|
Fármacos |
Laboratorio |
Comentarios |
|---|---|---|
|
AFN-1252 |
Affinium Pharmaceuticals |
AFN-1252 es una nueva clase de antibiótico con una potente capacidad de inhibición de la proteina transportadora enoil-acil reductasa (FabI) con capacidad para inhibir entre otros al Staphylococcus aureus y Staphylococcus epidermidis. |
|
Afuresertib (GSK-2110183) |
GSK |
Afuresertib es un inhibidor de la proteín-quinasa B (PKB) con actividad oncolítica. |
|
Andexanet alfa (PRT4445) |
Portola |
Andexanet alfa es un inhibidor reversible recombinante del factor de coagulación Xa (FXa) que se estudia como inhibidor reversible de la actividad anticoagulante del FXa. |
|
Beloranib |
Zafgen |
Beloranib es un inhibidor de la metionina aminopeptidasa 2 (MetAP 2) que se encuentra en fase II de ensayos clínicos para el tratamiento de la obesidad. |
|
Benralizumab |
Astrazeneca |
Benralizumab es un anticuerpo monoclonal antiinterleukina 5 (IL-5) indicado para el tratamiento del asma. |
|
Cadazolido |
Actelion |
Cadazolido es un antibiótico experimental del grupo de la oxazolidinona designado para el tratamiento de diarreas asociadas a Clostridium difficile. |
|
Cerdulatinib (PRT2070) |
Portola Pharmaceuticals |
Cerdulatinid es un doble inhibidor de tirosinaquinasa (Syk) y janusquinasa (JAK). Se está estudiando en pacientes con cánceres hematoloógicos geneticamente definidos y en pacientes que han fracasado en el tratamiento de estos cánceres por mutaciones adquiridas. |
|
Copanlisib BAY 80-6946) |
Bayer |
Copanlisib es un nuevo inhibidor de fosfatidilinositol-3 cinasas (PI3K), enzima implicada en el crecimiento y supervivencia de las células tumorales. |
|
Dasabuvir |
– |
Dasabuvir es inhibidor directo de la ARN-polimerasa ARN-dependiente (NS5B), indicado para el tratamiento de la infección por virus de la hepatitis C. |
|
Deleobuvir (BI-207127) |
Boehringer Ingelheim |
Deleobuvir es un inhibidor directo de la RNA-polimerasa (NS5B), que se encuentra en fase III de desarrollo clínico como agente antiviral. |
|
Eravaciclina (TP-434) |
Tetraphase Pharmaceuticals |
Eravacyclina es un agente antibacteriano indicado para el traramiento de infecciones intraabdominales e infecciones complicadas del tracto urinario. |
|
Filgotinib (GLPG-0634) |
AbbVie |
Filgotinib es un inhibidor selectivo de la tirosina protein-quinasa JAK1 indicado para el tratamiento de la artritis reumatoide y la enfermedad de Crohn. |
|
Galunisertib |
Eli Lilly |
Galunisertib es un inhibidor del receptor TGF-beta tipo-1 (ALK5; SKR4) con acitividad oncolítica. |
|
Lebrikizumab |
Roche |
Lebrikizumab es un anticuerpo monoclonal anti interleucina 13, (IL 13) indicado para el tratamiento personalizado de procesos asmáticos. |
|
Lenvatinib (E7080) |
Eisai |
Lenvatinib es un inhibidor selectivo de la tirosina-cinasa (ITC) indicado en el tratamiento de cáncer diferenciado de tiroides refractario al yodo radioactivo. |
|
Nivolumab (BMS-936558) |
Bristol-Myers Squibb |
Nivolumab es un inhibidor experimental del punto de control inmunitario PD-1 para el tratamiento de pacientes con cáncer de pulmón no microcítico (CPNM) altamente pretratados. |
|
Onartuzumab |
Roche |
Onartuzumab es un anticuerpo monoclonal Anti-HGFR (c-Met) con actividad onclolítica, debido a que el proto-oncogen c-Met tiene un papel relevante en el desarrollo y progresión del cáncer. |
|
Otlertuzumab |
Emergent |
Otlertuzumab es un anticuerpo monoclonal Anti-CD37 capaz de inducir apoptosis, que se está estudiando para el tratamiento de cánceres de células B. |
|
Palbociclib PD0332991) |
Pfizer |
Palbociclib es un inhibidor selectivo de las cinasas ciclin-dependientes (CDK) 4 y 6, implicadas en el crecimiento celular tumoral. |
|
Patritumab (U3-1287) |
Daiichi Sankyo |
Patritumab es un anticuerpo monoclonal anti HER3 con actividad oncolítica |
|
Plazomicina (ACHN-490) |
Achoagen |
Plazomicina es un nuevo antibiótico aminoglucosídico, derivado semisintético de la sisomicina, indicado para el tratamiento de las infecciones por gram-negativos |
|
Ponesimod |
Actelion |
Ponesimod es una molecula agonista selectiva del receptor S1P con actividad sobre los procesos inflamatorios mediados por células T. |
|
Romosozumab |
Amgen |
Romosozumab es un anticuerpo monoclonal humanizado antiesclerostina, glucoproteina de los osteocitos, indicado para el tratamiento de la osteoporosis. |
|
Sarilumab (SAR15319) |
Sanofi |
Sarilumab es un anticuerpo monoclonal totalmente humano dirigido contra el receptor de IL-6 (IL-6R) que interrumpe la señalización inflamatoria resultante mediada por citoquinas. |
|
Tralokinumab |
AstraZeneca |
Tralokinumab es un anticuerpo monoclonal anti-interleukina 13, indicado para el tratamiento del asma y de la fibrosis pulmonar idiopática. |
|
Ulodesina (BCX4208) |
BioCryst |
Ulodesina es un inhibidor de Purina nucleósido fosdforilasa (PNP) indicado para el tratamiento de la gota. |
Archive
Revista PAM: 377
Número 377, Octubre 2014
Vilanterol/Fluticasona RELVAR ELLIPTA® (GlaxoSmithKline)
ASMA Y EPOC
La EPOC (enfermedad pulmonar obstructiva crónica) es una patología caracterizada por una disminución progresiva y fundamentalmente no reversible del flujo aéreo. La limitación crónica al flujo aéreo se asocia con una respuesta inflamatoria anormal del pulmón relacionada, sobre todo, con el humo del tabaco como causa fundamental. Por su parte, el asma es una enfermedad inflamatoria crónica de las vías respiratorias, en cuya patogenia intervienen diversas células y mediadores de la inflamación, condicionada en parte por factores genéticos y que cursa con hiperrespuesta bronquial y una obstrucción variable al flujo aéreo, total o parcialmente reversible, ya sea por la acción medicamentosa o espontáneamente
Relvar Ellipta® está formado por una combinación a dosis fija constituida por vilanterol, un agonista de acción prolongada de los receptores β2 adrenérgicos estrechamente relacionado química y farmacológicamente con el salmeterol, y furoato de fluticasona, un glucocorticosteroide que ya estaba previamente comercializado en España. Dicha combinación ha sido autorizada para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista β2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada: pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas β2 inhalados de acción corta administrados «a demanda». También está indicado para el tratamiento sintomático de adultos con EPOC, con un FEV1 <70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora.
En general, los efectos observados tanto en asma como en EPOC son equiparables a los de otras combinaciones de agonistas β2 adrenérgicos de larga duración (formoterol, salmeterol) y glucocorticoides (propionato de fluticasona, budesonida, beclometasona), con una clara superioridad sobre el glucocorticoide solo. El perfil de seguridad de la combinación está en línea con el de otras combinaciones conocidas (nasofaringitis y cefalea, principalmente) y no se han observado eventos adversos específicos; en cualquier caso, la incidencia no es elevada y no es un motivo frecuente de suspensión del tratamiento. No obstante, se han registrado algunos casos mortales aislados de neumonía.
En definitiva, se trata de la primera combinación de un agonista β2 adrenérgicos de larga duración (LABA, long-acting beta-adrenoceptor agonist) y un glucocorticoide que está disponible para su uso tanto en asma como en EPOC y que solo requiere una única administración diaria en inhalación, lo cual, presumiblemente, podría facilitar la tasa de adherencia al tratamiento y, con ello, mejorar el curso y el pronóstico en estos pacientes. En este sentido, hasta ahora, al menos en España, las únicas combinaciones disponibles comercialmente de un LABA (formoterol, salmeterol) y glucocorticoides (beclometasona, budesonida, propionato de fluticasona) solo estaban autorizadas para el tratamiento del asma (con la excepción de salmeterol/fluticasona, que también lo está para EPOC) y todos ellos requieren la administración de dos dosis diarias (cada 12 h).
La EPOC es una patología caracterizada por una disminución progresiva y fundamentalmente no reversible del flujo aéreo. La limitación crónica al flujo aéreo se asocia con una respuesta inflamatoria anormal del pulmón relacionada, sobre todo, con el humo del tabaco como causa fundamental. Esta enfermedad respiratoria crónica engloba patologías como el enfisema1, la bronquitis crónica2 y la enfermedad de vías aéreas pequeñas.
Los parámetros funcionales respiratorios más habitualmente utilizados en la descripción clínica de la EPOC son:
- Capacidad Vital (CV): volumen máximo que el sujeto es capaz de inspirar y espirar en condiciones no forzadas, y corresponde a la suma VC + VRI +VRE:
- Volumen corriente (VC): cantidad de aire empleado en cada respiración (inspiración y espiración) normal (no forzada), lo que da idea del volumen de aire circulado en cada ciclo respiratorio. Habitualmente, el valor expresado representa el aire espirado, que no corresponde exactamente al inspirado, por ser un valor generalmente más estable que el inspirado. En término medio es de 0,5 litros.
- Volumen de Reserva Inspiratoria (VRI): máximo volumen de aire que puede ser inspirado. Se sitúa habitualmente en torno a 3 litros (con las evidentes diferencias entre grupos de edad, desarrollo, etc.).
- Volumen de Reserva Espiratoria (VRE): volumen máximo de aire que es espirado en condiciones normal (sin forzar la respiración). En término medio, se sitúa sobre 1,7 l.
- Capacidad Vital Forzada (CVF): es el equivalente a la capacidad vital (CV), pero en condiciones forzadas y con la máxima rapidez que el sujeto pueda.
- Volumen Espiratorio Forzado en 1 segundo (VEF1): volumen de aire exhalado durante el primer segundo de una espiración forzada (máxima). Se trata de un parámetro fundamental para el diagnóstico y control de la evolución clínica de la EPOC. Una variante de este parámetro es el VEF1%, la fracción correspondiente del VEF1 entre la Capacidad Vital (CV), expresándolo en términos porcentuales (VEF1% = [VEF1/CV] x 100). Actualmente, se acepta también como valor porcentual el correspondiente al cociente VEF1/CVF.
El pronóstico de los pacientes con EPOC depende del grado de obstrucción al flujo aéreo y del abandono del consumo de tabaco. La tasa de mortalidad a los 10 años de establecido el diagnóstico es superior al 50%. La evolución o historia natural de la EPOC sigue un curso lento, pero inexorable, en el que pueden sobrevenir episodios de exacerbación, con desarrollo de insuficiencia respiratoria e hipercapnia potencialmente graves, incluso mortales, que constituyen, por tanto, una urgencia médica en potencia.
La prevalencia de la EPOC en España es similar en líneas generales a la encontrada en otros países europeos y en los Estados Unidos. Concretamente, en España es del 10% de la población entre 40 y 80 años; es decir, la EPOC afectaría a más de 2 millones de personas, de los cuales un 15 % son varones y un 6% mujeres. En España, la EPOC representa la cuarta causa de mortalidad, con una tasa global de 33/100.000 habitantes, que aumenta a 176/100.000 habitantes en población de más de 75 años, e incluso algunos estudios señalan que pasará a ser la tercera causa de mortalidad para el 2020. Se trata, por tanto, de un problema clínico relevante, con importantes repercusiones socioeconómicas, lo que lo convierte en un importante problema de salud pública, que merece todo el interés de profesionales y autoridades sanitarias.
La mayoría de los casos de EPOC están asociados al consumo de cigarrillos, si bien sólo el 15-20% de los fumadores desarrollan esta patología debido a diferencias en susceptibilidad de naturaleza aún desconocida, aunque se piensa que tienen una base genética o quizás ambiental. Desde el punto de vista funcional, la EPOC se define por un aumento de la resistencia al flujo aéreo al que contribuyen:
- a) La disminución de la luz bronquial por el engrosamiento de la pared y la hipersecreción de mucinas.
- b) La contracción de la musculatura lisa de las vías aéreas.
- c) La pérdida de elasticidad del parénquima pulmonar.
En contraposición al asma bronquial, que es una inflamación eosinofílica crónica con obstrucción reversible de vías aéreas, la EPOC presenta un perfil de inflamación neutrofílica crónica con obstrucción no reversible. Los estudios histopatológicos muestran una implicación predominante de las vías respiratorias (bronquiolos) y del parénquima pulmonar, mientras que el asma implica inflamación de todas las vías respiratorias, aunque normalmente sin implicación del parénquima pulmonar. Existe una obstrucción de los bronquiolos, con fibrosis e infiltración con macrófagos y linfocitos T. Se produce la destrucción del parénquima pulmonar y un aumento de macrófagos y linfocitos T, con un mayor aumento de células CD8+ (citotóxicas) en comparación con las células CD4+ (coadyuvantes).
Cualquier opción terapéutica en la EPOC debe pasar por el abandono radical e inmediato del hábito tabáquico en el paciente, habida cuenta su decisivo papel en el origen y mantenimiento de la enfermedad. Junto con el abandono definitivo del tabaquismo, la oxigenoterapia continua domiciliaria cuando la situación lo requiera, son los dos elementos básicos para frenar la progresión de la enfermedad. El alivio sintomático y la mejoría de la calidad de vida se obtienen mediante el empleo de agentes broncodilatadores, la rehabilitación respiratoria y el soporte ventilatorio domiciliario.
La prevención de las exacerbaciones o reagudizaciones también es el objetivo de la utilización de broncodilatadores, así como los de la vacunación antigripal y del uso de corticosteroides en inhalación. Cuando las terapias farmacológicas y rehabilitadoras no son suficientes, se recurre a la cirugía de reducción pulmonar o, incluso, al trasplante. Cuando hay disnea o intolerancia al ejercicio físico, se recomienda el uso de agentes broncodilatadores de acción corta, en inhalación. La elección de agonistas beta2-adrenérgicos (salbutamol, terbutalina, etc.) o de anticolinérgicos (ipratropio) depende de la respuesta y de la incidencia de efectos adversos en cada paciente. En caso de exacerbaciones o disnea persistente con la terapia anterior, las opciones dependen del grado de limitación del flujo respiratorio:
- FEV1 ≥ 50%: Las alternativas son
- Agonista beta2-adrenérgico de larga duración (indacaterol, formoterol, salmeterol, etc.), manteniendo el uso de los de acción corta, a demanda. En caso de falta de respuesta adecuada, puede añadirse un corticosteroide en inhalación, o un anticolinérgico de acción prolongada en inhalación en caso de baja respuesta o intolerancia al corticosteroide.
- Anticolinérgico de acción prolongada (tiotropio, etc.), descontinuando el uso de anticolinérgicos de acción corta. En caso de respuesta inadecuada, puede combinarse con un agonista beta2-adrenérgico de larga duración y un corticosteroide, todos ellos en inhalación.
- FEV1 ≤ 50%:
- Agonista beta2-adrenérgico de larga duración (indacaterol, formoterol, salmeterol, vilanterol, etc.) más un corticosteroide en inhalación, o un anticolinérgico de acción prolongada en inhalación en caso de baja respuesta o intolerancia al corticosteroide.
- Anticolinérgico de acción prolongada (tiotropio, etc.), descontinuando el uso de anticolinérgicos de acción corta. En caso de respuesta inadecuada, puede combinarse con un agonista beta2-adrenérgico de larga duración y un corticosteroide, todos ellos en inhalación.
- Solo cuando la terapia inhalada sea insuficiente para mantener la calidad de vida de los pacientes, se debe recurrir a la terapia sistémica (oral):
- Corticosteroides: no se recomienda su uso continuado en EPOC, salvo en aquellos casos en que no puede suspenderse el tratamiento, tras una exacerbación.
