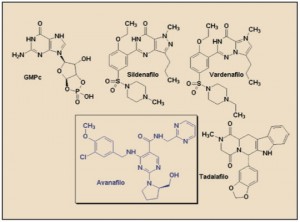
El avanafilo, en dosis de 100 y de 200 mg, utilizado a demanda, incrementó en 30 puntos sobre el placebo el porcentaje de relaciones sexuales satisfactorias y en 20 puntos el correspondiente a la penetración vaginal, sin diferencias estadísticamente significativas entre ambas dosis; una eficacia que parece mantenerse de forma permanente, al menos con el uso eventual a demanda durante un año. Los resultados obtenidos en pacientes diabéticos son algo inferiores a los observados en la población general.
Dado que la disfunción eréctil tiene prevalencia que aumenta sustancialmente con la edad, con tasas por encima del 40% a partir de los 60 años y del 50% a partir de los 70, la ausencia de experiencia clínica contrastada en pacientes mayores de 70 años limita el potencial del avanafilo que, por otro lado, no parece aportar ninguna ventaja sobre los fármacos actualmente disponibles en España.
j. ANTIINFECCIOSOS, USO SISTÉMICO
|
SOFOSBUVIR |
|---|
|
► SOVALDI® (Gilead) |
|
J05AE. ANTIINFECCIOSOS SISTÉMICOS Antivirales de acción directa: inhibidores de la proteasa. |
El sofosbuvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos.
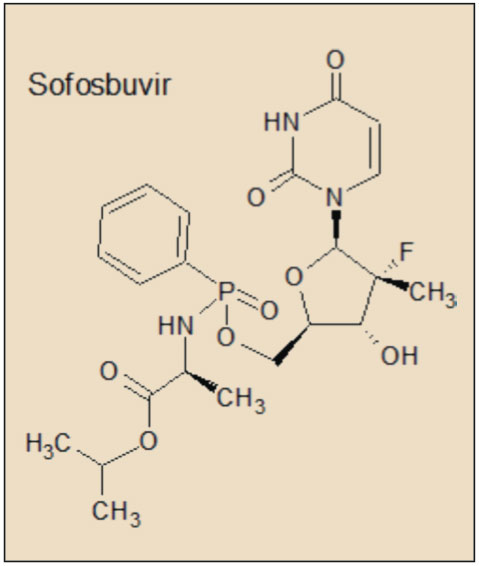{kind=link}
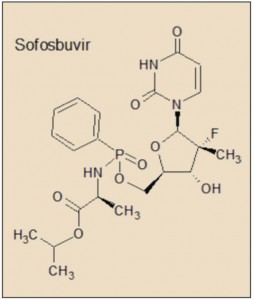
Las tasas globales de respuesta virológica sostenida en pacientes con VHC de genotipo 1 son, en combinación con peginterferón alfa-2a y ribavirina son muy elevadas (90% y 80% en pacientes sin y con cirrosis, respectivamente). Por otro lado, la utilización de pautas de 24 semanas de duración y sin interferón obtiene tasas del 50-70%, superiores a las obtenidas con pautas de interferón y ribavirina de 48 semanas. Por otro lado, se obtienen tasas cercanas al 80% en pacientes coinfectados con VIH utilizando pautas de 12 o 24 semanas de sofosbuvir y ribavirina (con o sin tratamiento previo para el VIH). En infecciones por VHC de genotipo 2, las tasas de respuesta están en torno al 95% en pacientes naïve y del 93% en aquellos inelegibles o intolerantes al interferón, con pautas de sofosbuvir y ribavirina de 12 semanas. Por su parte, en los casos ligados al genotipo 3 se requieren tratamientos de sofosbuvir y ribavirina de 24 semanas de duración, ya que las de 12 semanas producen resultados notablemente inferiores. Por último, los datos correspondientes a los genotipos 4, 5 y 6 – que son raros en la Unión Europea – son escasos, aunque no parecen diferir sustancialmente de los obtenidos para el genotipo 1.
Tras la primera oleada de antivirales específicos frente al VHC, con boceprevir y telaprevir, que ciertamente modificaron el pronóstico y las expectativas terapéuticas en este campo, el sofosbuvir forma parte de una segunda oleada, junto con simeprevir, daclastavir, ledipasvir y algunos otros, que aportan elevadas tasas de curación en todos los subtipos de pacientes, a través de regímenes más sencillos y mejor tolerados, con la particularidad de que van eliminando la otrora imprescindible presencia del interferón alfa en el tratamiento.
Estos nuevos fármacos, autorizados hace apenas unos meses, han tenido tal impacto que han dejado obsoleta rápidamente a la guía de práctica clínica de la European Association for the Study of the Liber (EASL) publicada en diciembre de 2013, obligando a una nueva edición en la que incluye las nuevas recomendaciones. Es importante no olvidar que la prevalencia de la hepatitis C en España se sitúa en entorno al 1-2% de la población, lo que supone que el número de personas infectadas está entre 500.000 y 1.000.000. Habida cuenta de que un 60-80% desarrollan la infección crónica y que un 20% de estos evoluciona a cirrosis hepática al cabo de 20-25 años, de los cuales un 80% acabará por padecer un cáncer de hígado, es evidente el interés sanitario y económico de un tratamiento curativo eficaz.
Sofosbuvir (Sovaldi®, de laboratorios GILEAD) fue elegido PREMIO PANORAMA 2014 «por su aportación al tratamiento – en combinación con otros medicamentos – de la hepatitis C, mejorando notablemente la respuesta viral sostenida del tratamiento actual al producir tasas de entre un 80% y un 95%, según el genotipo viral y las características fisiopatológicas de los pacientes. El nuevo fármaco incorpora un nuevo mecanismo de acción, que le permite ser combinado con otros fármacos antivirales activos; ha sido estudiado mediante amplios y rigurosos ensayos clínicos en poblaciones diversas de pacientes con hepatitis C, habiéndose mostrado también eficacia en aquellos coinfectados por VIH; adicionalmente, mejora el perfil toxicológico y de interacciones con relación a la terapia farmacológica estándar, acortando sustancialmente la duración de ésta. Estos aspectos innovadores del sofosbuvir revisten una especial importancia considerando la relevancia clínica (por el riesgo de cirrosis, cáncer hepático y/o la necesidad de trasplante de hígado) y la elevada prevalencia de la infección por VHC en España«.
|
SIMEPREVIR |
|---|
|
► OLYSIO® (Janssen Cilag) |
|
J05AE. ANTIINFECCIOSOS SISTÉMICOS Antivirales de acción directa: inhibidores de la proteasa. |
El simeprevir es un agente antiviral activo frente a virus de la hepatitis C, que actúa inhibiendo específicamente el complejo de serina proteasa NS3/4A, lo que impide la replicación viral. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos. El fármaco es especialmente activo frente a los genotipos 1a y 1b, aunque también tiene efectos significativos sobre los genotipos 2a, 4a y 6a; por el contrario, la proteasa de los genotipos 3a y 5a es menos susceptible.
{kind=link}
Los datos clínicos indican tasas de respuesta viral sostenida a las 12 semanas 30 o 40 puntos porcentuales por encima de las conseguidas solo con la terapia doble de peginterferón alfa y ribavirina: 80 vs. 50% en pacientes no tratados anteriormente (naïve), 79 vs. 37% en pacientes tratados y respondedores pero con recaída viral posterior, 76 vs. 9% en parcialmente respondedores e incluso 51 vs. 19% en no respondedores. Los resultados son también satisfactorios (aunque metodológicamente menos consistentes) en pacientes coinfectados por VIH y VHC. No obstante, la eficacia en pacientes infectados por VHC de genotipo 1a y con polimorfismo Q80K es solo marginal con respecto a la terapia doble. Este polimorfismo viral es frecuente en el genotipo 1a (19-48%) y se asocia con un cierto grado de resistencia al simeprevir.
Algunos datos sugieren resultados muy convincentes asociando simeprevir y sofosbuvir, tanto en pacientes pretratados no respondedores al peginterferón como en naïve y con independencia del nivel de fibrosis hepática. Precisamente, la eficacia de esta combinación, en ausencia de interferón, es muy apreciable al añadir el ahorro de la toxicidad del interferón y mejorar así la tolerabilidad del tratamiento.
Como se indicó con el sofosbuvir, la inclusión del simeprevir dejó obsoleta a la guía de práctica clínica de la European Association for the Study of the Liber (EASL) publicada en diciembre de 2013, promoviendo una nueva edición en la que incluía las nuevas recomendaciones. Igualmente, el simeprevir forma parte de una segunda oleada de antivirales frente al VHC, tras telaprevir y boceprevir, que aportan elevadas tasas de curación en todos los subtipos de pacientes, a través de regímenes más sencillos y mejor tolerados. Como preveíamos en Panorama Actual del Medicamento con ocasión de la comercialización del boceprevir y telaprevir, la situación actual evoca un evidente paralelismo farmacológico y terapéutico con la introducción a mediados de los años 90 del pasado siglo de los inhibidores de la proteasa del VIH (saquinavir, etc.), que revolucionaron la terapia del SIDA, permitiendo el desarrollo de los denominados TARGA (tratamientos antirretrovirales de gran actividad). Esperemos que sea así también para la hepatitis C, por el bien de todos.
|
ELVITEGRAVIR |
|---|
|
► STRIBILD® (Gilead) |
|
J05AX. ANTIINFECCIOSOS, USO SISTÉMICO Antivirales para tratamiento de infecciones por VIH y combinaciones. |
Es una combinación de tres agentes antirretrovirales, dos inhibidores de la transcriptasa inversa (emtricitabina y tenofovir) y un inhibidor de la integrasa (elvitegravir), junto con un potenciador farmacocinético (cobicistat), coformulados en un mismo medicamento que ha sido autorizado para el tratamiento de la infección por el virus de inmunodeficiencia humana tipo 1 (VIH-1) en adultos de 18 años de edad o mayores que nunca han recibido tratamiento antirretroviral o que están infectados por un VIH-1 sin mutaciones conocidas asociadas con resistencia a ninguno de los tres fármacos del medicamento. Elvitegravir es un inhibidor de la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Cobicistat es un análogo del ritonavir que es utilizado en esta combinación con el fin de potenciar los niveles de elvitegravir. Carece de efectos significativos sobre VIH y es un potente inhibidor de algunos isoenzimas del sistema citocromo P450 (CYP), especialmente sobre la subfamilia CYP3A y, en particular sobre la isoforma CYP3A4, principal vía metabólica del elvitegravir. Con ello, el cobicistat incrementa notablemente los niveles plasmáticos y la semivida de eliminación del elvitegravir y, con ello, sus efectos frente al VIH. Tenofovir y emtricitabina son dos inhibidores de la transcriptasa inversa análogos de nucleósidos o de nucleótidos (ITIAN), con capacidad de impedir la síntesis de DNA viral a partir de la cadena de ARN infectante.
{kind=link}
Los datos clínicos indican que el la combinación de elvitegravir, cobicistat, emtricitabina y tenofovir no es inferior a la combinación de efavirenz, emtricitabina y tenofovir (EET) o la de atazanavir potenciando con ritonavir y asociados a emtricitabina-tenofovir (ARET). En este sentido, las tasas de supresión virológica fueron del 87,6% vs. 84,1% (EET) a las 48 semanas y del 84,2% vs. 81,5% a las 96 semanas; en comparación con ARET, las correspondientes tasas fueron del 89,5% vs. 86,8% a las 48 semanas, y del 83,3% vs. 82,3% a las 96. En todos los casos, las diferencias no fueron estadísticamente significativas.
Sin duda alguna, uno de los objetivos terapéuticos esenciales de Stribild® es ofrecer una combinación eficaz en la primera línea del tratamiento de la infección por VIH en pacientes previamente no tratados, incluyendo por primera vez un inhibidor de la integrasa en una combinación coformulada una única unidad de dosis oral, que solo requiere una única administración diaria. Hasta ahora, las combinaciones disponibles solo contenían inhibidores de transcriptasa (la única combinación coformulada con inhibidores de la proteasa incluye simplemente al lopinavir potenciado por ritonavir; véase la tabla adjunta).
Actualmente, se considera que la falta de adherencia al mismo constituye la primera causa de fracaso terapéutico del tratamiento antirretroviral. De hecho, se considera que el uso de regímenes completos en comprimido único constituye la estrategia más eficiente para facilitar la adherencia. En definitiva, una innovación terapéutica de cierto interés que va claramente enfocada a prolongar la eficacia del tratamiento de primera línea, a través de una optimización de la adherencia de los pacientes, con una combinación innovadora de mecanismos de acción y un perfil aceptable de toxicidad.
|
DOLUTEGRAVIR |
|---|
|
► TIVICAY® (Glaxo SmithKline) |
|
J05AX. ANTIINFECCIOSOS, USO SISTÉMICO Antivirales para tratamiento de infecciones por VIH y combinaciones. |
Dolutegravir es un agente antiviral directo que actúa inhibiendo la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Está relacionado química y farmacológicamente con raltegravir y elvitegravir, previamente registrados. Ha sido autorizado para el tratamiento de adultos y adolescentes mayores de 12 años infectados por el Virus de Inmunodeficiencia Humana (VIH), en combinación con otros medicamentos antirretrovirales.
{kind=link}
Los datos clínicos confirman que las combinaciones con dolutegravir no son inferiores a las de raltegravir y elvitegravir en cuanto a eficacia virológica y clínica, y son superiores al efavirenz/tenofovir/emtricitabina y a las combinaciones incluyendo darunavir/ritonavir, en pacientes no tratados previamente. En cuanto a aquellos insatisfactoriamente tratados anteriormente, los que no había recibido un inhibidor de la integrasa, el dolutegravir fue superior estadísticamente al raltegavir, e incluso mostró una notable respuesta virológica (69%) en pacientes insatisfactoriamente tratados con combinaciones incluyendo raltegravir o elvitegravir.
La incorporación de fármacos de la clase de los inhibidores de la integrasa como tratamiento de primera línea de la infección por VIH da lugar a mejores resultados que otros grupos ya establecidos en la terapéutica, como los inhibidores de la proteasa (indinavir, etc.). Sin embargo, la aparición de la resistencia a los primeros inhibidores de la integrasa (raltegravir y elvitegravir) ha sido relativamente rápida. Por el contrario, el dolutegravir presenta una elevada barrera frente a las mutaciones del VIH en pacientes previamente no tratados con esta clase de antirretrovirales. Por ello, cuando se usa como parte de un tratamiento de primera línea, el dolutegravir es uno de los pocos antirretrovirales que no parecen producir mutaciones clínicamente relevantes, posiblemente debido a la mayor duración e intensidad del bloqueo de la integrasa, así como por la mayor limitación de la capacidad de replicación de las cepas de VIH que podrían hacerse resistentes al dolutegavir. Sin duda, el dolutegravir supone una cierta evolución en el relativamente reciente grupo de los inhibidores de la integrasa del VIH. Además, su perfil farmacocinético es más favorable que el de sus antecesores de este grupo, con una semivida celular prolongada que facilita una única administración diaria, y todo ello, unido a la utilidad en pacientes que previamente habían fracasado con raltegravir o elvitegravir, le aporta un significativo valor añadido y viene a consolidar la relevancia de los inhibidores de la integrasa como fármacos de primera consideración en el tratamiento de la infección por VIH.
|
VACUNA MENINGOCÓCICA GRUPO B |
|---|
|
► BEXSERO® (Novartis) |
|
J07AH. terapia antiinfecciosa sistémica Vacunas antibacterianas: meningococos. |
Se trata de una vacuna combinada de varias proteínas procedentes del serotipo B de la Neisseria meningitidis indicada para la inmunización activa de individuos a partir de 2 meses de edad frente a la enfermedad meningocócica invasiva causada por Neisseria meningitidis grupo B.
Los resultados serológicos sobre la capacidad inmunogénica de la nueva vacuna son claros y contundentes, con porcentajes de sujetos con títulos preventivos de anticuerpos por encima del 98%, medidos un mes después de la última dosis. Los estudios clínicos son de actividad bactericida de los anticuerpos generados y de la persistencia de estos, ya que es prácticamente imposible realizar un estudio clínico convencional de eficacia, dada la baja incidencia de la enfermedad invasiva por meningococo. Por ello, el beneficio real de esta nueva vacuna solo podrá evaluarse realmente tras su implantación en los programas oficiales de vacunación sistemática. En cualquier caso, la utilización del ensayo bactericida ha demostrado una estrecha correlación con la protección frente a la enfermedad invasiva meningocócica. La duración de la protección todavía no es conocida, aunque se ha podido observar que los títulos de anticuerpos para dos de los antígenos (PorA P1.4 y NHBA) declinan rápidamente (6 meses después de la primovacunación y 12 después de la dosis de recuerdo.
La enfermedad invasiva por meningococo B es un cuadro grave, con una mortalidad asociada superior al 8%, que afecta fundamentalmente a los niños pequeños, con un segundo pico de incidencia durante la adolescencia, y hasta ahora no existía ninguna vacuna eficaz para su prevención. Por ello, la vacuna está claramente dirigida para estos grupos de edad; no obstante, en caso de focos epidémicos es deseable vacunar a toda la población.
Al hecho de ser la primera vacuna capaz de inmunizar frente al serotipo B del meningococo, que es el más prevalente en España y en el resto de la Unión Europea, hay que agregar que se trata de la primera vacuna eficaz que no utiliza el sistema de conjugación de proteínas y polisacáridos capsulares empleado con tanto éxito para prevenir meningitis y septicemias de otros serotipos de meningococos (A, C, W, Y), de neumococos (13 serotipos) y de Heamophillus influenzae tipo b. Esto último también supone un avance farmacotécnico nada desdeñable, que abre nuevas líneas de investigación para el desarrollo de vacunas frente a bacterias y otros microorganismos patológicamente relevantes.
|
VACUNA HERPES ZÓSTER |
|---|
|
► ZOSTAVAX® (Sanofi Pasteur MSD) |
|
J07AH. Terapia antiinfecciosa sistémica Vacunas varicela zóster. |
Es una vacuna viva atenuada conteniendo virus varicela-zóster (VVZ) de la cepa Oka/Merck, autorizada para la prevención del herpes zóster y la neuralgia posherpética relacionada con herpes zóster, en individuos de 50 años de edad o mayores.