- Teofilina: Puede usarse en combinación con agonistas beta2-adrenérgicos y anticolinérgicos, teniendo siempre presente el amplio espectro de interacciones farmacológicas de este agente.
- Mucolíticos: Se considera aceptable su uso en pacientes con tos crónica productiva, siempre que produzcan una mejora sintomática apreciable. No se recomienda su uso para la prevención de las exacerbaciones.
- Inhibidores selectivos de la fosfodiesterasa 4 (PDE4): Roflumilast es un fármaco que es capaz de reducir la incidencia de exacerbaciones graves o muy graves en pacientes con EPOC e historial de exacerbaciones frecuentes.
ASMA
El asma es un síndrome que comparten manifestaciones clínicas similares en apariencia pero de causas diferenciadas. Ello supone que sea complejo el alcanzar una definición precisa y exacta de la enfermedad, existiendo varias definiciones para la misma. Se define como una enfermedad inflamatoria crónica de las vías respiratorias, en cuya patogenia intervienen diversas células y mediadores de la inflamación, condicionada en parte por factores genéticos y que cursa con hiperrespuesta bronquial y una obstrucción variable al flujo aéreo, total o parcialmente reversible, ya sea por la acción medicamentosa o espontáneamente (Cortijo, 2014a).
El asma en adultos se clasifica, por el grado de gravedad, en cuatro categorías: intermitente, persistente leve, persistente moderada y persistente grave. En los niños se definen dos patrones principales: asma episódica y asma persistente. El asma episódica puede ser ocasional o frecuente, dependiendo del número de crisis que presente. El asma persistente en el niño no puede considerarse como leve, sino que al menos es moderada o grave. Existe un fenotipo mixto EPOC-asma, caracterizado por la presencia de una obstrucción no completamente reversible al flujo aéreo acompañada de síntomas o signos de una reversibilidad aumentada de la obstrucción.
El asma es un problema de salud de elevada prevalencia, con importantes implicaciones en la esperanza y calidad de vida de las personas que la padecen y que genera un importante consumo de recursos sanitarios y unas fuertes pérdidas sociales. Según la Organización Mundial de la Salud (OMS), el asma es la séptima enfermedad más prevalente en el mundo, con cerca de 235 millones de afectados. La cifra de afectados en Europa es de alrededor de 29 millones. Además, supone la quinta causa de muerte en los países desarrollados. En el territorio español, la prevalencia de síntomas asmáticos en niños se ha mantenido constante en los niños de 13-14 años, mientras que ha sufrido un aumento significativo en el grupo de 6-7 años. En los adultos la prevalencia es inferior a la de los países anglosajones y centroeuropeos. El Estudio Europeo de Salud Respiratoria constató en nuestro país unas tasas del 4,7% en Albacete, 3,5% en Barcelona, 1,7% en Oviedo, 1,1% en Galdakao (Vizcaya) y 1% en Huelva. Un 52% de las personas con asma no habían sido diagnosticadas y hasta un 26% de éstas, a pesar de padecer síntomas frecuentes, no seguía ningún tratamiento.
El objetivo principal del tratamiento del asma es lograr y mantener el control de la enfermedad lo antes posible, además de prevenir las exacerbaciones y la obstrucción crónica al flujo aéreo y reducir su mortalidad.
Las exacerbaciones (ataques o crisis) de asma son episodios agudos o subagudos caracterizados por un aumento progresivo de uno o más de los síntomas típicos (disnea, tos, sibilancias y opresión torácica) acompañados de una disminución del flujo espiratorio (FEV1). Según la rapidez de instauración de las crisis, existen dos tipos: las de instauración lenta (normalmente en días o semanas) y las de instauración rápida (en menos de 3 horas), que deben identificarse por tener causas, patogenia y pronóstico diferentes. Las de instauración lenta (más del 80% de las que acuden a urgencias) se deben frecuentemente a infecciones respiratorias altas o a un mal control de la enfermedad.
Las exacerbaciones (ataques o crisis) de asma son episodios agudos o subagudos caracterizados por un aumento progresivo de uno o más de los síntomas típicos (disnea, tos, sibilancias y opresión torácica) acompañados de una disminución del flujo espiratorio (FEV1). Según la rapidez de instauración de las crisis, existen dos tipos: las de instauración lenta (normalmente en días o semanas) y las de instauración rápida (en menos de 3 horas), que deben identificarse por tener causas, patogenia y pronóstico diferentes. Las de instauración lenta (más del 80% de las que acuden a urgencias) se deben frecuentemente a infecciones respiratorias altas o a un mal control de la enfermedad por mala adhesión terapéutica; el mecanismo fundamental del deterioro es la inflamación y la respuesta al tratamiento es también lenta; mientras que las de instauración rápida se deben a alérgenos inhalados, ingestión de fármacos (AINE o fármacos betabloqueantes), alimentos (por los aditivos y conservantes) o estrés emocional; el mecanismo es el broncoespasmo y, aunque tienen una mayor gravedad inicial (con riesgo de intubación y muerte), la respuesta al tratamiento es mejor y más rápida. La intensidad de las exacerbaciones es variable, cursando en ocasiones con síntomas leves e indetectables por el paciente y en otras con episodios muy graves que ponen en peligro su vida.
El objetivo inmediato del tratamiento de una crisis es preservar la vida del paciente revirtiendo la obstrucción al flujo aéreo y la hipoxemia si está presente, de la forma más rápida posible, y posteriormente instaurar o revisar el plan terapéutico para prevenir nuevas crisis.
La pauta de tratamiento en la exacerbación leve debe incluir la administración de broncodilatadores agonistas β2-adrenérgicos de acción rápida (salbutamol o terbutalina), glucocorticoides orales y oxígeno (si es necesario). Los agonistas β2-adrenérgicos de acción corta inhalados son los fármacos broncodilatadores más eficaces y rápidos en el tratamiento de la exacerbación asmática. Si en las primeras 2 horas del tratamiento se constata una evolución favorable (desaparición de síntomas, FEV1 superior al 80% del teórico o del mejor valor personal del paciente) y ésta se mantiene durante 3-4 horas, no son necesarios más tratamientos. Sin embargo, en pacientes no controlados adecuadamente con combinaciones inhaladas de corticosteroides y agonistas β2 de acción corta administradas «a demanda», puede recurrirse a combinaciones similares en las que el agonista β2 sea de acción prolongada (salmeterol, formoterol, vilanterol, etc.).
El uso de glucocorticoides sistémicos acelera la resolución de las exacerbaciones. Excepto en crisis muy leves, deben administrarse siempre, especialmente si: a) no se consigue una reversión de la obstrucción de las vías respiratorias con agonistas β2-adrenérgicos de acción rápida inhalados; b) el paciente estaba tomando ya glucocorticoides orales; c) el paciente ha tratado ya su pérdida de control previa con otras opciones terapéuticas sin éxito; d) existen antecedentes de exacerbaciones previas que requirieron glucocorticoides orales.
ACCIÓN Y MECANISMO
Se trata de un medicamente administrado en forma de inhalación, formado por una combinación a dosis fija de un agonista de acción prolongada de los receptores β2 adrenérgicos (vilanterol) y de un glucocorticosteroide (fluticasona). Ha sido autorizado para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista β2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada: pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas β2 inhalados de acción corta administrados «a demanda». También está indicado para el tratamiento sintomático de adultos con EPOC, con un FEV1 <70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora.
Existen tres tipos de receptores adrenérgicos β: β1 (predominantes en el corazón), β2 (músculo liso) y β3 (relacionados con el metabolismo lipídico intracelular). La activación de todos los subtipos de receptores β desencadena un mecanismo de acción similar, mediante el cual la adenilciclasa ligada al receptor se activa y cataliza la conversión del trifosfato de adenosina (ATP) en adenosín monofosfato cíclico (AMPc), provocando, en el caso de los receptores β2 de las células musculares bronquiales, la relajación de éstas y, en consecuencia, una broncodilatación. Estudios de autorradiografía en pulmón humano han demostrado la presencia exclusiva de receptores β2 en el músculo liso de las vías aéreas, sobre todo en las vías periféricas, aunque existen también adrenoceptores β2 en el epitelio, la pared alveolar, las glándulas submucosas, el músculo liso vascular pulmonar, células con capacidad de liberar mediadores (mastocitos, basófilos, eosinófilos), y en los ganglios y las terminaciones nerviosas posganglionares colinérgicas.
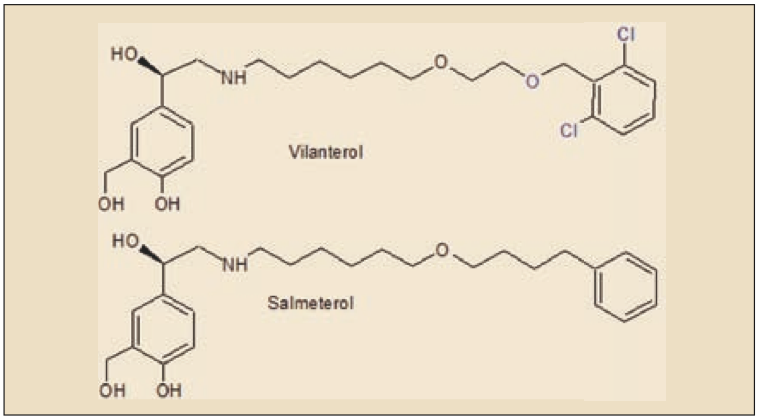
El vilanterol presenta una elevada afinidad hacia los receptores β2 adrenérgicos, similar a la del salmeterol y superior a la del formoterol e indacaterol; igualmente, la selectividad relativa para receptores β2 adrenérgicos (en relación a los β1 y β3) es equiparable a la del salmeterol y mayor que la del formoterol e indacaterol.
Por su parte, la fluticasona es un glucocorticosteroide que, como tal, tiene acciones antiinflamatorias e inmunomoduladoras. Tras unirse a los receptores intracelulares específicos, los glucocorticosteroides dan lugar a un factor de transcripción capaz de modular la expresión génica que induce, entre otras acciones, la síntesis de lipocortina-1, polipéptido que inhibe la fosfolipasa A2 (enzima clave en la producción de mediadores de la inflamación como prostaglandinas, leucotrienos, factor activador plaquetario) y disminuye la formación de citocinas tales como las interleucinas IL-5 e IL-3, y el factor de necrosis tumoral (TNF).
{kind=link}
ASPECTOS MOLECULARES
El vilanterol es un agonista β2 adrenérgico de acción prolongada, estrechamente relacionado estructural y farmacológicamente con el salbutamol y, especialmente, con el salmeterol que, como el vilanterol, es de acción prolongada. Químicamente, corresponde al 4-[(1R)-2-[6-[2-[(2,6-diclorofenil)metoxi]etoxi]hexilamino]-1-hidroxietil]-2-(hidroximetil)fenol.
Por su parte, la fluticasona es un glucocorticosteroide bien conocido, ya utilizado en medicamentos previamente autorizados, tanto solo como en combinación con broncodilatadores adrenérgicos. Se trata de un derivado halogenado (contiene es su estructura dos átomos de flúor); está estrechamente relacionado química y farmacológicamente con otros corticosteroides utilizados en asma y EPOC, tanto halogenados (beclometasona, mometasona), como no halogenados (ciclesónido, budesonida).
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de la combinación a dosis fijas de vilanterol y furoato de fluticasona han sido adecuadamente contrastadas tanto en asma como en EPOC mediante varios estudios multicéntricos, aleatorizados, doblemente ciegos y controlados con placebo, en los que se utilizó la formulación de furoato de fluticasona (FF; 50, 100 o 200 µg por pulsación, equivalentes a 46, 92 y 184 µg de fluticasona base) y trifenatato de vilanterol (VI; 25 µg por pulsación, equivalentes a 22 µg de vilanterol base) y el mismo dispositivo dosificador en inhalación que ha sido posteriormente comercializado.
Eficacia clínica en asma
Entre los diversos ensayos clínicos disponibles de fase 3 (confirmatorios de eficacia y de seguridad), los considerados como principales o pivotales son tres. Dichos ensayos clínicos fueron llevados a cabo sobre pacientes adolescentes (≥12 años) o adultos con asma persistente de moderada a grave, que estaban bajo tratamiento con corticosteroides inhalados, eventualmente asociados a un broncodilatador β2 adrenérgico de larga duración.
El estudio HZA106827 (Bleecker, 2014) comparó durante 12 semanas la combinación FF/VI 100/25 µg vs. el corticoide solo (FF 100 µg) y placebo (P), en un conjunto de 609 pacientes con valores de Volumen Espiratorio Forzado en 1 segundo (VEF1) entre el 40% y el 90% del predicho (mediana de 68%), un VEF1 valle (predosis) medio de 2.230 ml y de 2.850 postdosis broncodilatadora. Demográficamente, un 58% eran mujeres, la mediana de edad era de 39,7 años, (13% menores de 18 años), 94% blancos y una mediana de peso de 75,6 kg. Se utilizaron dos covariables primarias de eficacia consistentes en la variación entre los valores basales y los registrados al final del estudio del VEF1 valle (predosis) y la correspondiente a la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h tras la dosis).
Los resultados mostraron un aumento en el VEF1 valle (predosis) de 368 ml (FF/VI), de 332 ml (FF) y 196 ml (P), siendo la diferencia entre FF/VI y FF de 36 ml (IC95% -45 a 120; p= 0,405) y la de FF/VI vs. placebo (P) de 172 ml (IC95% 87 a 258; p< 0,001). Por lo que respecta a la variación de la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h tras la dosis), fue de 513 ml (FF/VI), de 398 ml (FF) y 212 ml (P), siendo la diferencia entre FF/VI y FF de 116 ml (IC95% -5 a 236; p= 0,06) y la de FF/VI vs. placebo (P) de 302 ml (IC95% 178 a 426; p< 0,001). Asimismo, la diferencia del porcentaje de días completos sin necesidad de utilizar medicación broncodilatadora de rescate, en relación con el placebo, fue del 19,3% (IC95% 13,0 a 26,6 p< 0,001) para FF/VI y del 10,6% (IC95% 4,3 a 16,8 p< 0,001) para FF; además, la diferencia del porcentaje de días completos sin síntomas, en relación con el placebo, fue del 18,0% (IC95% 12,0 a 23,9 p< 0,001) para FF/VI y del 12,1% (IC95% 6,2 a 18,1 p< 0,001) para FF.
El estudio HZA106829 (O’Byrne, 2014) comparó durante 24 semanas la combinación FF/VI 200/25 µg vs. furoato de fluticasona 200 µg cada 24 h (FF) y propionato de fluticasona 500 µg cada 12 h (PF), en un conjunto de 586 pacientes con VEF1 entre el 40% y el 90% del predicho. Un 59% eran mujeres, la mediana de edad era de 46,2 años, (4% menores de 18 años), 84% blancos y una mediana de peso de 79,9 kg. Los resultados mostraron un aumento en el VEF1 valle (predosis) de 388 ml (FF/VI), de 218 ml (FF) y 173 ml (FP), siendo la diferencia entre FF/VI y FF de 193 ml (IC95% 108 a 277; p< 0,001) y la de FF/VI vs. FP de 210 ml (IC95% 127 a 294; p< 0,001). Por lo que respecta a la variación de la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h tras la dosis), fue de 394 ml (FF/VI), de 201 ml (FF) y 183 ml (FP).
La diferencia del porcentaje de días completos sin necesidad de utilizar medicación broncodilatadora de rescate, en relación con FF solo, fue del 11,7% (IC95% 4,9 a 18,4 p< 0,001) y del 6,3% (IC95% 6,2 a 18,1 p< 0,001) frente a FP. Además, la diferencia del porcentaje de días completos sin síntomas fue del 8,4% (IC95% 2,0 a 14,8 p= 0,01) frente a FF y del 4,9% (IC95% -1,6 a 11,3 p= 0,137) frente a FP.