Los estudios clínicos disponibles muestran que la vacuna es relativamente efectiva para prevenir la enfermedad del herpes zóster, reduciendo su incidencia global en torno al 50%. Sin embargo, algunos estudios han mostrado que apenas un 50% de los sujetos vacunados presentan un aumento de al menos dos veces en el título basal de anticuerpos. Por ello, parece que tanto la inmunidad humoral como la celular son cruciales para controlar la reactivación de virus del herpes zóster; en cualquier caso, no se conoce cuál es el título de anticuerpos predictivo a largo plazo para la protección frente al desarrollo de herpes zóster.
Debido a la escasez de datos y la carga desconocida de la enfermedad en la mayoría de los países, la evidencia inicial de disminución de la protección a lo largo del tiempo y la incertidumbre de la edad óptima para la vacunación, el Strategic Advisory Group of Experts (SAGE) on Immunization de la Orgnización Mundial de la Salud no ha hecho ninguna recomendación sobre el uso rutinario de la vacuna contra el herpes zóster. Conviene, en cualquier caso, no olvidar que el número necesario a tratar para beneficiar (NNTB) es de 50.
l. ANTINEOPLÁSICOS Y TERAPIA INMUNOMODULADORA
|
DECITABINA |
|---|
|
► DACOGEN® (Janssen Cilag) |
|
L01BC. terapia antineoplásica y agentes inmunomoduladores Citostáticos: antimetabolitos análogos de la pirimidina. |
|
Medicamento Huérfano |
La decitabina es un agente antitumoral de tipo antimetabolito (es un análogo de la citidina), estructural y farmacológicamente relacionado con la azacitidina. La capacidad de la decitabina para limitar sustancialmente los procesos de metilación del ADN, parece ser determinante para permitir la recuperación de la expresión de ciertos genes supresores tumorales y para dificultar la conversión tumoral de los precursores celulares hematológicos. El medicamento ha sido autorizado para el tratamiento de pacientes adultos de 65 o más años de edad con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional. Dada la muy baja prevalencia de esta condición, el medicamento ha sido designado como huérfano.
{kind=link}
Los datos clínicos disponibles han mostrado una mayor eficacia que la citarabina en bajas dosis en términos de supervivencia global, con una mejora de 2,7 meses, así como en otras variables clínicamente significativas. Tales diferencias son consideradas como clínicamente relevantes, habida cuenta de que los pacientes sobre los que se utilizaron ambas eran de alto riesgo (un 40% tenían ≥75 años).
Es difícil establecer el papel de la decitabina en el tratamiento, aunque dada la notable escasez de alternativas existente en los pacientes con peor pronóstico, no cabe duda que supone una opción plenamente válida; asimismo, su perfil toxicológico no excesivamente problemático la convierte en una atractiva alternativa en terapias de combinación con otros agentes antileucémicos. Por otro lado, tal como señala el Informe de Posicionamiento Terapéutico de la AEMPS, los datos no permiten comparar clínicamente a la decitabina con la azacitidina, aunque el nivel de evidencia de la primera es mayor en pacientes mayores de 65 años con leucemia mieloide aguda, tanto primaria como secundaria.
|
BRENTUXIMAB VEDOTINA |
|---|
|
► ADCETRIS® (Takeda) |
|
L01XC. terapia antineoplásica y agentes inmunomoduladores Citostáticos: anticuerpos monoclonales. |
|
Medicamento Huérfano |
El brentuximab vedotina es un anticuerpo monoclonal conjugado con un agente citotóxico (vedotina), en el que el anticuerpo actúa como transportador seleccionando las células con presencia de la proteína CD30 en su superficie; ésta es expresada por los linfocitos B de la mayoría de los pacientes con linfoma de Hodgkin clásico (LHc) y en algunas formas de linfoma no Hodgkin, como el linfoma anaplásico de células grandes (LACG). Una vez en el interior celular, el fármaco accede al interior de los lisosomas donde sufre un proceso de escisión proteolítica liberando la vedotina (monometil auristatina E, cuya estructura aparece en la figura). La vedotina es un potente agente citotóxico que actúan interfiriendo con la polimerización de la tubulina; por ello, impide la formación del huso mitótico durante la división celular bloqueando el ciclo celular en fase G2/M, lo que provoca la activación de los mecanismos de apoptosis (muerte celular programada) y, en definitiva, la muerte celular.
{kind=link}
El medicamento ha sido autorizado para el tratamiento de pacientes adultos con linfoma de Hodgkin CD30+ en recaída o refractario después de trasplante autólogo de células madre o después de al menos dos tratamientos previos cuando el trasplante autólogo de células madre o la poliquimioterapia no es una opción terapéutica; también está indicado para el tratamiento de pacientes adultos con linfoma anaplásico de células grandes sistémico en recaída o refractario.
Los datos clínicos muestran importantes tasas de respuesta objetiva: 75% (34% completa) en los pacientes con linfoma de Hodgkin y 86% (59% completa) en aquellos con linfoma anaplásico de células grandes, con medianas de supervivencia libre de progresión de 5,3 y 14,3 meses, respectivamente. Por otro lado, la administración de brintuximab vedotina en pacientes con linfoma anaplásico refractario facilita que estos puedan beneficiarse de un ulterior trasplante de médula y, con ello, ofrece la oportunidad de curación del cuadro o, al menos, de una supervivencia prolongada. Asimismo, atendiendo a sus datos de eficacia clínica – aun asumiendo la notable incertidumbre derivada de la ausencia de brazo comparador – y no excesiva toxicidad, el tratamiento con brentuximab vedotina puede considerarse como tratamiento de elección en pacientes con linfoma de Hodgkin refractario/recidivante tras un trasplante autólogo de células madre o varios regímenes quimioterápicos.
Es importante destacar que el conjugado brentuximab-vedotina es el resultado de un diseño teórico muy interesante – aunque no nuevo – en el que se utiliza un agente que transporta el auténtico agente citotóxico (vedotina) y permite su incorporación selectiva a las células diana del tratamiento. En definitiva, se trata de una innovadora vía de actuación farmacológica, con tasas notables de respuesta en cuadros de muy difícil tratamiento, aunque la incertidumbre derivada de la ausencia de brazo comparador en los ensayos clínicos disponibles atempere sustancialmente el grado de interés innovador de este medicamento.
|
OFATUMUMAB |
|---|
|
► ARZERRA® (Glaxo SmtihKline) |
|
L01XC. terapia antineoplásica y agentes inmunomoduladores Citostáticos: anticuerpos monoclonales. |
|
Medicamento Huérfano |
Ofatumumab es un anticuerpo monoclonal humano (IgG1) que se une específicamente a la proteína CD20 presente en la membrana de los linfocitos B provocando su muerte, fundamentalmente a través de mecanismos citotóxicos activados por el complemento y por células dependientes de anticuerpos. Ha sido autorizado para el tratamiento de leucemia linfocítica crónica en pacientes que son refractarios a fludarabina y alemtuzumab. Dada la condición de enfermedad rara de esta indicación, el medicamento fue designado como huérfano.
Ofatumumab produce una respuesta objetiva clínicamente relevante: 49% de los pacientes doblemente refractarios a fludarabina y alemtuzumab obtuvieron una respuesta parcial al tratamiento con ofatumumab, con una supervivencia media libre de enfermedad de 6,4 meses y una supervivencia global de 13,9, sin que haya diferencias sustanciales entre los pacientes tratados previamente o no con rituximab.
Aunque los datos clínicos no son todo lo completos que serían de desear y los resultados no son espectaculares, hay que tener en cuenta que actualmente no existe ningún tratamiento establecido para los cuadros refractarios a fludarabina y alemtuzumab, por lo que supone un paso positivo en esta línea. Por otro lado, debe tenerse en cuenta que la leucemia linfocítica crónica es una enfermedad con un curso clínico muy heterogéneo; no obstante, se ha producido una notable mejora en la capacidad de establecer un pronóstico a través de la utilización de parámetros clínicos, biológicos y genéticos, lo que permite hacer un enfoque específico para cada situación. Sin embargo, esto, a su vez, supone un importante reto para elegir la estrategia terapéutica más adecuada entre las disponibles. En esto, la incorporación del ofatumumab viene a colaborar en los cuadros más refractarios, con una moderada pero significativa eficacia y pese a no ser farmacológicamente innovador, ya que el rituxumab también es selectivo para CD20, su eficacia en pacientes refractarios a este último le hace portador de un cierto valor.
|
PERTUZUMAB |
|---|
|
► PERJETA® (Roche) |
|
L01XC. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Citostáticos: anticuerpos monoclonales. |
El pertuzumab es un anticuerpo monoclonal humanizado recombinante dirigido contra el dominio de dimerización extracelular (subdominio II) de la proteína 2 del receptor de crecimiento epidérmico (HER2), lo que bloquea la formación de heterodímeros de HER2 con otros miembros de la familia HER activados por sus ligandos. En contraste con el pertuzumab, el trastuzumab se une al subdominio IV del dominio extracelular de HER2 y altera las interacciones independientes de los ligandos, pero no es eficaz en el bloqueo de la heterodimerización de HER2 con otros miembros de la familia HER (ErbB) activados por ligandos (EGFR, HER3 o HER4). El pertuzumab ha sido autorizado para el tratamiento en combinación con trastuzumab y docetaxel para el tratamiento de pacientes adultos con cáncer de mama HER2 positivo localmente recidivante irresecable o metastásico, que no han recibido tratamiento previo anti-HER2 o quimioterapia para la enfermedad metastásica.