El estudio HZA106837 (Bateman, 2014) comparó durante 24 a 78 semanas la combinación FF/VI 100/25 µg vs. el corticoide solo (FF 100 µg), en un conjunto de 2.019 pacientes con VEF1 entre el 50% y el 90% del predicho. Demográficamente, un 67% eran mujeres, la mediana de edad era de 41,7 años, 73% blancos y una mediana de peso de 74,2 kg. La duración media del cuadro asmático era de 15,5 años y un 57% había experimentado al menos un episodio de exacerbación grave del asma durante el último año (un 43% había tenido al menos dos). Como variable primaria de eficacia se determinó el riesgo estadístico de experimentar una exacerbación asmática grave (que requiriese la administración de corticosteroides sistémicos durante al menos tres días y/o la hospitalización del paciente). La tasa anual de exacerbaciones graves fue del 12,8% (IC95% 10,7 a 14,9) con FF/VI vs. 15,9% (IC95% 13,5 a 18,2), lo que implica una reducción del 20% en la tasa de riesgo (HR= 0,795; IC95% 0,642 a 0,985; p= 0,036)
Adicionalmente, un estudio (Woodcock, 2013) comparó durante 24 semanas la combinación FF/VI 100/25 µg vs. propionato de fluticasona y salmeterol 250/50 µg cada 12 h (PF/SA), en un conjunto de 806 pacientes asmáticos. Los resultados mostraron un aumento en el VEF1 valle (predosis) de 341 ml (FF/VI) vs. 377 ml (FP/SA), siendo la diferencia entre ellos de -37 ml (IC95% -88 a 15; p= 0,162). Por lo que respecta a la variación de la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h), fue de 281 ml (FF/VI) vs. 300 ml (FP/SA), con una diferencia de -19 ml (IC95% -73 a 34; p= 0,485).
Eficacia clínica en EPOC
Hay cuatro ensayos clínicos pivotales, dos de ellos de 6 meses de duración y otros dos de un año. En todos los casos fueron multicéntricos, aleatorizados, controlados y con grupos paralelos.
Los estudios HZC102870 y HZC102871 (Dransfield, 2013) son dos estudios clínicos replicados de un año de duración, realizados sobre 3.245 (1.622 y 1.623) pacientes con EPOC, mayores de 40 años, con un historial de tabaquismo de al menos diez años, cociente VEF1/CVF ≤70% del predicho y con al menos una exacerbación de la enfermedad en el último año. Los pacientes recibieron por vía inahalatoria una dosis diaria de vilanterol (25 µg) solo o asociado a 50, 100 o 200 µg furoato de fluticasona.
Demográficamente, en el estudio HZC102870, un 45% eran mujeres, la mediana de edad era de 63,7 años, 88% blancos y un índice de masa corporal medio de 27,1; los pacientes tenían una mediana VEF1 del 45,7%, un VEF1 valle (predosis) medio de 1.150 ml y de 1.289 postdosis broncodilatadora. Por su parte en el estudio HZC102870, un 41% eran mujeres, la mediana de edad era de 63,6 años, 82% blancos y un índice de masa corporal medio de 26,7; los pacientes tenían una mediana VEF1 del 45,2%, un VEF1 valle (predosis) medio de 1.143 ml y de 1.281 postdosis broncodilatadora
Los resultados del HZC102872 mostraron una tasa anual de exacerbaciones de 0,79 (FF200/VI), 0,90 (FF100/VI), 0,92 (FF50/VI) y 1,14 (VI), con lo que el riesgo relativo (RR) de exacerbación fue reducida en relación al vilanterol solo con la combinación a dosis fija en un 31% (RR= 0,64; IC95% 0,56 a 0,85; p< 0,001) con FF200/VI, en un 21% (RR= 0,79; IC95% 0,64 a 0,97; p= 0,024) con FF100/VI y en un 19% (RR= 0,81; IC95% 0,66 a 0,99; p= 0,040) con FF50/VI. Por su parte, los resultados del HZC102871 mostraron una tasa anual de exacerbaciones de 0,90 (FF200/VI), 0,70 (FF100/VI), 0,92 (FF50/VI) y 1,05 (VI), con lo que el riesgo relativo (RR) de exacerbación fue reducida en relación al vilanterol solo con la combinación a dosis fija en un 15% (RR= 0,85; IC95% 0,70 a 1,04; p= 0,109) con FF200/VI, en un 34% (RR= 0,66; IC95% 0,54 a 0,81; p< 0,001) con FF100/VI y en un 13% (RR= 0,87; IC95% 0,72 a 1,06; p= 0,181) con FF50/VI.
El estudio HZC112206 (Kerwin, 2013) es un estudio multicéntrico, aleatorizado, doblemente ciego, controlado con placebo y de grupos paralelos, realizado sobre 1.030 pacientes con EPOC moderado a grave, que recibieron durante 24 semanas una dosis diaria una combinación de 100 µg de furoato de fluticasona y 25 µg de vilanterol (FF100/VI), 50 µg de furoato de fluticasona y 25 µg de vilanterol (FF50/VI), 100 µg de furoato de fluticasona (FF), 25 µg de vilanterol (VI) o placebo (P).
Los resultados mostraron un aumento de la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h tras la dosis) de 200 ml (FF100/VI), de 218 ml (FF50/VI), 129 ml (FF), 80 ml (VI) y 26 ml (P); siendo estadísticamente significativas las diferencias entre FF100/VI, FF, VI y P. Por lo que respecta a la variación en el VEF1 valle (predosis), fueron de 151 ml (FF100/VI), de 166 ml (FF50/VI), 103 ml (FF), 70 ml (VI) y 37 ml (P); siendo estadísticamente significativas las diferencias entre FF100/VI y FF, VI y P, así como las registradas entre FF50/VI y VI, y P. Asimismo, las variaciones registradas en la puntuación del Chronic Respiratory Questionnaire Self-Administered Standarized (CRQ-SAS) de disnea fueron de 0,53 (FF100/VI), de 0,42 (FF50/VI), 0,37 (FF), 0,29 (VI) y 0,23 (P), siendo estadísticamente significativas las diferencias entre FF100/VI vs. FF y P; ninguna diferencia fue significativa entre FF50/VI y el resto de grupos.
Por último, el estudio HZC112207 (Martínez, 2013) es un estudio de características similares al anterior, realizado sobre 1.224 pacientes con EPOC moderado a grave, que recibieron durante 24 semanas una dosis diaria una combinación de 200 µg de furoato de fluticasona y 25 µg de vilanterol (FF200/VI), 100 µg de furoato de fluticasona y 25 µg de vilanterol (FF100/VI), 25 µg de vilanterol (VI) 200 µg de furoato de fluticasona (FF200), 100 µg de furoato de fluticasona (FF100) o placebo (P).
Los resultados mostraron un aumento de la media ponderada de los valores del VEF1 postdosis (entre 0 y 24 h tras la dosis) de 197 ml (FF200/VI), de 202 ml (FF1000/VI), 173 ml (VI), 29 ml (FF200), 34 (FF100) y -12 ml (P); siendo estadísticamente significativas las diferencias entre FF200/VI vs. FF200 y P, así como entre FF100/VI vs. FF100 y P. 197 ml (FF200/VI), de 202 ml (FF1000/VI), 173 ml (VI), 29 ml (FF200), 34 (FF100) y -12 ml (P); siendo estadísticamente significativas las diferencias entre FF200/VI vs. FF200 y P, así como entre FF100/VI vs. FF100 y P. Por lo que respecta a la variación en el VEF1 valle (predosis), fueron de 135 ml (FF200/VI), de 148 ml (FF1000/VI), 103 ml (VI), 12 ml (FF200), 48 (FF100) y 4 ml (P); siendo estadísticamente significativas las diferencias entre FF200/VI vs. FF200 y P, así como entre FF100/VI vs. FF100 y P. Además, las variaciones registradas en la puntuación del CRQ-SAS de disnea fueron de 0,31 (FF200/VI), 0,45 (FF100/VI), 0,28 (VI), 0,21 (FF200), 0,10 (FF100) y 0,21 (P), siendo estadísticamente significativas las diferencias entre FF100/VI vs. FF200, FF100 y P; ninguna diferencias fue significativa entre FF50/VI y el resto de grupos.
Seguridad
El perfil toxicológico de vilanterol/fluticasona es equiparable al de otras combinaciones a dosis fijas para inhalación de una agonista β2 adrenérgico de larga duración y un glucocorticosteroide. Los eventos adversos más frecuentemente descritos en los ensayos clínicos controlados y con una incidencia superior a la del placebo son nasofaringitis (12-16 vs. 6% con placebo), cefalea (7-8 vs. 5%) infecciones del tracto respiratorio inferior (8-10 vs. 1-2%) y candidiasis oral (7-8 vs. 0-1%). Los tres primeros presentan una incidencia muy parecida a la del vilanterol y a la de fluticasona por separado. Entre los eventos adversos más graves, cabe citar varios casos mortales de neumonía, así como un incremento de la incidencia de fracturas óseas, en ambos casos más comunes en los pacientes con EPOC que en aquellos con asma.
ASPECTOS INNOVADORES
Se trata de un medicamente administrado en forma de inhalación, formado por una combinación a dosis fija constituida por vilanterol, un agonista de acción prolongada de los receptores β2 adrenérgicos estrechamente relacionado química y farmacológicamente con el salmeterol, y furoato de fluticasona, un glucocorticosteroide que ya estaba previamente comercializado en España. Dicha combinación ha sido autorizada para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista β2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada: pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas β2 inhalados de acción corta administrados «a demanda». También está indicado para el tratamiento sintomático de adultos con EPOC, con un FEV1 <70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora.
La combinación ha sido extensamente estudiada mediante ensayos clínicos aleatorizados y controlados con placebo y/o comparadores activos. En pacientes asmáticos, incrementó el FEV1 193 ml más que el furoato de fluticasona sola y 210 ml más que el propionato de fluticasona, tras 24 semanas de tratamiento. Asimismo, el riesgo de experimentar exacerbaciones asmáticas graves fue reducido en un 20% anual en relación a la fluticasona sola. No se encontraron diferencias estadísticamente significativas frente a la combinación de propionato de fluticasona y salmenterol.
En pacientes con EPOC el riesgo relativo (RR) de exacerbación fue reducida en relación al vilanterol solo con la combinación a dosis fija en un 15-31%, 21-34% y 13-19% con las dosis de 200, 100 y 50 µg de furoato de fluticasona, respectivamente, aunque no pudo establecerse una relación entre la dosis de fluticasona y la respuesta clínica.
En general, los efectos observados tanto en asma como en EPOC son equiparables a los de otras combinaciones de agonistas β2 adrenérgicos de larga duración (formoterol, salmeterol) y glucocorticoides (propionato de fluticasona, budesonida, beclometasona), con una clara superioridad sobre el glucocorticoide solo. En algunos grupos – generalmente, con las dosis inferiores – no llega a alcanzarse la significación estadística en la superioridad de la combinación frente a los componentes por separado, aunque esta circunstancia no es inhabitual en otras combinaciones previamente autorizadas. En cualquier caso, en términos generales, la relevancia clínica del efecto diferencial de la combinación parece significativa para la mayoría del comité de evaluación (CHMP) de la Agencia Europea de Medicamentos (EMA), aunque algunos de sus miembros mantuvieron una posición discrepante, resaltando que en los pacientes asmáticos la superioridad de la combinación sobre los componentes por separado en relación al efecto broncodilatador y a la mejoría sintomática no había sido suficientemente demostrada; asimismo, en EPOC, tampoco aprecian un claro beneficio sintomático.
El perfil de seguridad de la combinación está en línea con el de otras combinaciones conocidas (nasofaringitis y cefalea, principalmente) y no se han observado eventos adversos específicos; en cualquier caso, la incidencia no es elevada y no es un motivo frecuente de suspensión del tratamiento. No obstante, se han registrado algunos casos mortales aislados de neumonía.
En definitiva, se trata de la primera combinación de un agonista β2 adrenérgicos de larga duración (LABA, long-acting beta-adrenoceptor agonist) y un glucocorticoide que está disponible para su uso tanto en asma como en EPOC y que solo requiere una única administración diaria en inhalación, lo cual, presumiblemente, podría facilitar la tasa de adherencia al tratamiento y, con ello, mejorar el curso y el pronóstico en estos pacientes (Caramori, 2014). En este sentido, hasta ahora, al menos en España, las únicas combinaciones disponibles comercialmente de un LABA (formoterol, salmeterol) y glucocorticoides (beclometasona, budesonida, propionato de fluticasona) solo estaban autorizadas para el tratamiento del asma (con la excepción de salmeterol/fluticasona, que también lo está para EPOC) y todos ellos requieren la administración de dos dosis diarias (cada 12 h).
La terapéutica farmacológica actual del asma y la enfermedad pulmonar obstructiva crónica (EPOC) se fundamenta principalmente en el uso de fármacos broncodilatadores de corta y/o larga duración de acción, como los β2-agonistas y anticolinérgicos administrados por vía inhalatoria, pudiendo también considerarse el uso de teofilina por vía oral. La utilización de esteroides inhalados solos o en combinación con β2-agonistas de larga duración de acción es también relevante, así como la posible utilización de los inhibidores de la isoenzima 4 de la fosfodiesterasa, como el roflumilast. Sin embargo, ninguno de los fármacos actualmente utilizados en el tratamiento del asma y de la EPOC ha demostrado capacidad para modificar favorablemente el deterioro inexorable de la función pulmonar y del estado general de estos pacientes (Cortijo, 2104b).
|
VALORACIÓN |
|
|---|---|
|
Vilanterol/Fluticasona ► RELVAR ELLIPTA® (GlaxoSmithKline) |
|
|
Grupo Terapéutico (ATC): R03AK. APARATO RESPIRATORIO. Medicamentos contra alteraciones obstructivas pulmonares: combinación de adrenérgicos con corticosteroides. |
|
|
Indicaciones autorizadas: Tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista β2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada: pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas β2 inhalados de acción corta administrados «a demanda». También está indicado para el tratamiento sintomático de adultos con EPOC, con un FEV1 <70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora. |
|
|
Valoración global: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad físico-química: Mejora las características farmacocinéticas, con incidencia en las condiciones de uso. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Salbutamol/Beclometasona |
Butosol |
Aldo Unión |
1981 |
|
Salmeterol/Fluticasona |
Seretide |
Glaxo SmithKline |
2001 |
|
Formoterol/Budesonida |
Symbicort |
AstraZeneca |
2002 |
|
Formoterol/Beclometasona |
Foster |
Chiesi |
2007 |
|
Vilanterol/Fluticasona |
Relvar Ellipta |
Glaxo SmithKline |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
Bibliografía
Dexmedetomidina DEXDOR® (Orion)
SEDOANALGESIA EN LA UCI
La sedoanalgesia es uno de los elementos fundamentales para el manejo de los pacientes en estado crítico. La mayoría de estos que son ingresados en una Unidad de Cuidados Intensivos (UCI) requiere una combinación de analgesia y sedación prolongadas para reducir su grado de estrés y facilitar su manejo clínico, lo que redundará en un mejor pronóstico.
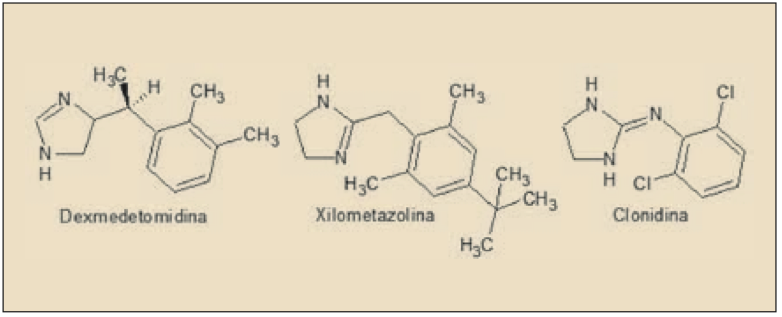
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, estrechamente relacionado con la clonidina y la oximetazolina, que ha sido autorizado para la sedación de pacientes adultos en la UCI que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond. Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta.
Los ensayos clínicos con dexmedetomidina controlados con midazolam y propofol han mostrado la no inferioridad frente a estos en la indicación autorizada, en particular en lo que se refiere al porcentaje del tiempo (60-65%) durante el cual se mantiene el nivel de sedación requerido para cada paciente, sin necesidad de medicación sedante de rescate; tampoco se han registrado diferencias significativas en cuanto a la duración media del uso de ventilación mecánica en la UCI.
La dexmedetomidina representa una forma algo diferente de sedación para la UCI que los habitualmente utilizados, el propofol y el midazolam. Produce un tipo de sedación consciente, con la que el paciente parece estar dormido pero es fácilmente despertado, manteniendo la cooperación y la comunicación con el personal sanitario. En cualquier caso, supone una nueva vía de actuación con unos perfiles de utilidad y de seguridad propios, lo que en un ámbito sin demasiadas opciones permite ampliar las disponibles para adecuarse a las condiciones específicas de cada paciente.
La sedoanalgesia es uno de los elementos fundamentales para el manejo de los pacientes en estado crítico. La mayoría de estos que son ingresados en una Unidad de Cuidados Intensivos (UCI) requiere una combinación de analgesia y sedación prolongadas para reducir su grado de estrés y facilitar su manejo clínico, lo que redundará en un mejor pronóstico.
El dolor en el paciente crítico es común (hasta el 40% de los pacientes refiere haber sufrido dolor en algún momento de su estancia en la UCI) y contribuye decisivamente a la aparición de trastornos del sueño, agotamiento, desorientación y agitación, y desencadena una respuesta neuroendocrina (respuesta al estrés), con taquicardia, aumento del consumo miocárdico de oxígeno, hipercoagulabilidad, inmunosupresión y aumento del catabolismo proteico, hasta el punto de que la respuesta al estrés es asociada con un aumento de la morbilidad y la mortalidad. Por ello, la sedoanalgesia constituye una parte decisiva de la asistencia integral en la UCI y, de hecho, los fármacos usados con este fin son, junto con los antiulcerosos y los antibacterianos, los más utilizados en el paciente crítico. En definitiva, una adecuada sedoanalgesia reduce la respuesta al estrés, limita la ansiedad, mejora la tolerabilidad a la ventilación mecánica y facilita los cuidados del paciente. Por consenso, se define la sedación prolongada como aquélla cuya duración excede las 72 horas (Estébanez, 2008).
Obviamente, las necesidades de sedación no son las mismas para todos los pacientes, ni para el mismo paciente a lo largo del día ni durante su evolución en la UCI, por lo que se debe individualizar el tratamiento en función de los requerimientos de analgesia y sedación que precise el paciente en cada momento. Normalmente, se consideran don niveles de sedación, el superficial y el profundo.
En la sedación superficial, (con una puntuación 0 a -3 en la Richmond Agitation Sedation Scale, RASS) el objetivo es mantener a los pacientes despiertos o con un grado de sedación que permita un fácil despertar, lo que posibilita una evaluación más fácil del dolor, un mejor contacto del paciente con el personal sanitario y la familia y una cooperación en técnicas como la fisioterapia respiratoria o la evaluación neurológica. Por su parte, la sedación profunda (RASS de -4 a -5) se busca en pacientes en ventilación mecánica en los que es importante inhibir el estímulo respiratorio, aquellos que requieren el uso de bloqueantes neuromusculares, pacientes con hipertensión endocraneal, estatus epiléptico o psicosis aguda; pacientes en los que se realiza limitación del esfuerzo terapéutico y aquellos en los que se realizan técnicas diagnósticas y/o terapéuticas muy agresivas.
Las características del paciente crítico en la UCI requieren idealmente fármacos sedantes con un rápido inicio de acción y una no menos rápida recuperación, un fácil ajuste de la dosificación, un amplio margen terapéutico y la ausencia de acumulación en el organismo, de interacciones farmacológicas y de efectos adversos; todo ello, a un bajo coste. Obviamente, tal medicamento no existe y hay que recurrir a un amplio colectivo de fármacos que cumplen con alguna de las condiciones antes mencionadas, pero no todas, para obtener una sedación prolongada con niveles de eficacia y seguridad razonables. Los utilizados habitualmente son las benzodiacepinas (midazolam, lorazepam, diazepam, etc.), anestésicos (propofol), analgésicos opiáceos (remifentanilo), agonistas α2 adrenégicos (clonidina, etc.) e hipnóticos barbitúricos (tiopental); entre ellos, los más utilizados actualmente son las benzodiazepinas y el propofol.
Las benzodiazepinas presentan unas excelentes propiedades farmacocinéticas para su utilización en el paciente crítico y su empleo es seguro, ya que poseen mayor margen terapéutico que otros hipnóticos y sedantes. Todos los miembros del grupo actúan sobre sobre determinadas subunidades del receptor GABAA que potencian los efectos del ácido γ-aminobutírico (GABA) sobre dicho receptor. En términos bioquímicos, estos fármacos aumentan la respuesta de ciertas neuronas al GABA, facilitando la apertura de los canales de iones cloruro que son activados por este aminoácido neurotransmisor, el cual media gran parte de la transmisión sináptica inhibitoria en el sistema nervioso central. Los fármacos de este grupo dan lugar a un aumento de la frecuencia de apertura del canal inducida por GABA, sin producir cambios en la conductancia o el tiempo medio de apertura. Este efecto es compatible con la observación de que tales fármacos no se fijan al sitio de unión de GABA en el receptor GABAA (sitio ortostérico), sino a un sitio modulador cuya ocupación ocasiona que el GABA incremente su afinidad por el receptor cuando el poro está abierto. Este mecanismo alostérico explica el bajo grado de toxicidad aguda de estos hipnóticos, ya que no pueden ocasionar la entrada de iones cloruro en ausencia de GABA. En términos farmacoterapéuticos, los efectos asociados a la activación de este receptor son ansiolíticos, hipnóticos, amnésicos, miorrelajantes y anticonvulsivantes. Sin embargo, carecen de capacidad analgésica y antiemética. La benzodiazepina más utilizada en sedación prolongada en Europa es el midazolam, pero en principio cualquier miembro del grupo con acción rápida, como el lorazepam, podría utilizarse teóricamente en esta indicación.
Las benzodiazepinas producen disminución de la presión arterial por vasodilatación, además de depresión respiratoria en relación con la dosis, la velocidad de administración y el estado del paciente. Su uso prolongado puede dar lugar al desarrollo de tolerancia. Además, pueden producir conducta agresiva u hostil por desinhibición o un estado inicial paradójico de nerviosismo antes de que se establezca el efecto ansiolítico o sedante. Por el contrario, un aspecto favorable muy interesante de las benzodiazepinas es que ofrecen la posibilidad de revertir su efecto farmacológico mediante la utilización de flumazenilo, un antagonista específico de los receptores, aunque su empleo rutinario en sedación prolongada no está recomendado por el riesgo de aparición de un síndrome de abstinencia.
El propofol o disoprofol tiene propiedades sedantes, hipnóticas y antieméticas, pero carece de efectos analgésicos significativos. Su acción anestésica es consecuencia de su interacción con un lugar alostérico para anestésicos generales en el receptor GABAA, que es diferente al de a las benzodiazepinas, facilitando la apertura del canal de cloruro. Presenta un inicio de acción rápido y una vida media corta. En infusión continua no plantea problemas de acumulación (aunque se ha observado que el tiempo de despertar desde la suspensión del fármaco está relacionado con el tiempo de sedación). Sus características farmacocinéticas permiten un fácil control del nivel de sedación, al igual que una temprana recuperación del nivel de consciencia tras el cese de su administración (unos 10 minutos), lo que facilita la evaluación neurológica de los pacientes. Además, si se asocia a morfina mejora el control de la presión intracraneal en pacientes con traumatismos craneoencefálico graves.
Induce hipotensión por reducción de las resistencias vasculares periféricas sin modificar el gasto cardiaco, lo cual ocurre con más frecuencia tras la administración en bolo, en pacientes hipovolémicos o con inestabilidad hemodinámica. Provoca una profunda depresión respiratoria y bradicardia por depresión del reflejo barorreceptor, en particular, durante la inducción, efecto que es potenciado por los opioides.
Los barbitúricos y, en particular el tiopental (el único utilizado en clínica para esta indicación), actúan de forma similar al propofol (sitio alostérico acoplado a los receptores GABAA del canal de cloruro neuronal), provocando una depresión reversible del tejido nervioso. Producen hipotensión (en bolo) y depresión miocárdica, además de predisponer a la infección y al íleo paralítico, por lo que no se recomiendan para la sedación de rutina de los pacientes críticos, reservándose para aquellos con estatus epiléptico y en pacientes con hipertensión intracraneal refractaria
Entre los analgésicos opiáceos, el más utilizado en sedación prolongada es el remifentanilo. Actúa como un agonista puro de los receptores µ(mu) opiáceos y tanto el inicio de su acción como la duración de ésta son breves. Como es obvio, su acción fundamental es la analgesia, incrementando el umbral del dolor, alterando la percepción del mismo e inhibiendo las vías ascendentes del estímulo doloroso. La sedación con remifentanilo reduce la necesidad de ventilación mecánica y el tiempo de extubación en relación a la sedación estándar y sus propiedades farmacocinéticas lo convierten en un buen agente para su utilización en estrategias de sedación secuencial y dinámica
La utilización de agonistas alfa-2 adrenérgicos no es nueva en sedación, aunque su empleo proviene en origen del ámbito de la veterinaria, donde algunos fármacos de este grupo (xilazina, detomidina) se han venido utilizando durante décadas para inducir la analgesia y la sedación en diversas especies de animales. Todos ellos derivan de la clonidina, aunque la farmacología de este grupo es más compleja de lo que a primera vista pudiera parecer.
Existen tres tipos de receptores o isorreceptores alfa-2 (∝2) adrenérgicos: a, b y c. La acción estimulantes o agonista sobre los receptores ∝2a produce sedación, hipnosis, analgesia e inhibe la secreción de insulina; por su parte, la activación de los receptortes ∝2b produce analgesia espinal y vasoconstricción en la arterias periféricas, mientras que la de los receptores ∝2c da lugar a una modulación del procesamiento sensorial cognitivo y del estado de ánimo, induce una estimulación locomotriz y regula la liberación de noradrenalina por la médula adrenal. Todos estos tipos de receptores poseen localización pre-, post- y extrasinápticas, lo que determina que cada agente activo sobre estos tenga un perfil farmacodinámico particular, no necesariamente extrapolable al resto. Sea como fuere, las localizaciones presinápticas de estos receptores tienen una particular relevancia, por que actúan como autorreceptores, es decir forman parte de un sistema de retroalimentación (feedback) negativo que regula inhibitoriamente la liberación de noradrenalina.
La estimulación a nivel cerebral y espinal de los receptores ∝2 produce una inhibición de la activación (firing) neuronal, que conduce a hipotensión, bradicardia, sedación y analgesia. Reduce la entrada de calcio en las terminaciones nerviosas, lo que puede contribuir a su efecto inhibitorio sobre la liberación de neurotransmisores. En otros órganos y tejidos, produce una reducción de la salivación, y de la secreción y motilidad gástrica; inhibe la liberación de renina, incrementa la tasa de filtración glomerular renal (y con ello la eliminación renal de sodio y agua), reduce la presión intraocular y la secreción de insulina por el páncreas.
Con independencia del fármaco utilizado para producir una sedación prolongada, existe un amplio capítulo de complicaciones asociadas. La primera de ellas es la infrasedación, que desprotege al paciente crítico, haciéndole experimentar miedo, ansiedad, trastornos del sueño, desorientación y agitación, lo que se asocia a un peor pronóstico; todo ello conlleva un incremento del riesgo para la autorretirada (arrancamiento) de dispositivos y de las necesidades de cuidados director de enfermería, lo que se asocia con un aumento del consumo de recursos profesionales y de costes. Además, el aumento del consumo de oxígeno y de la actividad del sistema autónomo con el aumento del trabajo miocárdico puede ser especialmente peligrosos en determinados pacientes críticos (traumatismo craneoencefálico, insuficiencia respiratoria, shock, etc.)
Por el contrario, el empleo de dosis elevadas o de pautas que combinan sedantes puede dar lugar a la sobresedación del paciente, que se asocia a una prolongación del tiempo de ventilación mecánica y, por lo tanto, a las complicaciones relacionadas con la misma (neumonía, hemorragia digestiva, bacteriemia, trombosis venosa profunda, etc.) y a un aumento de la duración de la estancia en la UCI y en el hospital, a un mayor consumo de recursos sanitarios y a la dificultad para monitorizar la evolución neurológica. Asimismo, estos pacientes tienen mayor frecuencia de sueños paranoides, pesadillas y alucinaciones, lo que puede dar lugar a secuelas psicológicas graves.
Por su parte, el desarrollo de tolerancia (requerimiento de dosis progresivamente mayores de sedantes y analgésicos para mantener el mismo nivel de sedoanalgesia) se relaciona con fenómenos de regulación a la baja (down-regulation) de los receptores celulares y da lugar a un aumento de la dificultad para conseguir y/o mantener un nivel adecuado de sedación y, en ocasiones, obliga al empleo de dosis elevadas de sedantes o a la combinación de diferentes fármacos, aumentando el riesgo de complicaciones asociadas
Finalmente, al iniciar la retirada de la sedoanalgesia los pacientes pueden desarrollar síntomas de abstinencia o deprivación1, especialmente si han desarrollado previamente tolerancia o han recibido dosis altas de sedantes durante más de 3-5 días. Los síntomas de abstinencia varían según el fármaco empleado, la edad del paciente, la función cognitiva y la situación clínica. Para ello, en el caso de benzodiazepinas y/u opioides, debe evitarse la suspensión brusca de la pauta, con un descenso progresivo de la dosis de infusión en un 20-40% diario, para continuar con un descenso del 10% cada 12-24 horas guiado por la respuesta clínica, evitando disminuir la dosis en más del 10% en aquellos pacientes con factores de riesgo para el desarrollo de síntomas de abstinencia (especialmente aquellos que han recibido dosis altas durante más de una semana).
ACCIÓN Y MECANISMO
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, autorizado para la sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (Richmind Agitation Sedation Score, RASS).
La dexmedetomidina presenta un amplio abanico de actividades sobre el funcionamiento orgánico: sedación, inducción del sueño, ansiolisis, amnesia, analgesia, hipotensión, bradicardia, etc. Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta. Cuando el fármaco se une y estimula los receptores ∝2 adrenérgicos a este nivel neuronal, se produce una inhibición de la adenil ciclasa, responsable de la formación de AMPc. La reducción de los niveles intraneuronales de AMPc se traduce en un aumento de las actividades anabólicas y catabólicas; simultáneamente, se produce una salida del potasio intracelular a través de los canales de potasio activados por calcio, y una inhibición de la entrada de calcio en las terminaciones nerviosas, todo lo cual produce a una hiperpolarización de la membrana neuronal que suprime la activación del locus ceruleus y la actividad de las vías noradrenérgicas ascendentes. El locus ceruleus es también el origen de las vías adrenérgicas médulo-espinales descendentes, de particular relevancia en los mecanismos reguladores de la transmisión de impulsos nociceptivos (implicados en la sensación de dolor). De hecho, los efectos analgésicos se sitúan fundamentalmente a nivel del asta dorsal de la médula espinal.
La inhibición de la liberación de noradrenalina en el locus ceruleus por la dexmedetomidina impide el control inhibitorio del núcleo preóptico ventrolateral, que libera ácido gamma(γ)aminobutírico (GABA) y galanina, que incrementan la propia inhibición del locus ceruleus y del núcleo tuberomamilar. Esta inhibición también reduce la liberación de histamina y su efecto sobre las neuronas de diversas áreas subcorticales, que se traduce en un efecto hipnótico adicional, tal como ocurre durante el sueño fisiológico.
Los efectos sedantes e hipnóticos de la dexmedetomidina son dependientes de la dosis, hasta el punto de desarrollar una actividad anestésica con dosis elevadas. La amnesia asociada al fármaco es mucho menos marcada que la relacionada con las benzodiazepinas, en particular con midazolam.
La dexmedetomidina suprime los escalofríos (frecuentes durante los periodos postoperatorios), posiblemente por su actividad agonista ∝2b adrenérgica a nivel del centro termorregulador hipotalámico. A pesar de su potente efecto sedante, la dexmedetomidina está asociada como mínimos efectos depresores respiratorios, incluso con dosis muy superiores a las empleadas en sedación.