Los datos clínicos indican que el pertuzumab incrementa en 6,1 meses la duración de la supervivencia libre de progresión tumoral, con una reducción de la tasa de mortalidad a los 30 meses de casi 10 puntos porcentuales (28,1% vs. 37,9%). Los resultados fueron relevantes considerando diferentes aspectos de los pacientes y lo fueron particularmente en pacientes mayores de 65 años.
Los tumores HER2 positivos representan aproximadamente el 20% de todos los cánceres de mama y, además, confieren a estos un comportamiento más agresivo y, por tanto, empeoran el pronóstico general. Por consiguiente, la acción selectiva del trastuzumab vino a revolucionar el tratamiento y, de hecho, se ha convertido en un estándar para este tipo de tumores. Aunque está farmacológicamente relacionado con el trastuzumab, el pertuzumab presenta un mecanismo parcialmente diferente, lo que permite complementar – y ampliar – el bloqueo de la vía de señalización tumoral HER; en este sentido, el pertuzumab impide la formación de heterodímeros con otros miembros de la familia (HER1 o EGFR, HER3 y HER4), amplificando los efectos del trastuzumab. En definitiva, el pertuzumab supone una relevante incorporación para el tratamiento del cáncer de mama HER2 positivo, al incrementar significativamente la supervivencia de los pacientes ampliando el perfil farmacológico del trastuzumab, todo ello con un perfil de seguridad aceptable.
|
AFATINIB |
|---|
|
► GIOTRIF® (Boehringer Ingelheim) |
|
L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Otros citostáticos: inhibidores directos de la proteína cinasa |
El afatinib es un antineoplásico que actúa inhibiendo de forma potente, irreversible y selectiva determinadas proteína cinasas implicadas en la proliferación celular tumoral, en particular las cinasas que forman parte de los receptores de la familia ErbB y, en particular, el EGFR o receptor del Factor de Crecimiento Epidérmico (EGFR, HER-1, erbB1, c-erbB). Ha sido autorizado para el tratamiento en monoterapia de adultos naïve a inhibidores de tirosina cinasa (TKI) del Receptor del Factor de Crecimiento Epidérmico (EGFR) con cáncer de pulmón no microcítico localmente avanzado o metastásico, con mutaciones activadoras del EGFR.
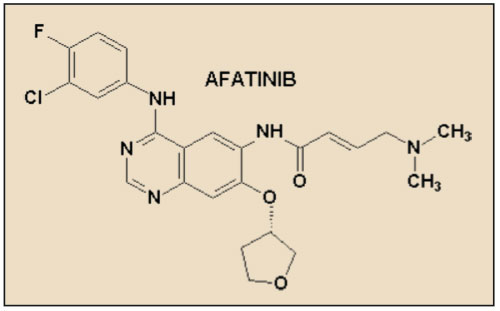{kind=link}
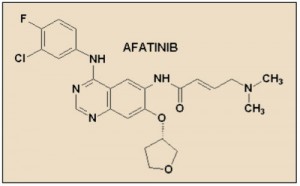
Los ensayos clínicos indican que el fármaco es capaz de retrasar significativamente la progresión tumoral en estos pacientes, al menos en comparación con la quimioterapia estándar, con valores medianos de 11,1 frente a 5,6-5,9 meses. La principal diferencia se aprecia en los pacientes con genotipo tumoral Del-19 (13,7 vs. 5,6 meses), mientras que para L858R la diferencia es menor (9,6-10,8 vs. 5,6-8,1) hasta el punto de que en uno de los estudios no alcanza la significación estadística (aunque con una evidente tendencia); en el caso de otras mutaciones los resultados son poco consistentes. Tampoco alcanza significación estadística la diferencia entre ambos tratamientos en el caso de pacientes no asiáticos (mayoritariamente caucásicos).
En términos de retraso de la progresión tumoral, afatonib mejora los conseguidos con la quimioterapia estándar y parecen ser también algo mejores que los de erlotinib y gefitinib, aunque – como es obvio – las comparaciones indirectas son cuestionables; con todo, afatinib ha demostrado inhibir in vitro a cinasas con las mutaciones T790M y T854A, causantes de resistencia tumoral a los inhibidores reversibles de cinasas erlotinib y gefitinib.
Sea como fuere, no se aprecia ninguna mejora demostrable con afatinib en términos de supervivencia global, comparación con otros inhibidores de tirosina cinasas (TKI) o incluso la quimioterapia estándar, lo que viene a reafirmar el papel que, al menos por el momento, se suele reservar a la mayoría de los miembros de esta serie: retrasar la progresión tumoral y mejorar la calidad de vida de los pacientes, más que incrementar la duración total de la vida de estos. En este sentido, las mutaciones específicas del EGFR (o de otras cinasas asociadas a receptores) parecen constituir biomarcadores predictivos del beneficio terapéutico de los TKI (al menos en términos de retraso de la progresión tumoral), lo que justifica la necesidad de determinar el genotipo tumoral previamente al uso de estos fármacos en cáncer de pulmón no microcítico y, posiblemente, en otras indicaciones donde se emplean.
|
AXITINIB |
|---|
|
► INLYTA® (Pfizer) |
|
L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Otros citostáticos: inhibidores directos de la proteína cinasa |
El axitinib es un agente antineoplásico, que actúa inhibiendo potente y selectivamente determinadas proteína cinasas implicadas en la proliferación celular tumoral y, en particular, las cinasas que forman parte de los receptores 1, 2 y 3 del factor de crecimiento endotelial vascular VEGF (VEGFR-1, 2 y 3), que están implicados en los procesos de neovascularización y la progresión tumoral. Ha sido autorizado para el tratamiento de pacientes adultos con carcinoma avanzado de células renales tras fracaso a un tratamiento previo con sunitinib o citocinas.
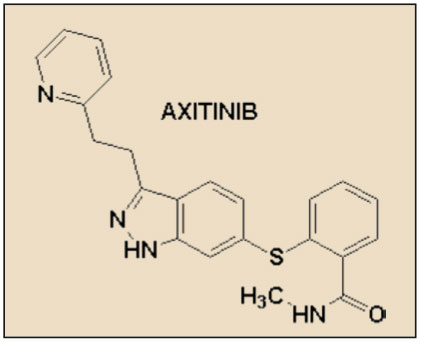{kind=link}
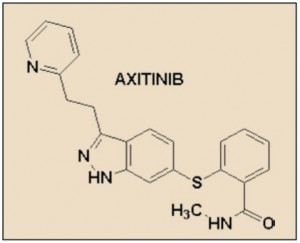
En términos globales de supervivencia libre de progresión tumoral, el axitinib parece ser superior a sorafenib, con 8,3 vs. 5,7 meses, una diferencia estadísticamente significativa de 2,6 meses; sin embargo, en términos de supervivencia global, tal diferencia prácticamente desparece y, en cualquier caso, se hace estadísticamente insignificante (20,1 vs. 19,2 meses). Asimismo, cuando se consideran exclusivamente los pacientes previamente tratados con sunitinib (supervivencia libre de progresión tumoral de 4,8 vs 3,4 meses, con diferencia estadísticamente significativa de 1,4 meses), los resultados clínicos son notablemente peores que los obtenidos tras usar citocinas (12,1 vs. 6,5 meses, con diferencia estadísticamente significativa de 5,6 meses). También la supervivencia global es inferior en los pacientes tratados previamente con sunitinib que en aquellos tratados con citocinas (15,2 vs. 29,4 meses con axitinib y 16,5 vs. 27,8 meses con sorafenib).
Sea como fuere, el axitinib muestra una respuesta clínica en pacientes previamente tratados con TKI que, aunque modesta, es significativamente mejor que la del sorafenib (4,8 vs. 3,4), bastante similar a la obtenida con everolimús (5,4 meses) en este tipo de pacientes y, aunque las comparaciones indirectas de datos clínicos deben valorarse siempre con mucha prudencia, parece sugerir una cierta similitud en la respuesta clínica. Sin embargo, las mejoras leves, aunque estadísticamente significativas, en las variables clínicas utilizadas más habitualmente (como la supervivencia libre de progresión tumoral) no deben ser el único criterio en este tipo de pacientes, sino que deben complementarse con aspectos relativos a la calidad de vida, el balance entre el control de síntomas clínicos, incidencia y gravedad de efectos adversos, y evitar interrupciones o ajustes posológicos innecesarios. En este sentido, en ausencia de terapias curativas (como es el caso del cáncer metastásico renal), el mantenimiento de la calidad de vida es una objetivo fundamental del tratamiento y elegir un fármaco como segunda línea de tratamiento que sea capaz de ello es, sin duda, una prioridad en la investigación clínica.
|
CRIZOTINIB |
|---|
|
► XALKORI® (Pfizer) |
|
L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Otros citostáticos: inhibidores directos de la proteína cinasa |
Crizotinib es un inhibidor específico y selectivo de la tirosina cinasa de tipo ALK y sus variantes oncogénicas (EML-ALK, NPM–ALK), así como del Receptor del Factor de Crecimiento de los Hepaticitos (c-Met/HGFR) y enzimas RON en varias líneas celulares humanas. Como consecuencia de ello, crizotinib inhibe la proliferación celular, migración, invasión y motilidad en células tumorales y endoteliales, lo que induce un efecto antitumoral y antiangiogénico. El medicamento ha sido autorizado para el tratamiento de adultos con carcinoma de pulmón no microcítico avanzado, positivo para la cinasa del linfoma anaplásico (ALK), previamente tratado.
{kind=link}
Los datos clínicos indican tasas de respuesta objetiva tumoral del 53% al 65%, una supervivencia sin progresión tumoral que va de 7,7 a 9,2 meses y una supervivencia global estimada entre 20,3 y 29,6 meses. Los datos son muy similares a los obtenidos con la quimioterapia de referencia (pemetrexed o docetaxel).
El crizotinib supone una innovación al actuar selectivamente sobre cuadros clínicos que pueden ser adecuadamente diagnosticados gracias a la existencia de marcadores biológicos, como es el caso de la cinasa del sarcoma anaplásico (ALK), lo que permite obtener unos resultados clínicos relevantes. Precisamente, la detección de marcadores moleculares específicos está permitiendo mejorar la respuesta terapéutica del cáncer de pulmón no microcítico. Así, por ejemplo, al considerar las mutaciones específicas del receptor del factor de crecimiento epidérmico (EGFR), la utilización de erlotinib y de gefitinib mejora notablemente los resultados en este tipo de cáncer. En esta misma línea, la presencia de una traslocación en el gen que codifica la cinasa asociada al linfoma anaplásico (ALK) ha sido identificada como un marcador biológico potencialmente útil para el tratamiento selectivo de determinados inhibidores de tirosina cinasas, como es el caso del crizotinib. De hecho, la translocación genética que implica la ALK ocurre en el 2-5% de todos los casos de cáncer de pulmón no microcítico y predominantemente en personas jóvenes (menores de 50 años) con adenocarcinomas y no fumadoras. Actualmente, están en fase de desarrollo otros diversos inhibidores específicos de cinasas procedentes de genes mutados.
|
DABRAFENIB |
|---|
|
► TAFINLAR® (Pfizer) |
|
VEMURAFENIB |
|
► ZELBORAF® (Roche) |
|
L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Otros citostáticos: inhibidores directos de la proteína cinasa |
El vemurafenib y el dabrafenib son agentes antineoplásicos, que actúan inhibiendo potente y selectivamente determinadas proteína cinasas particularmente implicadas en la proliferación celular tumoral. En concreto, inhiben selectivamente las diversas formas mutadas de la serina-treonina cinasa BRAF, relacionadas con la promoción de la proliferación celular en ausencia de factores de crecimiento. En este sentido, han sido autorizados para el tratamiento en monoterapia de pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600 positiva.
{kind=link}
La eficacia clínica de dabrafenib y vemurafenib ha sido contrastada frente a la dacarbazina, mostrando valores de supervivencia en ausencia de progresión tumoral de 6,9 meses en ambos casos (frente a 1,6 y 2,9 meses con dacarbazina, en los dos ensayos clínicos con vemurafenib y dabrafenib, respectivamente). Esto sugiere una clara superioridad de los dos inhibidores de proteína cinasas sobre la dacarbazina. Otro tanto puede indicarse para las variables de supervivencia global, con valores medianos de 13,6 vs. 9,7 meses (vemirafenib vs. dacarbazina) y 18,2 vs. 15,6 (dabrafenib vs. dacarbazina). En este sentido, cabe agregar que las variables de eficacia clínica de supervivencia global y supervivencia libre de progresión muestran una notable correlación en melanoma (Flaherty, 2014).
Puede afirmarse que los inhibidores de la cinasa mutada BRAF vemurafenib y dabrafenib, junto con el ipilimumab, están cambiando el panorama de tratamiento del melanoma metastásico. De hecho, estas nuevas terapias han demostrado una mejora a largo plazo en la evolución del paciente, un beneficio que no ofrece la quimioterapia convencional. No obstante, es muy importante optimizar la secuencia y/o la combinación de los tratamientos actuales, con el fin de aumentar el número de pacientes potencialmente beneficiados con un aumento significativo de su supervivencia en condiciones de calidad de vida razonablemente buena.
|
ENZALUTAMIDA |
|---|
|
► XTANDI® (Astellas) |
|
L02BB. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Terapia endocrina: antiandrógenos. |
La enzalutamida es un antagonista de los receptores androgénicos, indicado en el tratamiento de hombres adultos con cáncer de próstata metastásico resistente a la castración cuya enfermedad ha progresado durante o tras el tratamiento con docetaxel.
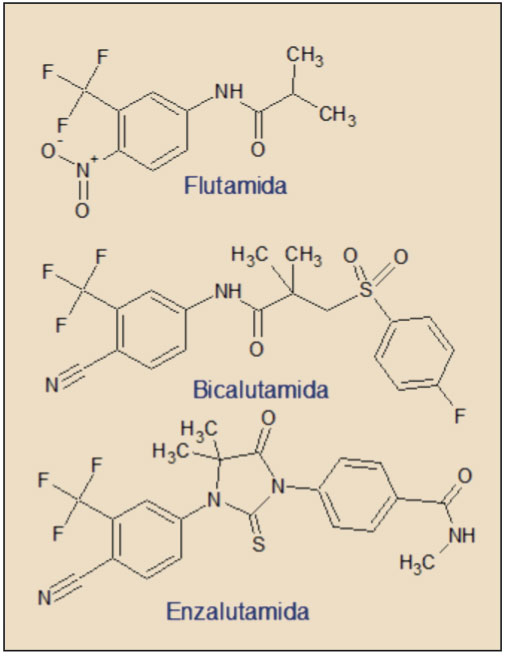{kind=link}
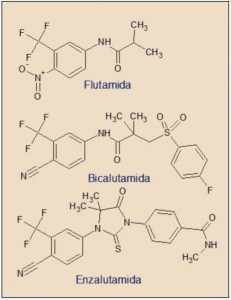
Aunque la enzalutamida está estrechamente relacionada con otros antagonistas del receptor anndrogénico (flutamida, bicalutamida), la enzalutamida tiene entre cinco a ocho veces mayor afinidad que la bicalutamida hacia el receptor androgénico. Pero, lo más importante es que además reduce la translocación nuclear del receptor androgénico activado, resultando en una disminución significativa de la cantidad de receptores presente en el citoplasma, y dificulta la unión del receptor activado con el ADN celular e interfiere con el reclutamiento del co-activador, todo lo cual resulta en una disminución del crecimiento celular y la producción de apoptosis celular y, en definitiva conduce a una reducción del volumen tumoral prostático. En este sentido, puede considerarse como una evolución farmacológica dentro de este grupo (Aragón-Ching, 2014).
Los datos clínicos indican una clara superioridad del fármaco frente al placebo, con una mejora de 4,8 meses en la supervivencia global. Aunque pueda parecer un periodo no excesivamente grande, es clínicamente relevante dada la condición prácticamente terminal de los pacientes con progresión tumoral tras recibir docetaxel. El resto de variables clínicas, incluyendo calidad de vida, muestran una superioridad incontestable.
Una vez que se produce la resistencia hormonal (incremento de la población de células tumorales independientes de la activación androgénica), no está claro si los regímenes posteriores con agentes antiandrogénicos ejercerían el mismo beneficio, ya que no existen comparaciones entre los medicamentos actualmente disponibles.
En definitiva, un nuevo fármaco del grupo de los antagonistas androgénicos, bioquímicamente evolucionado y clínicamente útil, que puede facilitar un moderado alargamiento de la supervivencia en pacientes con cuadros progresivos tras la terapia de segunda línea (con docetaxel) y, por tanto, se consolida como una alternativa a las opciones actualmente disponibles para estos pacientes: cabazitaxel y abiraterona, a utilizar en función del historial clínico y farmacoterapéutico de cada paciente.
|
LIPEGFILGRASTIM |
|---|
|
► LONQUEX® (Teva) |
|
L02BB. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Terapia endocrina: antiandrógenos. |
El lipegfilgrastim es una forma glucosilada del pegfilgrastim, un derivado del factor estimulante de colonias de granulocitos (G-CSF). Ha sido autorizado para la reducción de la duración de la neutropenia y de la incidencia de neutropenia febril en pacientes adultos con tumores malignos tratados con quimioterapia citotóxica (con excepción de leucemia mieloide crónica y síndromes mielodisplásicos).