Los efectos cardiovasculares tienen un carácter bifásico. Tras la administración iv inicial, se produce un aumento de la presión arterial y una bradicardia refleja, como consecuencia de una acción agonista ∝2b adrenérgica en las células musculares lisas vasculares. Este efecto inicial dura entre 5 y 10 minutos, tras los que se produce una ligera caída de la presión arterial como consecuencia de la inhibición del tono simpático central; asimismo, los receptores ∝2b adrenérgicos presinápticos también son activados, lo que da lugar a una reducción de la liberación de noradrenalina, todo lo cual conduce a una reducción de la presión arterial y el ritmo cardiaco.
{kind=link}
ASPECTOS MOLECULARES
La dexmedetomidina es el enatiómero farmacológicamente activo de la medetomidina. Químicamente, es el 5-[(1S)-1-(2,3-dimetilfenil)etil]-1H-imidazol y está estrechamente relacionado estructural y farmacológicamente con otros agentes agonistas de los receptores α2 adrenérgicos, como clonidina, apraclonidina, brimonidina, nafazolina, tramazolina y, en particular, con xilometazolina y oximetazolina (empleados fundamentalmente con descongestivos nasales). Aunque es considerado fundamentalmente como un agonista de los receptores ∝2 adrenérgicos, dada la presencia de un núcleo de imidazolina en su estructura también desarrolla actividad agonista sobre los receptores imidazolínicos (I), de los que existen tres tipos (I1 implicado en la actividad simpaticolítica de los antihipertensivos imidazolínicos, como la clonidina; I2, una zona de unión alostérica de la monoamino oxidasa, implicada en la modulación del dolor; I3, regula la secreción de insulina por las células beta pancreáticas).
EFICACIA Y SEGURIDAD CLÍNICAS
Existe un buen número de ensayos clínicos controlados con placebo y con comparadores activos que han permitido contrastar la eficacia y la seguridad clínicas de la dexmedetomidina en la indicación autorizada. En el informe público de evaluación de la Agencia Europea de Medicamentos (EMA, 2011) se recogen tres ensayos clínicos principales (pivotales) controlados con comparador activo (propofol y midazolam), todos ellos de fase 3, multicéntricos, multinacionales, aleatorizados y doblemente ciegos, realizados en pacientes sometidos a sedación continua durante más de 24 h (máximo de 14 días) en una Unidad de Cuidados Intensivos (UCI) y con ventilación mecánica.
La dexmedetomidina fue administrada en infusión IV (sin bolo inicial) con un ritmo de 0,8 µg/kg/h la primera hora, reajustándose en función de las necesidades entre 0,25 y un máximo de 1,4 µg/kg/h para mantener el nivel de sedación requerido (expresado como puntuación RASS) para cada paciente. Las dosis de propofol utilizadas variaron entre 0,3 y 4,0 mg/kg/h y las de midazolam entre 0,03 y 0,20 mg/kg/h.
Como criterio de valoración de la eficacia se definieron dos variables co-primarias. La primera fue la proporción de tiempo durante el cual se mantuvo el nivel de sedación requerido para cada paciente sin necesidad de recurrir a medicación sedante adicional (de rescate), mientras que la segunda variable consistió en la duración media de la estancia en la UCI (estudio 3005011) o en la duración media de la ventilación mecánica (estudios 3005012 y 3005013).
En el primero de estos estudios (3005011; n= 79), la mediana del porcentaje del tiempo durante el que se mantuvo la sedación requerida fue del 55,4% (IC95% 48,9 a 65,0) con dexmedetomidina vs. 57,2% (IC95% 47,4 a 66,9) con propofol o midazolam. En aquellos cuyo objetivo era mantener una puntuación RASS entre 0 y -3, estos porcentajes fueron del 67,6 vs. 63,7%, mientras que en aquellos cuyo objetivo era una puntuación de -4 la dexmedetomidina fue claramente inferior a los comparadores (30,7 vs. 63,0%; p= 0,0006). En cuanto a la duración media de la estancia en la UCI fue de 175 vs 187 h (los valores de las medianas fueron de 137 y 132 horas, respectivamente).
En el segundo estudio (3005012; n= 498) se comparó a la dexmedetomidina con propofol, obteniéndose unos valores para el porcentaje de tiempo durante el que se mantuvo el nivel de sedación requerido del 62,4 vs. 65,5% (diferencia ajustada de -2,22; IC95% -7,05 a 2,60), mientras que la mediana de la duración de la ventilación mecánica fue de 96,5 vs. 117,5 h. Un 73% vs. 64% de los pacientes requirieron al menos una vez sedación adicional de rescate (midazolam).
En el tercer estudio (3005013; n= 500) la comparación se hizo con midazolam, registrándose unos valores para el porcentaje de tiempo con el nivel de sedación requerido del 59,9 vs. 54,9% (diferencia ajustada de 4,97; IC95% -0,46 a 10,40), siendo las medianas de la duración de la ventilación mecánica de 123,0 vs. 164,0 h. Un 44% vs. 45% de los pacientes requirieron al menos una vez sedación adicional de rescate (propofol).
En cuanto al perfil de seguridad de la dexmedetomidina, los eventos adversos más comunes descritos son hipotensión, bradicardia e hipertensión (inicial). El porcentaje de pacientes con eventos adversos relacionados con el tratamiento fue (en los ensayos clínicos controlados) del 36% (dexmedetomidina), 25% (midazolam), 20% (propofol) y 27% (placebo), siendo de carácter grave en el 3,0%, 1,0%, 1,7% y 1,5%, respectivamente. Se suspendió el tratamiento por la incidencia de eventos adversos en el 8,2%, 8,7%, 8,4% y 3,6%, respectivamente. Los eventos adversos más prevalentes descritos fueron bradicardia, con 24% (12% con midazolam y 10% con propofol), hipotensión (22%, 17% y 14%), taquicardia (15%, 17% y 1,1%) e hipertensión arterial (12%, 15% y 0%).
ASPECTOS INNOVADORES
La dexmedetomidina es un fármaco agonista de los receptores alfa-2 (∝2) adrenérgicos, estrechamente relacionado con la clonidina y la oximetazolina, que ha sido autorizado para la sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal, correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (Richmind Agitation Sedation Score, RASS). Los efectos neurológicos del fármaco parecen estar mediados por un fenómeno de hiperpolarización de las neuronas noradrenérgicas en el locus ceruleus cerebral, el principal centro de modulación del estado de alerta y que da lugar a una inhibición de la liberación de noradrenalina en dicho núcleo, lo que impide el control inhibitorio del núcleo preóptico ventrolateral, que libera ácido gamma(γ)aminobutírico (GABA) y galanina, que incrementan la propia inhibición del locus ceruleus y del núcleo tuberomamilar. Esta inhibición también reduce la liberación de histamina y su efecto sobre las neuronas de diversas áreas subcorticales, que se traduce en un efecto hipnótico adicional, tal como ocurre durante el sueño fisiológico.
Los ensayos clínicos controlados con midazolam y propofol han mostrado la no inferioridad frente a estos en la indicación autorizada, en particular en lo que se refiere al porcentaje del tiempo (60-65%) durante el cual se mantiene el nivel de sedación requerido para cada paciente, sin necesidad de medicación sedante de rescate; tampoco se han registrado diferencias significativas en cuanto a la duración media del uso de ventilación mecánica en la UCI. Sin embargo, sí se apreció un uso más frecuente de la medicación sedante de rescate con dexmedetomidina que con propofol (73 vs. 64%), pero similar al midazolam (60 vs. 57%). No obstante, cuando se utiliza en pacientes en los que se requiere un nivel de sedación más profundo (puntuación -4 RASS), la desxmedetomidina fue claramente inferior a propofol y midazolam.
Desde el punto de vista de la seguridad, el nuevo fármaco presenta un perfil coherente con so condición farmacológica de agonista ∝2 adrenérgico, siendo los eventos adversos más comunes la hipotensión (con hipertensión inicial) y la bradicardia; en cualquier caso, se trata de fenómenos conocidos y manejables clínica y farmacológicamente.
Un meta-análisis (Fraser, 2013) que incluyó los resultados de seis ensayos clínicos sugiere que el uso de dexmedetomidina y propofol reducen significativamente la duración de la estancia en la UCI, en relación a las benzodiazepinas (midazolam, lorazepam, etc.), así como la necesidad de la ventilación mecánica de los pacientes (diferencia de 1,9 días, IC95% 1,7 a 2,1; p< 0,0001), pero sin diferencias en cuanto a la prevalencia de delirio y a la mortalidad.
Otro meta-análisis (Xia, 2013), confeccionado a partir de los datos de 10 ensayos clínicos controlados, mostró que la dexmedetomidina parece ofrecer ventajas sobre el propofol en términos de la duración de la estancia en la UCI (-0,81 días; IC95% -1,48 a -0,15) y un 60% de reducción en el riesgo de delirio (RR= 0,40; IC95% 0,22 a 0,74), pero no encontró diferencias estadísticamente significativas en cuanto a la duración de la ventilación mecánica (0,53 horas; IC95% -2,66 a 3,72) o mortalidad (RR= 0,83; IC95% 0,32 a 2,12). La dexmedetomidina fue asociada con un mayor riesgo de hipertensión (RR= 1,56; IC95% 1,11 a 2,20).
Se trata de una nueva línea de actuación farmacológica en sedación humana, ya que en veterinaria este tipo de agentes se venía utilizando desde hace décadas; de hecho, la dexmedetomidina es un análogo de la etomidina, profusamente utilizada en veterinaria. En un sentido estricto, no se trata de un medicamento reciente, toda vez que ya se intentó su autorización por la Agencia Europea de Medicamentos (EMA) en 1998, pero ante las objeciones mayores presentadas por ésta, el expediente de registro fue retirado. Posteriormente, con una profunda revisión de dicho expediente e incluyendo nuevos y más rigurosos ensayos clínicos, fue presentado de nuevo en 2010, siendo autorizado el medicamento en 2011 para toda la Unión Europea.
La dexmedetomidina representa una forma algo diferente de sedación para la UCI que los habitualmente utilizados, el propofol y el midazolam (en Estados Unidos se utiliza más habitualmente el lorazepam entre las benzodiazepinas para esta indicación). Produce un tipo de sedación consciente, con la que el paciente parece estar dormido pero es fácilmente despertado, manteniendo la cooperación y la comunicación con el personal sanitario. En cualquier caso, supone una nueva vía de actuación con unos perfiles de utilidad y de seguridad propios, lo que en un ámbito sin demasiadas opciones permite ampliar las disponibles para adecuarse a las condiciones específicas de cada paciente.
|
VALORACIÓN |
|
|---|---|
|
Dexmedetomidina ► DEXDOR® (Orion) |
|
|
Grupo Terapéutico (ATC): N05CM. SISTEMA NERVIOSO. Psicolépticos. Hipnóticos y sedantes: otros. |
|
|
Indicaciones autorizadas: Sedación de pacientes adultos en la UCI (Unidad de Cuidados Intensivos) que requieran un nivel de sedación no más profundo que despertarse en respuesta a la estimulación verbal (correspondiente a un grado de 0 a -3 en la Escala de Sedación y Agitación de Richmond (RASS)). |
|
|
Valoración global: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad molecular: Mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma o similar indicación terapéutica. |
⇑ |
Bibliografía
Dolutegravir TIVICAY® (GlaxoSmithKline)
VIH/SIDA
El objetivo esencial de la terapia antirretroviral (TAR) es maximizar la supresión permanente de la replicación del VIH, con el fin de limitar el desarrollo de resistencia viral y de restaurar la función inmunológica, reduciendo con ello la morbilidad y la mortalidad asociadas, mejorando la calidad de vida del paciente y previniendo la transmisión del VIH. En este sentido, el estándar terapéutico de supresión de la replicación viral es aquel que asegure al máximo una carga viral plasmática (CVP) estable por debajo de 50 copias/ml.
Dolutegravir es un agente antiviral directo que actúa inhibiendo la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Está relacionado química y farmacológicamente con raltegravir y elvitegravir, previamente registrados. Ha sido autorizado para el tratamiento de adultos y adolescentes mayores de 12 años infectados por el Virus de Inmunodeficiencia Humana (VIH), en combinación con otros medicamentos antirretrovirales.
Presenta un perfil farmacocinético es más favorable que el de sus antecesores de este grupo, con una semivida celular prolongada que facilita una única administración diaria, sin necesidad de bloqueantes enzimáticos adicionales (como el cobicistat, en el caso del elvitegravir) y además es metabolizado mayoritariamente por una vía ajena a los isoenzimas del citocromo p450, lo que limita notablemente el número y la relevancia de las interacciones con otros fármacos, incluyendo la mayoría de los antirretrovirales. Todo ello, unido a la utilidad en pacientes que previamente habían fracasado con raltegravir o elvitegravir, le aporta un significativo valor añadido y viene a consolidar la relevancia de los inhibidores de la integrasa como fármacos de primera consideración en el tratamiento de la infección por VIH.
{kind=link}
El VIH es un retrovirus, esto es, un virus ARN, que depende de un enzima, la transcriptasa inversa, para dar lugar a una cadena de ADN que, por la acción de otro enzima, una integrasa, se inserta en el genoma del hospedador. Forma parte de los lentivirus, una subfamilia cuyo nombre alude a la forma insidiosa (lenta e inicialmente desapercibida) en que se desarrolla su infección.
En 1983 se identificó el primer tipo de VIH (VIH-1), el más distribuido a nivel mundial, y en 1986 un segundo (VIH-2), de distribución más restringida. Se han descrito numerosos subtipos, algunos de los cuales han evolucionado mayoritariamente de forma pandémica, pero también se han identificado cepas recombinantes intersubtipo. Esta gran variabilidad que presenta es uno de los inconvenientes para conseguir un tratamiento eficaz. Tanto el VIH-1 como el VIH-2 son capaces de provocar el sida, si bien el VIH-2 se asocia más frecuentemente que el VIH-1 con el desarrollo de las formas neurológicas del síndrome.
El VIH-1 puede presentar cepas más o menos patógenas, con distinto tropismo celular; las cepas del grupo O son naturalmente resistentes a inhibidores de la transcriptasa inversa no análogos de los nucleósidos; en los distintos subgrupos del grupo M, algunas cepas del subtipo G son menos sensibles in vitro a inhibidores de la proteasa; y algunas mutaciones que condicionan la resistencia a inhibidores de la transcriptasa inversa no nucleósidos son más frecuentes, por ejemplo, en el subgrupo C que en el B. El VIH-2 no es sensible a los inhibidores de la transcriptasa inversa no análogos de los nucleósidos y, para algunos subtipos, la determinación de carga viral (criterio fundamental en el manejo actual de la infección por el VIH) puede dar resultados falsamente negativos.
Los VIH son virus de geometría esférica, con un diámetro medio aproximado de 100 nm (Figura 2). La capa más externa o cubierta está formada por una membrana lipídica derivada de la célula hospedadora, con un alto contenido en lipoproteínas. En dicha membrana se insertan antígenos de histocompatibilidad (HLA) del hospedador, que le permitirán una primera unión a la célula hospedadora y multitud de complejos heterodiméricos de glucoproteínas, compuestos por trímeros de la glicoproteína de superficie gp120 y de la glicoproteína transmembrana gp41. Por debajo de la membrana lipídica se encuentra la matriz proteica, formada por la proteína p17, que recubre a la cápsula o cápside propiamente dicha, constituida por la proteína p24. Dentro de la cápside se encuentran el material genético –que se presentará como dos hebras idénticas de ARN monocatenario–, la nucleoproteína y algunas enzimas (entre otras, la transcriptasa inversa). La transcriptasa inversa va a facilitar la formación, a partir del ARN, de una doble cadena de ADN, que se insertará en el material genético de la célula hospedadora.
En el genoma del VIH se incluyen varios genes: los gag, que codifican proteínas estructurales del núcleo y de la matriz, y los genes env, que codifican las glicoproteínas de la cubierta viral que intervienen en el reconocimiento de los receptores presentes en la superficie de las células «diana». También contiene genes pol, que codifican enzimas esenciales para el proceso de replicación viral y, entre ellas, la transcriptasa inversa, que permite la conversión del ARN viral en ADN, la integrasa, que facilita la incorporación del ADN viral al ADN de los cromosomas de las células huésped (provirus), y la proteasa, que facilita la escisión de las grandes proteínas precursoras inactivas gag y pol en sus formas activas.