El lipegfilgrastim ha demostrado su eficacia reduciendo la incidencia de neutropenia grave y de neutropenia febril, en comparación con placebo; asimismo, ha demostrado no ser inferior al pegfilgrastim en esta indicación. En definitiva, los perfiles de eficacia y de seguridad de lipegfilgrastim son similares a los del pegfilgrastim, sin que se aprecie ninguna mejora o innovación sobre este último.
|
PIRFENIDONA |
|---|
|
► ESBRIET® (InterMune) |
|
L04AX. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores: otros. |
|
Medicamento Huérfano |
La pirfenidona es un agente antifibrótico y antiinflamatorio que ha sido autorizado como medicamento huérfano para el tratamiento de la fibrosis pulmonar idiopática leve a moderada en adultos. Se desconoce su mecanismo concreto de acción sobre la fibrosis pulmonar, debido a que desarrolla múltiples acciones a nivel celular y tisular; en general, reduce la acumulación de células y citocinas proinflamatorias en la fibrosis pulmonar.
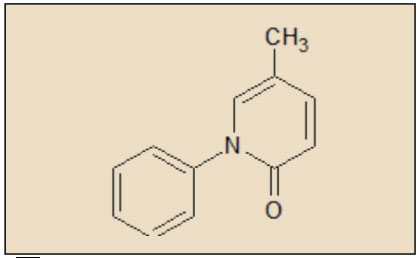{kind=link}
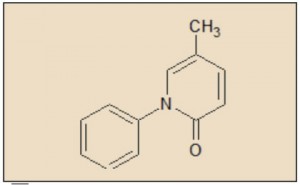
El fármaco ha demostrado producir en estos pacientes un notable y significativo descenso del 48% del riesgo de muerte por cualquier causa, que alcanza el 68% en la muerte relacionada con la fibrosis pulmonar. Aunque es cierto que los pacientes incluidos en los ensayos clínicos tenían un nivel leve a moderado de gravedad en su fibrosis pulmonar idiopática, la magnitud del efecto observado es lo suficientemente relevante como para tomarla seriamente en consideración, en especial si se tiene en cuenta que hasta el momento no existe ningún tratamiento que haya demostrado ejercer ningún efecto significativo en este aspecto.
Con todo, conviene no olvidar que la enfermedad sigue sin tener curación y que, lamentablemente, el paciente está abocado a la muerte; pero, por este mismo motivo, la disponibilidad de un tratamiento capaz de retrasar – siquiera modestamente – la progresión de la enfermedad debe ser valorada favorablemente. En la actualidad, se aconseja su uso en pacientes con CVF> 50% y DLCO> 35% y puede considerarse como tratamiento de primera línea para la fibrosis pulmonar idiopática leve-moderada. La duración recomendable del tratamiento es de 12 meses como mínimo y si la respuesta fuese clínicamente satisfactoria, parece lógico que deba continuarse el tratamiento.
|
POMALIDOMIDA |
|---|
|
► IMNOVID® (Celgene) |
|
L04AX. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores: otros. |
|
Medicamento Huérfano |
La pomalidomida es un agente antitumoral análogo de talidomida y lenalidomida, con propiedades inmunomoduladoras y antiangiogénicas. Ha sido autorizada para el tratamiento en combinación con dexametasona de pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento. Dada la condición de enfermedad rara que tiene el mieloma múltiple, la pomalidomida fue designada como medicamento huérfano.
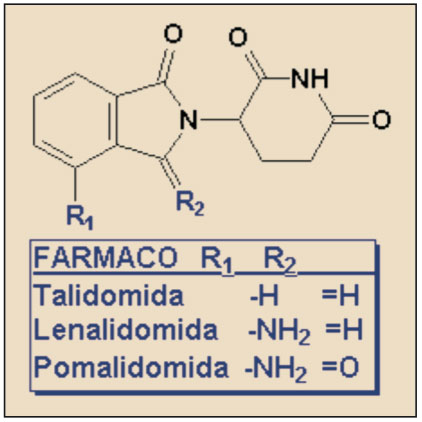{kind=link}
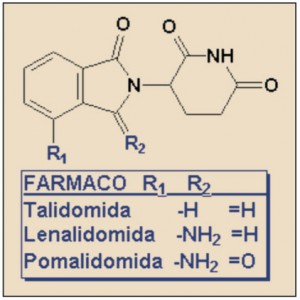
Los datos clínicos han establecido una diferencia estadísticamente significativa a favor de la combinación pomalidomida-dexametasona vs. dexametasona en altas dosis de casi dos meses (15,7 vs. 8,0 semanas) para la supervivencia libre de progresión tumoral, y mejoras también significativas en la supervivencia global. Aunque los datos numéricos no son obviamente espectaculares, una mejora de estas características debe valorarse favorablemente teniendo en cuenta que se ha conseguido en pacientes con refractarios o refractarios y recidivantes tras al menos dos tratamientos previos, en los que se incluía bortezomib y lenalidomida, considerados como piezas clave en el tratamiento del mieloma múltiple.
Los datos tanto in vitro como clínicos muestran que la pomalidomida presenta una actividad antimielomatosa mayor que la talidomida y la lenalidomida, con un perfil toxicológico similar o incluso algo más favorable. La importancia de este hecho viene reflejada por la circunstancia de que tanto la FDA como la EMA utilizaron un sistema acelerado de autorización, al considerar que la pomalidomida podría constituirse en referencia terapéutica en estos cuadros avanzadas y multirresistentes de mieloma, tradicionalmente con muy mal pronóstico. En este sentido, a pesar de las mejoras incorporadas en el tratamiento del mieloma, especialmente con el bortezomib y la lenalidomida (tras la talidomida), la mayoría de los casos acaban por hacerse resistentes y, por ello, la llegada de la pomalidomida puede considerarse un cierto avance en este campo, al haber mostrado eficacia sobre dichos cuadros resistentes. Así y todo, la magnitud global de la respuesta – y el lado oscuro de su toxicidad – no permite considerar a la pomalidomida como un avance sustancial en este campo, aunque sí viene a aportar alguna mejora en relación a sus antecesores.
n. SISTEMA NERVIOSO
|
PERAMPANEL |
|---|
|
► FYCOMPA® (Eisai) |
|
N03AX. SISTEMA NERVIOSO. Antiepilépticos: otros. |
Perampanel es un agente antiepiléptico que actúa como antagonista selectivo y no competitivo de los receptores ionotrópicos de glutamato de tipo AMPA en las neuronas postsinápticas. Ha sido autorizado para el tratamiento concomitante de las crisis de inicio parcial con o sin generalización secundaria en pacientes con epilepsia de 12 años y mayores.
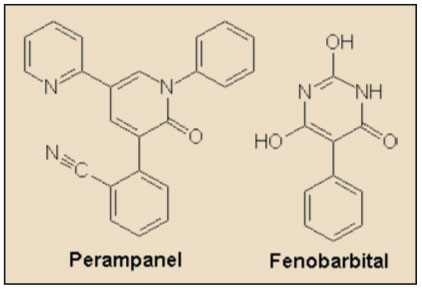{kind=link}
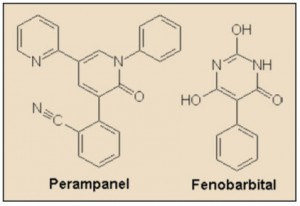
Mejora en 6-20 puntos porcentuales al placebo, con las dosis de 8 y 12 mg/día, en la reducción de la frecuencia de crisis epilépticas en pacientes refractarios a dos o tres tratamientos previos. Los resultados son incluso mejores cuando se considera específicamente los cuadros de crisis parciales complejas o aquellos con generalización secundaria, con 13 a 25 puntos porcentuales de diferencia. En términos de pacientes respondedores (considerando a aquellos que experimentan una reducción del al menos un 50% en el número de crisis), la diferencia con el placebo se sitúa en torno los 12-18 puntos. Otras variables también muestran una tendencia favorables al perampanel, aunque enocasiones tal diferencia es marginal o, incluso, estadísticamente no significativa. Sin embargo, la tasa de respondedores obtenida en los ensayos clínicos no alcanza ni siquiera al 40% de los pacientes estudiados.
El fármaco presenta un mecanismo de acción que, aunque conocido, no formaba parte del arsenal farmacológico disponible en clínica. El antagonismo de los receptores AMPA también está presente en algunos de los fármacos actualmente en uso, como fenobarbital, lamotrigina o topiramato, pero en estos solo tiene un papel marginal. Esto supone, de hecho, que el perampanel es el primer miembro efectivo de este grupo y, por ello, permite ampliar las opciones terapéuticas, algo siempre valorable un campo patológico donde más del 30% de los pacientes está inadecuadamente controlado. Sin embargo, la ausencia de estudios clínicos directamente comparativos entre los nuevos fármacos antiepilépticos no permite situar de forma nítida al perampanel en el arsenal antiepiléptico.
|
DEXMEDETOMIDINA |
|---|
|
► DEXDOR® (Orion) |
|
N05CM. SISTEMA NERVIOSO. Psicolépticos. Hipnóticos y sedantes: otros. |
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (µ2) adrenérgicos, estrechamente relacionado con la clonidina y la oximetazolina, que ha sido autorizado para la sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (Richmind Agitation Sedation Score, RASS).
{kind=link}
Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta y que da lugar a una inhibición de la liberación de noradrenalina en dicho núcleo, lo que impide el control inhibitorio del núcleo preóptico ventrolateral, que libera ácido gamma(γ)aminobutírico (GABA) y galanina, que incrementan la propia inhibición del locus ceruleus y del núcleo tuberomamilar. Esta inhibición también reduce la liberación de histamina y su efecto sobre las neuronas de diversas áreas subcorticales, que se traduce en un efecto hipnótico adicional, tal como ocurre durante el sueño fisiológico.
Los ensayos clínicos controlados con midazolam y propofol han mostrado la no inferioridad de la dexmedetomidina frente a estos últimos en la indicación autorizada, en particular en lo que se refiere al porcentaje del tiempo (60-65%) durante el cual se mantiene el nivel de sedación requerido para cada paciente, sin necesidad de medicación sedante de rescate; tampoco se han registrado diferencias significativas en cuanto a la duración media del uso de ventilación mecánica en la UCI. Sin embargo, sí se apreció un uso más frecuente de la medicación sedante de rescate con dexmedetomidina que con propofol (73 vs. 64%), pero similar al midazolam (60 vs. 57%). No obstante, cuando se utiliza en pacientes en los que se requiere un nivel de sedación más profundo (puntuación -4 RASS), la desxmedetomidina fue claramente inferior a propofol y midazolam.
Se trata de una nueva línea de actuación farmacológica en sedación humana, ya que en veterinaria este tipo de agentes se venía utilizando desde hace décadas; de hecho, la dexmedetomidina es un análogo de la etomidina, profusamente utilizada en veterinaria. En definitiva, la dexmedetomidina representa una forma algo diferente de sedación para la UCI que los habitualmente utilizados, el propofol y el midazolam. Produce un tipo de sedación consciente, con la que el paciente parece estar dormido pero es fácilmente despertado, manteniendo la cooperación y la comunicación con el personal sanitario. En cualquier caso, supone una nueva vía de actuación con unos perfiles de utilidad y de seguridad propios, lo que en un ámbito sin demasiadas opciones permite ampliar las disponibles para adecuarse a las condiciones específicas de cada paciente.
|
LISDEXANFETAMINA |
|---|
|
► ELVANSE® (Shire) |
|
N06BA. SISTEMA NERVIOSO. Psicoanalépticos. Psicoestimulantes usados en el síndrome de déficit de atención con hiperactividad y nootropos: simpaticomiméticos de acción central. |
La lisdexanfetamina es un profármaco inactivo de la dextroanfetamina, autorizado como parte de un programa de tratamiento integral para el trastorno por déficit de atención – hiperactividad (TDAH) en niños a partir de 6 años, cuando la respuesta al tratamiento previo con metilfenidato se considere clínicamente inadecuada. Actúa, como el resto de las anfetaminas, inhibiendo la recaptación presináptica de noradrenalina y dopamina, aumentando la concentración de estos neurotransmisores en el espacio sináptico. Obviamente, guarda una estrecha relación estructural y farmacológica con el metilfenidato, considerando como referencia del grupo en esta indicación.
{kind=link}
La eficacia de la lisdexanfetamina ha sido contrastada tanto frente a placebo como con relación al metilfenidato (formulado como sistema OROS®, Osmotic Release Oral System), con diferencias favorables estadísticamente significativas, si bien la comparación con metilfenidato solo se hizo en tratamiento de 7 semanas. En cualquier caso, supone una aportación de cierto interés para aquellos pacientes que no responden satisfactoriamente al metilfenidato, aunque con un perfil toxicológico similar (incluso con una incidencia ligeramente mayor de anorexia e insomnio) que obliga a mantener un control de peso y altura en los niños y adolescentes. La utilización de un profármaco inactivo de dextroanfetamina se ha justificado por facilitar un efecto más gradual y persistente que la dextroanfetamina y limitar su potencial de abuso, al retardar y amortiguar el efecto euforizante ligado con una rápida elevación de los niveles sanguíneos de dextroanfetamina.
|
NALMEFENO |
|---|
|
► SELINCRO® (Lundbeck) |
|
N07BB. SISTEMA NERVIOSO. Fármacos usados en dependencia alcohólica. |
El nalmefeno es un modulador del sistema opioide endógeno, con efectos antagonistas sobre los receptores µ y d, y agonista parcial sobre los k, contrarrestando los efectos reforzantes del consumo de alcohol sobre los circuitos neurológicos de recompensa, reduciendo dicho consumo, al modular las funciones del sistema cortico-mesolímbico cerebral. Ha sido autorizado para la reducción del consumo de alcohol en pacientes adultos con dependencia del alcohol que presentan un nivel de consumo de alcohol de alto riesgo, sin síntomas de abstinencia físicos y que no requieran una desintoxicación inmediata.
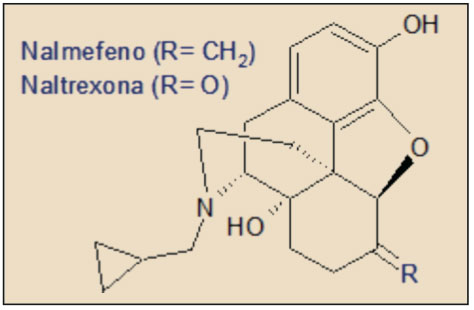{kind=link}
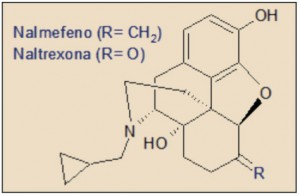
Los resultados de los estudios clínicos controlados con placebo muestran leves diferencias con el placebo, reduciendo respecto de éste entre 2,7 y 3,7 días de consumo elevado de alcohol cada mes, y entre 10 y 18 g/día de alcohol ingerido. Aunque esta diferencia es pequeña y, en algún caso, no alcanza la significación estadística, al hacer un análisis estratificado de los datos y considerando exclusivamente a los pacientes con riesgo alto o muy alto de consumo (más de 60 g/día para varones y más de 40 para mujeres), se aprecia en los pacientes tratados con nalmefeno una reducción de más del 50% en el número de días de consumo elevado de alcohol al mes (de 23 a 10-11 días/mes) y en la cantidad total de alcohol ingerida (de 102-113 a 43-44 g/día). Extrapolando estos resultados, supondría una reducción de 160 días adicionales por años sin consumo excesivo de alcohol. Esto supone que la eficacia del nalmefeno es claramente mejor entre los bebedores de alto o muy alto riesgo alcohólico.
Se echa claramente de menos la comparación con otros agentes empleados en la misma indicación y, desde luego, con la naltrexona. Asimismo, hay una alta tasa de abandono en los estudios clínico, que alcanzan el 43% de los asignados originalmente al nalmefeno y al 34% del grupo placebo, lo cual resta fiabilidad estadística a los resultados finales obtenidos. Ciertamente, no parece que el nalmefeno mejore el tratamiento de la dependencia alcohólica, dominado fundamentalmente por acamprosato y naltrexona. Sin embargo, la forma de utilización a demanda puede calificarse como novedoso, ya que mejora las limitaciones de las pautas posológicas fijas. En este sentido, el nalmefeno fue tomado el 52% de los días y la tasa de cumplimiento adecuado del tratamiento (más del 80% de los días requeridos) fue del 68%; un aspecto a considerar para aquellos pacientes que tienen dificultades en seguir un tratamiento estricto.
|
TAFAMIDIS |
|---|
|
► VYNDAQEL® (Pfizer) |
|
N07XX. SISTEMA NERVIOSO. Otros medicamentos. |
|
Medicamento Huérfano |
El tafamidis es un agente capaz de prevenir la disociación de los tetrámeros naturales o mutados de transtiretina en sus monómeros, impidiendo la formación de las fibrillas amiloides causantes de la amiloidosis transtiretina. La transtiretina (TTR) se produce principalmente en el hígado y, en ciertas cantidades, en el plexo coroideo y la retina. Desempeña un papel vital en el plasma humano y líquido cefalorraquídeo, actuando como un portador secundario de la tiroxina (T4), el único portador de la vitamina A mediante la proteína de unión a retinol en plasma y el principal transportador de la T4 en el líquido céfalo-raquídeo. La TTR es una de las aproximadamente 30 proteínas amiloidogénicas humanas relacionados con enfermedades causadas por fibrillas de amiloide. El tadamidis ha sido autorizado para el tratamiento de ésta en pacientes adultos, específicamente para aquellos afectados con polineuropatía sintomática en estadio 1 para retrasar la alteración neurológica periférica.