Además de los genes mencionados, el ARN del VIH contiene otros genes, como tat, rev, vif, vpu, nef y vpx, que codifican proteínas reguladoras. En concreto, el gen tat codifica una proteína (Tat) que es expresada de forma precoz tras la infección y regula la expresión de otros genes del VIH. Por su parte, la proteína Rev facilita el paso del ARN mensajero desde el núcleo al citoplasma; la proteína Vpr parece estar implicada en la detención del ciclo celular y también capacita al ADN transcrito a partir del ARN viral a acceder al núcleo de células que no se encuentren en fase de división. La proteína Vpu es requerida para una liberación correcta de las partículas virales, en tanto que la Vif parece estar implicada en la infectividad del VIH. La proteína Nef tiene, entre otras, la misión de provocar una regulación a la baja de la expresión de los receptores CD4 en la superficie de las células huésped, facilitando así la gemación del virus en las fases finales del ciclo replicador. El virus se apodera de la maquinaria celular y silencia la replicación de numerosos genes celulares en favor de la replicación de los propios.
El VIH es un microorganismo extraordinariamente sensible al medio, no puede sobrevivir fuera del torrente sanguíneo o del tejido linfático y es fácilmente destruido por cualquier detergente o desinfectante al uso. Por este motivo, la transmisión del VIH entre personas ha de producirse a través de algún fluido biológico en el que pueda sobrevivir el VIH, fundamentalmente la sangre y algunas secreciones (vaginal, espermática, etc.), que entre en íntimo contacto con estructuras potencialmente receptoras, como los vasos sanguíneos, o pequeñas erosiones en la piel o las mucosas. Las principales vías de transmisión del VIH son la sexual, la parenteral y la vertical (madre-hijo), siendo la vía sexual la que origina más del 80% de los contagios a nivel mundial.
El VIH presenta tropismo por los macrófagos y los linfocitos T CD4. Las células infectadas por el VIH pueden transferir el virus a las células del sistema inmune local, presentes en el epitelio vaginal o en la mucosa anorrectal. En el caso de las relaciones heterosexuales –actualmente, la forma de transmisión más común–, el primer tejido en ser infectado es la mucosa cervical, donde pueden infectarse las células dendríticas y los linfocitos T CD4+. La proporción de linfocitos T CD4+ infectados en sangre periférica es mínima (1-10%), pero su capacidad de producir viriones es muy alta y, a partir de estos, la infección puede difundirse hacia los nódulos linfáticos regionales y, posteriormente, ser distribuida por todo el organismo a través del torrente circulatorio.
Ya en los nódulos linfáticos se produce una intensa replicación del VIH que en unos casos provoca la lisis de las células infectadas, pero en otros queda como una infección latente en los macrófagos y los linfocitos T en reposo. Justamente, son estos últimos los que actúan como «reservorios naturales» del VIH y dificultan notablemente la respuesta inmunológica del organismo y, en la misma medida, la adopción de medidas terapéuticas eficaces desde el mismo inicio de la infección. Sólo una pequeña proporción de los linfocitos infectados (1%) replica activamente el VIH en determinado momento, permaneciendo latente en la inmensa mayoría de ellos. Aún no se conoce bien de qué forma estas células con infección latente pasan a la replicación activa, pero los factores implicados en este proceso podrían constituir nuevas dianas terapéuticas, ya que el virus latente carece de efectos patológicos.
Apenas 10-12 días después del contagio ya se puede detectar el ARN viral en la sangre, lo que implica que el paciente está en condiciones de contagiar a otra persona. Tras la primera replicación viral después de la infección, la carga viral puede llegar a alcanzar valores de hasta 100 millones de copias por mL de sangre. Esta elevada viremia provoca una rápida respuesta del sistema inmunológico. La respuesta inmune frente al VIH-1 se produce en la vertiente humoral (con una intensa producción de anticuerpos contra las proteínas reguladoras y estructurales, así como producción de complemento e interferones) y en la celular (activación de poblaciones linfocitarias T colaboradoras, citotóxicas y de estirpe NK o natural killer). Se produce una intensa expansión clonal de linfocitos CD8+ con actividad citotóxica, dirigida frente a diversas proteínas del VIH-1, y que originan una cierta supresión de la replicación del VIH y una drástica disminución de la viremia –en varios órdenes de magnitud – pudiendo incluso hacerse indetectable en la sangre (generalmente, menos de 50 copias de ARN por mL).
Entre 3 y 5 semanas después de la infección, aparecen anticuerpos, lo que determina la seroconversión, ya que a partir de este momento el paciente se transforma en seropositivo. Aunque esta potente respuesta antiviral llega a contener la replicación vírica, es incapaz de erradicar el virus del organismo. En la fase crónica de la infección, estas respuestas se mantienen. Existe una persistente activación inmunológica por parte del virus y un control parcial de la masiva replicación viral por parte del sistema inmune durante largos periodos de tiempo. El pronóstico y la evolución de la infección dependerán del equilibrio entre la virulencia del virus y la respuesta inmunológica del huésped.
En los estadios finales de la enfermedad se produce un daño en el control inmunológico, con caída de linfocitos CD4+ y elevación de la carga viral, así como disminución de anticuerpos frente a diversas estructuras virales. La destrucción de los CD4+, que ocupan un lugar preeminente en la organización y la activación del sistema inmune, origina un deterioro funcional de otras poblaciones celulares. Esta situación define lo que denominamos SIDA, que va a caracterizarse por la aparición de infecciones oportunistas y tumores.
Existe un pequeño grupo de pacientes (5-10 %) con infección por el VIH-1 que presenta una progresión lenta o una ausencia de progresión hacia estas fases finales. Estos pacientes de progresión lenta mantienen una carga viral basal reducida y cifras de linfocitos CD4+ mantenidas por encima de 500/mm3 en ausencia de terapia específica. Probablemente, este grupo de población es un colectivo heterogéneo, en cuya protección están implicados diversos factores inmunológicos (potentes y variadas respuestas citotóxicas), virológicos (cepas virales con defectos estructurales) y genéticos (defectos en ciertos correceptores del VIH; por ejemplo, CCR5, CXCR4) que permiten esta evolución benigna de la infección.
Según las cifras de 2008 publicadas por la OMS, hay actualmente 33,4 millones de afectados por el VIH. Se estima que durante ese año resultaron infectadas por el virus 2,7 millones de personas, y que 2 millones murieron de SIDA. El África subsahariana continúa siendo la región más afectada por el VIH. En 2008, dicha región tenía el 67% de las infecciones por VIH a nivel mundial, 68% de los nuevos casos en los adultos y el 91% de las nuevas infecciones en los niños. Según el último informe publicado por el Registro Nacional de Sida, en 2011 se diagnosticaron en España 2.763 nuevos casos de infección por VIH, lo que supone una tasa de 84,1/millón de habitantes. El 83% eran hombres y la mediana de edad fue de 35 años.
El proceso infeccioso del virus de la deficiencia humana (VIH-1) comienza tras su inoculación por una replicación extraordinariamente rápida del virus en el tejido linfoide, que se contrarresta por la aceleración de las tasas de producción y destrucción de las células de defensa inmunitaria. Más de un 30% de las partículas virales presentes en el plasma de los pacientes se renuevan diariamente, lo que implica una vida media viral de menos de dos días, mientras que el ritmo diario de sustitución de los linfocitos CD4+ plasmáticos es de un millón, lo que supone entre 10 y 100 veces más de lo normal.
La primoinfección por el VIH-1 es sintomática en más de la mitad de los casos, pero puede pasar desapercibida ya que sus síntomas son los de una virosis común (como un resfriado o una gripe). Como en esta fase todavía no hay anticuerpos (período ventana), lo que puede determinarse es la carga viral plasmática (CVP), detectable a partir de la primera semana. La CVP en la infección aguda suele estar muy elevada – más de un millón (6 log10) de copias de ARN viral – y se relaciona con la intensidad de las manifestaciones clínicas, detectándose la seroconversión entre una y dos semanas más tarde. El término infección aguda, que implica el diagnóstico antes de la seroconversión (menos de 30 días), no debe confundirse con el de infección reciente, aquella que tiene menos de seis meses de evolución (menos de 180 días).
Las manifestaciones clínicas del SIDA solo aparecen cuando el sistema inmunitario pierde la batalla frente a la infección. La facilidad de mutación del VIH explica la escasa eficacia de la monoterapia antirretroviral, dado que su rapidez de replicación garantiza la aparición y diseminación de cepas resistentes a varios fármacos. En cualquier caso, la progresión de la infección a SIDA es más rápida en los pacientes inicialmente sintomáticos y se asocia a factores iniciales de la infección, como la gravedad de la sintomatología en la infección aguda (mayor riesgo a mayor número de síntomas), cuantía del descenso inicial del número de linfocitos CD4+ (mayor riesgo si es inferior a 500 células/μL), nivel de la CVP basal o a partir del cuarto mes (mayor progresión si es mayor de 5 log10 o 100.000 copias/mL), a la cuantía de ADN proviral inicial (mayor progresión si es mayor de 3,4 log10 copias/millón de células mononucleares en sangre periférica), a la infección por virus X4 o con tropismo D/M, a las infecciones por más de un virus VIH-1 y al perfil genético de los individuos infectados.
A la fase aguda le sigue una fase asintomática, de duración variable pero raramente inferior a 18 meses, durante la que el VIH continúa replicándose en diferentes compartimentos orgánicos, contrarrestando la respuesta inmunológica natural del organismo y provocando un estado inflamatorio de carácter crónico. Una pequeña proporción de pacientes son capaces de mantener una viremia indetectable durante años, lo que parece indicar que en ellos funciona adecuadamente el sistema inmunológico y son capaces de enfrentarse eficazmente al VIH a través de mecanismos estrictamente fisiológicos pero todavía desconocidos. Sin embargo, en la gran mayoría de los pacientes infectados por el VIH, a lo largo del periodo asintomático se va produciendo una progresiva reducción de los recuentos de linfocitos T CD4+ y de la eficacia del sistema inmunológico. Cuando se ha alcanzado un grado significativo de deterioro del tejido linfoide, fruto de la replicación viral y del estado inflamatorio crónico, la replicación viral vuelve a crecer y la difusión del VIH se generaliza por todo el organismo, dando lugar a recuentos de linfocitos T CD4+ por debajo de 200/μL, momento a partir del cual se considera la aparición del sida propiamente dicho, en el que el riesgo de nuevas infecciones, reactivación de infecciones latentes, aparición de infecciones oportunistas, o de algunas neoplasias es muy elevado y que, en última instancia, acabará provocando la muerte del paciente si no se adoptan las medidas adecuadas (Cuéllar, 2014).
Los microorganismos oportunistas causantes de infecciones más frecuentemente asociados a la inmunodeficiencia provocada por el VIH son varias especies de Mycobacterium (M. tuberculosis, complejo avium-intracellulare, etc.), Pneumocystis jiroveci (antes P. carinii), Candida albicans, Herpes simple y zóster, Citomegalovirus, Criptosporidium, Giardia, Isospora, etc. (Figura 3). En cuanto a las neoplasias emergentes como consecuencia del fracaso inmunológico, las más comúnmente relacionadas con el VIH son el sarcoma de Kaposi, ciertos linfomas y otros trastornos linfoproliferativos (como la leucoencefalopatía multifocal progresiva: LMP) ligados infecciones oportunistas por el virus del herpes humano de tipo 8 (VHH-8). No es infrecuente el desarrollo de una encefalopatía progresiva, a veces inducida por el propio VIH (que es capaz de infectar y replicarse en las células de la microglía, en el SNC).
En los pacientes infectados por el VIH se observan con frecuencia distintos síndromes clínicos (afecciones oculares, renales, hematológicas, alteraciones endocrinas y del metabolismo, neurológicas, gastrointestinales, pulmonares, adenopatías, síndrome febril, anorexia, afecciones cardiacas y manifestaciones reumatológicas) de diferente etiología, ya sean relacionados con la propia infección, con la aparición de nuevas infecciones o con el tratamiento antirretroviral. En definitiva, se estima que, en ausencia de tratamiento, el tiempo medio transcurrido entre la infección y la muerte del paciente es de 11 años.
El objetivo esencial de la terapia antirretroviral (TAR) es maximizar la supresión permanente de la replicación del VIH, con el fin de limitar el desarrollo de resistencia viral y de restaurar la función inmunológica, reduciendo con ello la morbilidad y la mortalidad asociadas, mejorando la calidad de vida del paciente y previniendo la transmisión del VIH. En este sentido, el estándar terapéutico de supresión de la replicación viral es aquel que asegure al máximo una carga viral plasmática (CVP) estable por debajo de 50 copias/ml.
Actualmente, según el Documento de Consenso Gesida/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana (Berenguer, 2013) el inicio del tratamiento antirretroviral debe basarse en las manifestaciones clínicas, el número de linfocitos CD4+, la carga viral plasmática (CVP) y la presencia de otras patologías concomitantes.
En el caso de infección sintomática, se recomienda iniciar el tratamiento antirretroviral en todos los casos, pero en caso de infección asintomática el inicio del tratamiento está justificado:
- Cuando el nivel de linfocitos CD4+ sea inferior a 500 células/μL.
- En pacientes con determinadas comorbilidades (cirrosis hepática, hepatitis crónica por VHC, CVP >105 copias/mL, proporción de linfocitos CD4+ inferior al 14%, edad ≥55 años, riesgo cardiovascular elevado y trastornos neurocognitivos, etc.), incluso aunque el número de linfocitos CD4+ sea mayor de 500 células/μL.
Asimismo, en pacientes asintomáticos e independientemente del recuento de linfocitos CD4+, se recomienda el inicio del tratamiento antirretroviral en:
- Parejas serodiscordantes con alto riesgo de transmisión por vía sexual. En ningún caso ello debe suponer la abstención de otras medidas para impedir la transmisión del VIH.
- En mujeres gestantes, para prevenir la transmisión materno fetal.
- En la nefropatía VIH.
- En la hepatitis B que requiere tratamiento.
Los fármacos antirretrovirales disponibles actualmente son:
- Inhibidores de la transcriptasa inversa
- a. Inhibidores de la transcriptasa inversa análogos de nucleósidos o de nucleótidos (ITIAN): Fue el primer grupo disponible de fármacos activos contra VIH. Los primeros eran análogos de los nucleótidos naturales (ITIAN, inhibidores de la transcriptasa inversa análogos de nucléosidos o nucleótidos), con capacidad de impedir la síntesis de DNA viral a partir de la cadena de ARN infectante. Actúan como finalizadores de cadena: la transcriptasa inversa los incorpora como eslabones en la cadena de ADN en formación, pero es incapaz de unir a ellos el eslabón siguiente. Obstaculizan así la incorporación del ADN viral a la dotación genética de la célula infectada. La zidovudina fue el primer medicamento antirretroviral, es el más experimentado y se considera todavía la base del tratamiento. Como consideración muy general, hay dos perfiles distintos de efectos adversos dentro del grupo: la zidovudina y (con menor intensidad) la lamivudina tienen toxicidad hematológica (anemia y neutropenia) y gastrointestinal (náuseas). Didanosina y estavudina presentan, en grados diferentes, riesgo de neuropatía periférica y pancreatitis. Por su parte, abacavir produce reacciones hipersensibilidad en un 3% de los pacientes. El tenofovir fue el primero de una subclase de los ITIAN, los derivados nucleotídicos (a diferencia de los nucleosídicos), que presenta la peculiaridad de acortar el proceso bioquímico intracelular de fosforilación (un paso imprescindible para la activación farmacológica de estos medicamentos), además de facilitar su paso a través de las membranas celulares. La emtricitabina presenta un marcado parecido con la lamivudina, estructural, farmacológico y clínico.
- b. Inhibidores de la transcriptasa inversa no nucleósidos (ITINN): Su estructura química está muy alejada a la de los ITIAN. Los fármacos de este grupo que están actualmente disponibles son nevirapina, efavirenz, etravirina y rilpivirina. No parecen presentar resistencia cruzada con los derivados nucleosídicos (zidovudina, especialmente), aunque la resistencia frente a la propia nevirapina aparece de forma relativamente rápida, salvo que se utilice asociada con otros antirretrovirales (dos es el número mínimo recomendado). Los inhibidores no nucleosídicos de la transcriptasa inversa se unen al enzima en una zona relacionada aunque diferente de la utilizada por los ITIAN.