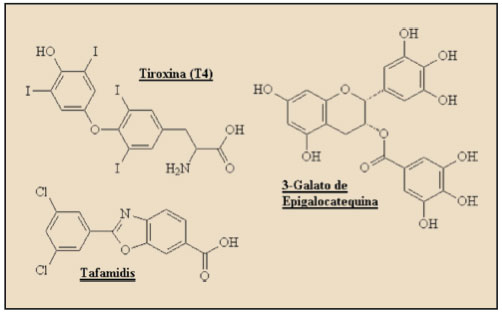{kind=link}
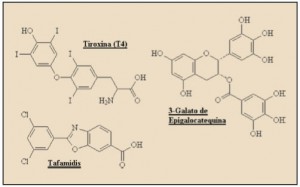
Los datos clínicos muestran tasas de mejoría o estabilización de los síntomas neurológicos en el 45% vs. 30% con placebo, aunque solo se refiere a pacientes con un estadio poco avanzado de la enfermedad (estado 1, en el que no requieren ayuda para caminar); asimismo, el tratamiento con tafamidis muestra efectos favorables sobre la calidad de vida de los pacientes y su estatus nutricional. Por otro lado, los datos apuntan a la posible utilidad en pacientes con mutaciones infrecuentes – aquellas diferentes de la Val30Met – pero no parece que se obtengan resultados significativos en los biomarcadores cardiacos en los pacientes con cardiopatía infiltrante derivada de la amiloidosis transtiretina.
Aunque los resultados clínicos son modestos y está por ver la influencia definitiva sobre la evolución global de la enfermedad, no cabe duda de que es el primer medicamento específicamente indicado en la amiloidosis transtiretina, una enfermedad rara que hasta ahora no tenía más posibilidades terapéuticas que el trasplante de hígado, aunque de hecho éste sigue siendo el estándar terapéutico. Con todo, es el primer tratamiento farmacológico etiológico de esta patología y ello no debe ser minusvalorado. Es importante resaltar la incorporación de un mecanismo de acción farmacológico nuevo, potencialmente útil en otros cuadros de amiloidosis asociados al depósito de fibrillas formadas por proteínas anómalamente plegadas. La utilización de estabilizadores estructurales de proteínas abre la puerta a múltiples posibles indicaciones, entre las que la amiloidosis transtiretina es un ejemplo característico.
r. APARATO RESPIRATORIO
|
VILANTEROL/FLUTICASONA |
|---|
|
► RELVAR ELLIPTA® (Pfizer) |
|
R03AK. APARATO RESPIRATORIO. Medicamentos contra alteraciones obstructivas pulmonares: combinación de adrenérgicos con corticosteroides. |
Se trata de un medicamente administrado en forma de inhalación, formado por una combinación a dosis fija constituida por vilanterol, un agonista de acción prolongada de los receptores β2 adrenérgicos estrechamente relacionado química y farmacológicamente con el salmeterol, y furoato de fluticasona, un glucocorticosteroide que ya estaba previamente comercializado en España. Dicha combinación ha sido autorizada para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista β2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada: pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas β2 inhalados de acción corta administrados «a demanda». También está indicado para el tratamiento sintomático de adultos con EPOC, con un FEV1 <70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora.
{kind=link}
En pacientes asmáticos, la combinación es capaz de incrementar el FEV1 193 ml más que el furoato de fluticasona sola y 210 ml más que el propionato de fluticasona, tras 24 semanas de tratamiento. Asimismo, el riesgo de experimentar exacerbaciones asmáticas graves es reducido en un 20% anual en relación a la fluticasona sola. No se encontraron diferencias estadísticamente significativas frente a la combinación de propionato de fluticasona y salmenterol. En pacientes con EPOC el riesgo relativo de exacerbación es reducido en relación al vilanterol solo con la combinación a dosis fija en un 15-31%, 21-34% y 13-19% con las dosis de 200, 100 y 50 µg de furoato de fluticasona, respectivamente, aunque no pudo establecerse una relación entre la dosis de fluticasona y la respuesta clínica. En general, los efectos observados tanto en asma como en EPOC son equiparables a los de otras combinaciones de agonistas β2 adrenérgicos de larga duración (formoterol, salmeterol) y glucocorticoides (propionato de fluticasona, budesonida, beclometasona), con una clara superioridad sobre el glucocorticoide solo.
En definitiva, se trata de la primera combinación de un agonista β2 adrenérgicos de larga duración (LABA, long-acting beta-adrenoceptor agonist) y un glucocorticoide que está disponible para su uso tanto en asma como en EPOC y que solo requiere una única administración diaria en inhalación, lo cual, presumiblemente, podría facilitar la tasa de adherencia al tratamiento y, con ello, mejorar el curso y el pronóstico en estos pacientes. En este sentido, hasta ahora, al menos en España, las únicas combinaciones disponibles comercialmente de un LABA (formoterol, salmeterol) y glucocorticoides (beclometasona, budesonida, propionato de fluticasona) solo estaban autorizadas para el tratamiento del asma (con la excepción de salmeterol/fluticasona, que también lo está para EPOC) y todos ellos requieren la administración de dos dosis diarias (cada 12 h).
s. ÓRGANOS DE LOS SENTIDOS
|
OCRIPLASMINA |
|---|
|
► JETREA® (Alcon Cusí) |
|
S01AX. ÓRGANOS DE LOS SENTIDOS. Oftalmológicos: otros. |
La ocriplasmina es un enzima proteolítico, capaz de actuar sobre laminina, fibronectina y colágeno en el vítreo ocular, produciendo una desagregación de la matriz proteica responsable de la adherencia vitreomacular y de la tracción vitreomacular. Ha sido autorizada para su administración intravítrea en el tratamiento de la tracción vitromacular, incluidos los casos en que ésta se presenta asociada a un agujero macular de diámetro menor o igual a 400 micrones.
Los datos clínicos indican que la ocriplasmina resuelve completamiente la adherencia vitreomacular al cabo de un mes de forma significativamente mejor que el placebo (26,5 vs. 10,1%), lo que se manifiesta en un mayor porcentaje de pacientes que experimentan mejoras en su agudeza visual (23,7 vs. 11,2% mejoraron al menos 2 líneas de lectura). Se trata, sin duda alguna, de mejoras clínicas modestas y clínicamente no demasiado relevantes (la mejora en la agudeza visual, incluso en la minoría de pacientes que alcanzaron el criterio secundario de eficacia, fue muy limitada). Por otro lado, aunque se constató un menor número de pacientes sometidos a vitrectomía durante los seis meses posteriores al tratamiento (17,7 vs. 26,6%), este parámetro es cuestionable por los diversos criterios empleados de la elección para vitrectomía.
Sea como fuere, resuelve la adherencia entre el humor vítreo y la retina, lo que disminuye la necesidad de vitrectomía a corto plazo, aunque está por ver si lo hace en términos absolutos. En cualquier caso, ofrece una alternativa más cómoda y segura que la vitrectomía para los pacientes con agujeros maculares pequeños y en las fases iniciales sintomáticas de adherencia vitreomacular.
v. VARIOS
|
FLOBETAPIR (18F) |
|---|
|
► AMYVID® (Lilly) |
|
V09AX. VARIOS. Radiofármacos para el diagnóstico. Sistema Nervioso Central: otros |
(18F) Florbetapir es un radiofármaco autorizado para la obtención de imágenes mediante tomografía por emisión de positrones (PET) de la densidad de placa neurítica de β-amiloide en el cerebro de pacientes adultos con deterioro cognitivo que están siendo evaluados por enfermedad de Alzheimer y otras causas de deterioro cognitivo.
{kind=link}
Se han obtenido valores de sensibilidad del 92%, es decir, la prueba es capaz de detectar al menos al 92% de los sujetos que presentan cantidades relevantes de placas de β-amiloide en la corteza cerebral, mientras que posee una sensibilidad del 100%, lo que se traduce en que es capaz de excluir a los sujetos sanos de falsos positivos, lo que proporciona un elevado grado de seguridad en el diagnóstico diferencial, todo ello basado en una elevada correlación estadística existente entre la determinación semicuantitativa mediante imagen PET con florbetapir y las pruebas inmunohistoquímicas cuantitativas.
La presencia de placas de β-amiloide no implica un diagnóstico definitivo de enfermedad de Alzheimer, ya que estas placas pueden estar presentes en pacientes con otros trastornos neurológicos o en personas de edad avanzada con la cognición normal; sin embargo, la ausencia de dichas placas sí puede descartar la posibilidad de padecer la enfermedad de Alzheimer, lo cual proporciona una notable utilidad en el diagnóstico diferencial. Con todo, está por ver que la implantación de la prueba PET con florbetapir consiga una mejora inmediata en el tratamiento de los pacientes con enfermedad de Alzheimer; tampoco se ha establecido la utilidad del fármaco en la predicción de la enfermedad en cualquier paciente con deterioro cognitivo ni en la monitorización de la respuesta de los pacientes a los tratamientos actuales.
Ciertamente, las imágenes PET con florbetapir del β-amiloide ofrecen interesantes expectativas, ya que permiten la medición directa de uno de los componentes fundamentales que contribuyen a la disminución cognitiva relacionada con la enfermedad de Alzheimer. Sin embargo, quedan aún muchas lagunas sobre su papel en la práctica clínica, que tendrán que ser llenados durante los próximos años por los estudios de utilidad clínica y el valor añadido.