- Inhibidores de la proteasa (IP): Interfieren una etapa vital en el ensamblaje de nuevos viriones y la diseminación del virus. Una particularidad de la dotación genética del VIH es codificar muchas de sus proteínas vitales como precursores que tienen que ser recortados tras la síntesis para obtener las cadenas proteicas útiles. El corte de las cadenas precursoras está catalizado por una proteasa específica. Entre los componentes virales que no se forman sin la proteasa están la propia proteasa, la transcriptasa inversa y varias proteínas estructurales. Los inhibidores de la proteasa imitan la estructura química de los puntos de la cadena peptídica donde el enzima produce los cortes, bloqueando así la acción. La potencia antiviral es superior a la de los inhibidores de la transcriptasa inversa, pero son también muy susceptibles a las resistencias. Actualmente, están disponibles en España indinavir, ritonavir, saquinavir, nelfinavir, fosamprenavir, lopinavir, tipranavir y darunavir.
- Inhibidores de la fusión (IF). La enfuvirtida ha sido el primer agente de este grupo en recibir la autorización de comercialización. Actúa bloqueando la penetración del VIH-1 en los linfocitos T CD4+, al inhibir el proceso por el que la cubierta viral se funde con la membrana de los linfocitos, impidiendo la penetración viral en las células diana para el VIH en el sistema inmunológico humano. El mecanismo específico transcurre a través de la asociación con la subunidad glucoprotéica gp41 del VIH-1. Esta asociación impide el cambio conformacional requerido en la gp41 para fusionarse con la membrana de los linfocitos T CD4+.
- Inhibidores de la integrasa (InInt): El grupo está formado actualmente por raltegravir, elvitegravir y dolutegravir, cuya característica común es ser inhibidores de la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Este proceso de integración genómica es indispensable para la replicación viral, dado que ésta depende de la maquinaria celular de duplicación del ADN, y se produce en cuatro pasos consecutivos: ensamblaje, procesamiento en 3′, transferencia de hebras y reparación. De estos cuatros pasos, los tres primeros son catalizados por la integrasa. Los fármacos de este grupo actúan específicamente inhibiendo el tercer paso, la transferencia de hebras de la integrasa.
- Inhibidores del co-receptor CCR5. Maraviroc es el único fármaco comercializado en España que actúa inhibiendo el co-receptor humano de quimiocinas CCR5, impidiendo con ello la penetración del VIH con tropismo CCR5 (virus R5). Maraviroc carece de actividad frente a VIH con tropismo CXCR4 (virus X4) o con tropismo dual CCR5/CXCR4. Estos co-receptores son requeridos, junto con el CD4, para facilitar la unión de la membrana celular con la cápside del VIH y, en consecuencia, la penetración del contenido del virus en el citoplasma de la célula infectada. Las cepas que infectan a monocitos y macrófagos, que son las primeras seleccionadas por el VIH, son R5 o monocitotropas; después el virus evoluciona cambiando su tropismo hacia un tropismo CXC4 (X4), que son las que infectan preferentemente a los linfocitos. Maraviroc se une a la región proteica del co-receptor CCR5 exterior a la membrana, provocando un cambio estable de su conformación y haciéndola inasequible para las quimiocinas del VIH. Por tanto, se trata de una forma de inhibición alostérica no competitiva. De esta manera, el VIH con tropismo CCR5 no es capaz de actuar sobre tal co-receptor, impidiendo la unión del VIH a la célula y, consecuentemente, la infección por el VIH.
El tratamiento de elección de la infección por el VIH-1 en el momento actual consiste en una combinación de al menos tres fármacos que incluyan dos ITIAN asociado a un IP/r, un ITINN o un InInt. Con la mayoría de estas combinaciones se puede conseguir una CVP <50 copias/mL en más del 70% de los casos a las 48 semanas. La elección de una u otra familia se realiza según sus potenciales ventajas, entre otras:
- Interacciones farmacológicas (de menos a más): InInt, ITINN, IP/r;
- Mayor barrera genética: IP/r;
- Menor coste: ITINN (además el momento idóneo del uso de los ITINN de primera generación es el tratamiento inicial, ya que en las pautas de rescate tienen menos actividad que otras familias de FAR).
En resumen, puede utilizarse la combinación de 2 ITIAN + 1 ITINN, 2 ITIAN + 1 IP/r ó 2 ITIAN + InInt como tratamiento de inicio. Una pauta con dos ITIAN (preferentemente tenofovir/emtricitabina) y un inInt (raltegravir, dolutegravir o elvitegravir) tendría la ventaja de una mayor concentración en las secreciones genitales y podría reducir más rápidamente la CVP durante las primeras 4-8 semanas en comparación con los IP o ITINN, lo que podría facilitar la reducción de la transmisión del VIH. En cualquier caso, se debe efectuar siempre una prueba de resistencias y un tropismo viral al diagnóstico de la infección aguda o reciente, se vaya a iniciar el tratamiento o no. Si no se dispone del resultado del estudio de resistencias es preferible comenzar con una pauta basada en un IP/r hasta tener los resultados. La monoterapia con un IP/r no se recomienda como tratamiento de inicio, como tampoco se deben usar pautas sin un ITIAN. Lo que es fundamental considerar es que si se inicia el tratamiento antirretroviral, éste debe administrarse por tiempo indefinido y en aquellos que no estén incluidos en los anteriores criterios, se recomienda reevaluarlos a partir de los 6 meses, cuando la infección pasa a ser crónica.
Asimismo, las combinaciones de tres fármacos constituye el tratamiento de inicio de elección de la infección crónica por el VIH. El tratamiento antirretroviral es recomendado siempre en los pacientes sintomáticos, en las embarazadas, en las parejas serodiscordantes con alto riesgo de transmisión, en la hepatitis B que requiera tratamiento y en la nefropatía relacionada con el VIH. En los pacientes asintomáticos el inicio del tratamiento depende en la cifra de linfocitos CD4+, la carga viral plasmática, la edad y las comorbilidades del paciente. El esquema terapéutico debe incluir dos inhibidores de la transcriptasa inversa análogos de nucleósido o nucleótido y un tercer fármaco (inhibidor de la transcriptasa inversa no nucleósido, inhibidor de la proteasa potenciado o inhibidor de la integrasa). Se han seleccionado por consenso combinaciones concretas de fármacos, en concreto, las combinaciones de ITIAN de elección para regímenes de inicio son tenofovir/emtricitabina (TDF/FTC) o abacavir/lamivudina (ABC/3TC), recomendándose el uso de fármacos coformulados en un mismo medicamento.
Debe tenerse presente, no obstante, que la aparición de resistencias es un fenómeno inevitable cuando el VIH continúa replicando bajo presión selectiva de fármacos. La detección de las mutaciones de resistencias por métodos genotípicos es muy útil en el fracaso virológico. Las opciones terapéuticas tras el fracaso virológico son limitadas, pero actualmente puede conseguirse el objetivo de una CVP indetectable. Afortunadamente, la toxicidad es un factor cada vez menos limitante del tratamiento.
ACCIÓN Y MECANISMO
Dolutegravir es un agente antiviral directo que actúa inhibiendo la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Ha sido autorizado para el tratamiento de adultos y adolescentes mayores de 12 años infectados por el Virus de Inmunodeficiencia Humana (VIH), en combinación con otros medicamentos antirretrovirales.
El proceso de integración genómica es indispensable para la replicación viral, dado que ésta depende de la maquinaria celular de duplicación del ADN, y se produce en cuatro pasos consecutivos: ensamblaje, procesamiento en 3′, transferencia de hebras y reparación; de ellos, los tres primeros son catalizados por la integrasa y el dolutegravir actúa específicamente inhibiendo el tercer paso, la transferencia de hebras de la integrasa, con una concentración inhibitoria para el 50% (IC50%) de 0,2 a 2 nM, frente a valores de 7-10 nM para elvitegravir.
El dolutegravir presenta una elevada barrera frente al desarrollo de resistencia primaria por las cepas clínicas de VIH, hasta el punto de que, por el momento, no se haya descrito la emergencia de cepas resistentes en pacientes que previamente no habían tratados con inhibidores de la integrasa. La presencia de la mutación que conduce al cambios en la secuencia peptídica de la integrasa, por sustitución del aminoácido glutamina (Q, Gln) por otro (generalmente, arginina, R, Arg) en la posición 148 (Q148R), puede reducir sustancialmente la susceptibilidad al dolutegravir, aunque solo si va acompañada de al menos otras dos mutaciones secundarias (G140A/C/S, E138A/K/T o L74I); de hecho, por sí sola no afecta significativamente a la susceptibilidad del VIH al dolutegravir (la mutación Q148R está presente en el 40% de las cepas aisladas de pacientes resistente al raltegravir). Como ocurre con otros inhibidores de la integrasa, la administración conjunta de dolutegravir con otros antirretrovirales reduce la aparición de cepas de VIH resistentes al fármaco.
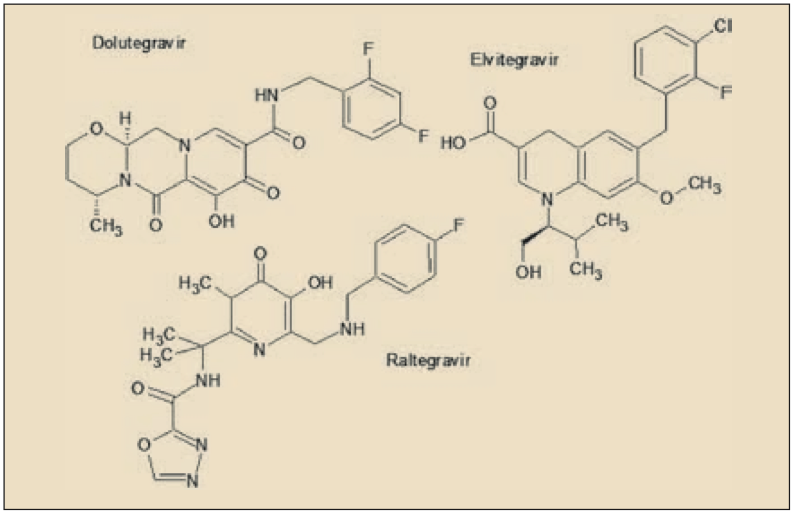{kind=link}
ASPECTOS MOLECULARES
El dolutegravir está estrechamente relacionado con sus antecesores farmacológicos, raltegravir y elvitegravir. Como estos, presenta una estructura pseudopeptídica que emula químicamente una fracción proteica y con ello es capaz de actuar de señuelo bioquímico para la integrasa del VIH, conduciendo a su bloqueo.
En términos químicos (denominación IUPAC), el dolutegravir es la (4R,12aS)-N-[(2,4-difluorofenil)metil]-7-hidroxi-4-metil-6,8-dioxo-3,4,12,12a-tetrahidro-2H-pirido[5,6]pirazino[2,6-b][1,3]oxazina-9-carboxamida
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del dolutegravir en la indicación autorizada han sido adecuadamente contrastadas mediante cinco ensayos clínicos principales (pivotales) de fase 3, tres de ellos controlados con comparadores activos y otros dos abiertos. Dos de los estudios controlados (SPRING-2 y SINGLE) y uno de los abiertos (FLAMINGO) se llevaron a cabo con pacientes no tratados previamente (naïve), mientras que los dos restantes (SAILING, controlado, y VIKING-3, abierto) lo fueron con pacientes insatisfactoriamente tratados anteriormente para la infección por VIH.
Pacientes naïve
El estudio SPRING-2 (ING113086; Raffi, 2013) es un estudio multinacional (Europa, Rusia, Estados Unidos, Canadá y Australia), aleatorizado, doblemente ciego y controlado con un comparador activo, en el que se comparó un régimen antiretroviral conteniendo dolutegravir frente a otro con raltegravir. El estudio fue realizado con 822 pacientes adultos con infección por VIH y con una carga viral sérica ≥ 1.000 copias/ml de ARN de VIH que no habían sido tratados con anterioridad por este motivo; el 86% eran varones, un 85% blancos, con una mediana de edad de 36 años, una carga viral media de 4,55 log10 (35.500) copias/ml y con un 28% de los pacientes con más de 100.000 (5 log10).
Los pacientes recibieron por vía oral dolutegravir (50 mg/24 h) o raltegravir (400 mg/12 h), junto con un tratamiento de base con tenofovir y emtricitabina o de abacavir y lamivudina, a criterio del investigador. Como variable primaria de eficacia se determinó el porcentaje de pacientes con una carga viral inferior a 50 copias/ml (1,7 log10) al final de 96 semanas de tratamiento.
Los resultados mostraron unas tasas del 81% con la combinación de dolutegravir vs. 76% con la de raltegravir, con una diferencia ajustada de 4,5 puntos (IC95% -1,1 a 10,0), mostrando la no inferioridad del dolutegravir con respecto al raltegravir, Se produjo un fallo en la respuesta virológica en el 5% vs. 10% y la mediana del incremento observado en el recuento de linfocitos CD4+ fue de 276 células/mm3 vs. 264.
El estudio SINGLE (ING114467; Walmsley, 2013) es un estudio multinacional (Europa, Estados Unidos, Canadá y Australia), aleatorizado, doblemente ciego y controlado con comparador activo, en el que se comparó un régimen antiretroviral conteniendo dolutegravir frente a efavirenz/tenofovir/emtricitabina. El estudio fue realizado con 833 pacientes adultos con infección por VIH y con una carga viral sérica ≥ 1.000 copias/ml de ARN de VIH que no habían sido tratados con anterioridad; el 84% eran varones, un 68% blancos (y un 24% negros), con una mediana de edad de 35 años, una carga viral media de 4,55 log10 (35.500) copias/ml y con un 32% de los pacientes con más de 100.000 (5 log10). Los pacientes recibieron por vía oral dolutegravir (50 mg/24 h) junto con abacavir y lamivudina, o un comprimido conteniendo efavirenz, tenofovir y emtricitabina.
Los resultados a las 48 semanas de tratamiento mostraron unas tasas del 88% con la combinación de dolutegravir vs. 81% con efavirenz/tenofovir/emtricitabina, con una diferencia ajustada de 7,4 puntos (IC95% 2,5 a 12,3), mostrando la superioridad del régimen de dolutegravir. Estas diferencias fueron de 83 vs. 76% en los pacientes con carga viral inicial mayor de 100.000 copias (5 log10) y de 90 vs 85% en aquellos con ≤ 100.000 copias/ml. La mediana de tiempo transcurrido hasta la supresión viral fue de 28 vs. 84 días (p< 0,001) y el aumento del recuento de linfocitos CD4+ fue de 267 vs. 208 (p< 0,001).
El estudio FLAMINGO (Clotet, 2014) es un estudio abierto, multinacional, en el que se comparó dolutegravir (50 mg/24 h) frente a darunavir/ritonavir (800/100 mg/24 h), junto con un tratamiento de base con tenofovir y emtricitabina o de abacavir y lamivudina, a criterio del investigador. El estudio fue realizado con 484 pacientes adultos con infección por VIH y con una carga viral sérica ≥ 1.000 copias/ml de ARN de VIH que no habían sido tratados con anterioridad. Los resultados a las 48 semanas de tratamiento mostraron unas tasas del 90% con la combinación de dolutegravir vs. 83% con la de darunavir/ritonavir, con una diferencia ajustada de 7,1 puntos (IC95% 0,9 a 13,2; p= 0,025), mostrando la superioridad del régimen de dolutegravir.
Pacientes con fracasos antirretrovirales previos, pero no tratados con inhibidores de integrasa
El estudio SAILING (ING111762; Cahn, 2013) es un estudio multinacional, aleatorizado, doblemente ciego y controlado con un comparador activo, en el que se comparó un régimen antiretroviral conteniendo dolutegravir frente a otro con raltegravir. El estudio fue realizado con 715 pacientes adultos con infección por VIH y con una carga viral sérica ≥ 400 copias/ml de ARN de VIH que habían sido tratados con antirretrovirales pero no de la clase de los inhibidores de integrasa; el 68% eran varones, un 50% blancos, con una mediana de edad de 43 años y un 16% coinfectados con hepatitis C.
Los pacientes recibieron por vía oral dolutegravir (50 mg/24 h) o raltegravir (400 mg/12 h), junto con un tratamiento de base con tenofovir y emtricitabina o de abacavir y lamivudina, a criterio del investigador. Los resultados mostraron unas tasas del 71% con la combinación de dolutegravir vs. 64% con la de raltegravir, con una diferencia ajustada de 7,4 puntos (IC95% 0,7 a 14,2; p= 0,03), mostrando la superioridad del dolutegravir sobre raltegravir. Estas diferencias fueron de 62 vs. 47% en los pacientes con carga viral inicial mayor de 50.000 copias (4,7 log10) y de 75 vs 71% en aquellos con ≤ 50.000 copias/ml. Se produjo un fallo en la respuesta virológica en el 1,1% vs. 4,7 %, con una diferencia ajustada de -3,7 puntos (IC95% -6,1 a -1,2; p= 0,003).
Pacientes con fracasos antirretrovirales previos incluyendo inhibidores de integrasa
El estudio VIKING-3 (ING112574; Castagna, 2014) es un estudio multinacional, abierto, de un único brazo de tratamiento, en el que se estudió un régimen antiretroviral conteniendo dolutegravir en pacientes insatisfactoriamente tratados con combinaciones incluyendo a otros inhibidores de la integrasa. El estudio fue realizado con 183 pacientes adultos con infección por VIH y evidencia genotípica de reistencia a raltegravir y/o elvitegravir, y con una carga viral sérica ≥ 500 copias/ml; el 77% eran varones y con una mediana de edad de 48 años.
Los pacientes recibieron por vía oral dolutegravir (50 mg/12 h) junto con un tratamiento de base previo, excluyendo al raltegravir o elvitegravir. Los resultados mostraron una variación de la carga viral el 8º día del tratamiento de -1,43 log10 (IC95% -1,3 a -1,5; p= 0,03); un 69% de los pacientes redujeron su carga viral a menos de 50 copias/ml a las 24 semanas de tratamiento.
Seguridad
Los eventos adversos más comúnmente descritos en los estudios clínicos controlados con la combinación de dolutegravir con otros antirretrovirales fueron diarrea (16%), náuseas (15%) y cefalea (14%), sin diferencias sustanciales con relación a la terapia de base. El porcentaje de pacientes que suspendieron el tratamiento por la aparición de eventos adversos fue del 2%, frente a un 2-4% con raltegravir y 10% con efavirenz.
ASPECTOS INNOVADORES
Dolutegravir es un agente antiviral directo que actúa inhibiendo la integrasa del VIH, un enzima codificado por el virus que facilita la integración (inserción) del genoma del VIH en el genoma de la célula huésped (linfocitos T). Está relacionado química y farmacológicamente con raltegravir y elvitegravir, previamente registrados. Ha sido autorizado para el tratamiento de adultos y adolescentes mayores de 12 años infectados por el Virus de Inmunodeficiencia Humana (VIH), en combinación con otros medicamentos antirretrovirales.
Los datos clínicos disponibles proceden de amplios y rigurosos ensayos clínicos, confirmando que las combinaciones con dolutegravir no son inferiores a las de raltegravir y elvitegravir en cuanto a eficacia virológica y clínica, y son superiores al efavirenz/tenofovir/emtricitabina y a las combinaciones incluyendo darunavir/ritonavir, en pacientes no tratados previamente. En cuanto a aquellos insatisfactoriamente tratados anteriormente, los que no había recibido un inhibidor de la integrasa, el dolutegravir fue superior estadísticamente al raltegavir, e incluso mostró una notable respuesta virológica (69%) en pacientes insatisfactoriamente tratados con combinaciones incluyendo raltegravir o elvitegravir.
Desde el punto de vista de la seguridad, el dolutegravir presenta un perfil aceptable, con una baja tasa de suspensiones del tratamiento por motivos toxicológicos. Los eventos más comunes son náuseas, diarrea y cefalea.
Un amplio meta-análisis (Messiaen, 2013) ha venido a concluir que la incorporación de fármacos de la clase de los inhibidores de la integrasa como tratamiento de primera línea de la infección por VIH da lugar a mejores resultados que otros grupos ya establecidos en la terapéutica, como los inhibidores de la proteasa (indinavir, etc.). Sin embargo, la aparición de la resistencia a los primeros inhibidores de la integrasa (raltegravir y elvitegravir) ha sido relativamente rápida. En particular, la mutación Q148R y otras mutaciones Q148 aparecen en el 40% en los VIH aislados de pacientes con fracaso virológico con raltegravir.
Por el contrario, el dolutegravir presenta una elevada barrera frente a las mutaciones del VIH en pacientes previamente no tratados con esta clase de antirretrovirales. Por ello, cuando se usa como parte de un tratamiento de primera línea, el dolutegravir es uno de los pocos antirretrovirales que no parecen producir mutaciones clínicamente relevantes. Esta mayor capacidad frente a las mutaciones responsables de la resistencia del VIH parece que podría deberse a la mayor duración e intensidad del bloqueo de la integrasa, así como por la mayor limitación de la capacidad de replicación de las cepas de VIH que podrían hacerse resistentes al dolutegavir (Mesplede, 2014). Sin duda, el dolutegravir supone una cierta evolución en el relativamente reciente grupo de los inhibidores de la integrasa del VIH.
Además, su perfil farmacocinético es más favorable que el de sus antecesores de este grupo, con una semivida celular prolongada que facilita una única administración diaria, sin necesidad de bloqueantes enzimáticos adicionales (como el cobicistat, en el caso del elvitegravir) y además es metabolizado mayoritariamente por una vía ajena a los isoenzimas del citocromo p4501, lo que limita notablemente el número y la relevancia de las interacciones con otros fármacos, incluyendo la mayoría de los antirretrovirales (Fantauzzi, 2014). Todo ello, unido a la utilidad en pacientes que previamente habían fracasado con raltegravir o elvitegravir, le aporta un significativo valor añadido y viene a consolidar la relevancia de los inhibidores de la integrasa como fármacos de primera consideración en el tratamiento de la infección por VIH.
|
VALORACIÓN |
|
|---|---|
|
Dolutegravir ► TIVICAY® (GlaxoSmithKline) |
|
|
Grupo Terapéutico (ATC): JO5AX. TERAPIA ANTIINFECCIOSA SISTÉMICA. Antivirales de acción directa: otros |
|
|
Indicaciones autorizadas: Tratamiento de adultos y adolescentes mayores de 12 años infectados por el Virus de Inmunodeficiencia Humana (VIH), en combinación con otros medicamentos antirretrovirales. |
|
|
Valoración global: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad clínica: Posibilidad de asociar con otros tratamientos actualmente en vigor, utilidad en cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
⇑ |
|
Novedad físico-química: Mejora las características farmacocinéticas, con incidencia en las condiciones de uso. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Raltegravir |
Isentress |
MerckSharpDohme |
2008 |
|
Elvitegravir |
Stribild** |
Gilead |
2013 |
|
Dolutegravir |
Tivicay |
GlaxoSmithKline |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
** Asociado a cobicistat, emtricitabina y tenofovir.
Bibliografía
Vacuna meningocócica del grupo B BEXSERO® (Novartis)
ENFERMEDAD MENINGOCÓCICA INVASIVA
Las bacterias que más frecuentemente pueden causar actualmente meningitis son Neisseria meningitidis (meningococo) y Streptococcus pneumoniae (neumococo). Neisseria meningitidis es un coco Gram negativo que incluye 13 serogrupos (A, B, C, D, H, I, K, L, X, Y, Z, 29-E y W-135), aunque los que se asocian con mayor frecuencia a enfermedad son A, B, C, W-135 e Y. En la Unión Europea, el 74% de los cuadros meningocócicos invasivos son causados por el serotipo B, especialmente en niños (88% de los casos); otro 14% (8,1% de los niños) está producido por el serotipo C.
Bexsero® es una vacuna combinada de varias proteínas procedentes del serotipo B de la Neisseria meningitidis indicada para la inmunización activa de individuos a partir de 2 meses de edad frente a la enfermedad meningocócica invasiva causada por Neisseria meningitidis grupo B. Los resultados serológicos sobre la capacidad inmunogénica de la nueva vacuna son claros y contundentes, con porcentajes de sujetos con títulos preventivos de anticuerpos por encima del 98% medidos un mes después de la última dosis. El perfil de seguridad de la vacuna es similar al de otras vacunas convencionales.
La enfermedad invasiva por meningococo B es un cuadro grave, con una mortalidad asociada superior al 8%, que afecta fundamentalmente a los niños pequeños, con un segundo pico de incidencia durante la adolescencia, y hasta ahora no existía ninguna vacuna eficaz para su prevención. Por ello, la vacuna está claramente dirigida para estos grupos de edad; no obstante, en caso de focos epidémicos es deseable vacunar a toda la población. Al hecho de ser la primera vacuna capaz de inmunizar frente al serotipo B del meningococo, que es el más prevalente en España y en el resto de la Unión Europea, hay que agregar que se trata de la primera vacuna eficaz que no utiliza el sistema de conjugación de proteínas y polisacáridos capsulares empleado con tanto éxito para prevenir meningitis y septicemias de otros serotipos de meningococos (A, C, W, Y), de neumococos (13 serotipos) y de Heamophillus influenzae tipo b. Esto último también supone un avance farmacotécnico nada desdeñable, que abre nuevas líneas de investigación para el desarrollo de vacunas frente a bacterias y otros microorganismos patológicamente relevantes.
Las bacterias que más frecuentemente pueden causar actualmente meningitis son Neisseria meningitidis (meningococo) y Streptococcus pneumoniae (neumococo); por el contrario, hace años el Haemophilus influenzae tipo B era la causa más común de meningitis en niños menores de un mes (raramente en adultos), pero la vacunación sistemática ha reducido drásticamente su incidencia. Por su parte, la meningitis producida por bacterias gramnegativas suele ser producida por Escherichia coli o especies de Klebsiella o Enterobacter y es más común en pacientes inmunodeprimidos, tras cirugía o traumatismo cráneo-encefálico, o con bacteriemia. Por su parte, la meningitis estafilocócica puede ser secundaria a traumatismos cráneo-encefálicos penetrantes, bacteriemia o intervenciones neuroquirúrgicas. Los casos provocados por Listeria puede darse a cualquier edad, especialmente en pacientes con insuficiencia renal crónica, enfermedades hepáticas, trasplantados e inmunodepreimidos.
Neisseria meningitidis es un coco Gram negativo que incluye 13 serogrupos (A, B, C, D, H, I, K, L, X, Y, Z, 29-E y W-135), aunque los que se asocian con mayor frecuencia a enfermedad son A, B, C, W-135 e Y. La clasificación serológica se basa en la composición de la cápsula que recubre la membrana externa que, junto con las proteínas principales de esta última, constituyen los componentes antigénicos principales. La cápsula está constituida por un lipooligosacárido que interviene activamente en la patogenia de la enfermedad meningocócica, ya que actúa como una auténtica endotoxina.
La infección meningocócica invasiva se observa en todo el mundo y su reservorio es exclusivamente humano, provocando alrededor de 1,2 millones de casos anualmente. El mecanismo de transmisión es directo y con mayor frecuencia se produce de un portador asintomático a un individuo susceptible, que a partir de un caso clínico. El contacto indirecto es muy improbable debido a la labilidad del meningococo, muy sensible a la desecación y a otros agentes externos.
En la Unión Europea, los últimos datos epidemiológicos publicados (ECDC, 2013) indican el registro de 3.808 casos de enfermedad meningocócica invasiva en los 29 países de la Unión Europea durante 2011, con una tasa de notificación de 0,77 casos por 100.000 habitantes, que en la mayoría de los países está por debajo de 1:100.000, con una distribución claramente estacional de casos y un pico máximo en enero. Los niños son los más afectados (12,3 casos por 100.000 niños), seguido de adolescentes y jóvenes (15-25 años).
En la Unión Europea, el 74% de los cuadros meningocócicos invasivos son causados por el serotipo B, especialmente en niños (88% de los casos); otro 14% (8,1% de los niños) está producido por el serotipo C. Actualmente, se aprecia un ligero repunte de los cuadros relacionados con el serotipo Y, aunque su incidencia sigue siendo muy baja. Por lo que se refiere a la forma de presentación clínica del cuadro invasivo meningocócico, el 43% fue la meningitis1. En cuanto a la tasa de mortalidad por enfermedad meningocócica invasiva en la Unión Europea, se sitúa en torno al 8%, sin que parezca existir relación entre ésta y el serotipo del meningococo causante; en cualquier caso, la mortalidad suele ser mayor entre los pacientes ancianos y en aquellos con presentación clínica en forma de septicemia.
La meningitis meningocócica tiene un carácter endémico, intercalándose cada cierto tiempo (5-10 años) como brotes epidémicos. Desde 1979 (en que la incidencia ascendió a 17 casos/100.000 habitantes) no se ha producido en España ningún brote agudo que pueda ser calificado realmente como epidémico.
El serogrupo más frecuente en España es también el B, aunque la menor circulación del serogrupo B a principio de la década de los 90 del siglo pasado fue interpretada erróneamente como aumento del serogrupo C. Con la generalización de la vacuna frente al meningococo C y de los otros grupos (salvo el B) y el consecuente descenso de su incidencia, actualmente la enfermedad meningocócica por el serogrupo B es, con gran diferencia, la causa más frecuente de meningitis y es responsable de 7 de cada 10 casos de meningitis en España.
Las cepas de Neisseria meningitidis pueden poseer pequeños filamentos (pili) y prolongaciones no flagelares de la membrana externa que se expanden hacia el exterior, los cuales favorecen la adhesión de la bacteria a las células del epitelio y, con ello, su infectividad. También se encuentra un lipooligosacárido, que actúa como una endotoxina, como componente de la membrana externa que es determinante para la virulencia de las cepas de meningococo.
Los meningococos se encuentran en la nasofaringe del 5-20% de la población española, dependiendo de la edad y otras variables, y se diseminan a través de las gotitas respiratorias y el contacto cercano. A pesar de su amplia distribución y por razones aún no bien comprendidas, sólo una pequeña fracción (menos del 1%) de los portadores acaba desarrollando la enfermedad invasiva (meningitis o septicemia). Una vez que estos microorganismos se depositan sobre la superficie de una mucosa, se produce una adherencia a la superficie de la célula eucariótica, la ingestión por la célula, el transporte el interior de fagosomas, la eyección a la submucosa y, ocasionalmente, la invasión del torrente sanguíneo.
Los meningococos carentes de cápsula se adhieren mejor a las células epiteliales que los capsulados, y tanto éstos como los no capsulados se adhieren con mayor facilidad a la mucosa faríngea que a la bucal. Además del grado de encapsulación y la presencia de pili, intervienen otros factores en el estado de portador meningocócico, por ejemplo, ciertas co-infecciones víricas (como la gripe) o el consumo de tabaco, que favorecen la colonización faríngea por parte del meningococo. En el torrente sanguíneo, la cápsula bacteriana resiste el ataque de los neutrófilos, de las células del sistema reticuloendotelial y de la vía clásica del complemento. Los receptores de los pili y otros componentes de la superficie bacteriana en los plexos coroideos (un lugar de inflamación precoz en el sistema nervioso central) facilitan la penetración en el espacio subaracnoideo.
La infección neurológica se desarrolla debido a los niveles relativamente bajos de anticuerpos, complemento y leucocitos existentes en el líquido céfalo-raquídeo (LCR). Los componentes de la superficie bacteriana, el sistema del complemento y las citocinas inflamatorias atraen a los neutrófilos hacia el espacio subaracnoideo y se produce el exudado inflamatorio que, al crecer, lesiona los pares craneales, destruye las vías de circulación del LCR (causando hidrocefalia) e induce una vasculitis y tromboflebitis (produciendo isquemia). Las prostaglandinas y las citocinas generados por la lesión de las membranas celulares y la rotura de la barrera hematoencefálica inducen edema cerebral, agravado posteriormente por la isquemia cerebral y el síndrome de secreción inadecuada de hormona antidiurética. La presión intracraneal aumenta, la presión arterial baja (causando shock séptico) y el paciente puede fallecer a causa de las complicaciones sistémicas o de la isquemia cerebral masiva.
Como se ha indicado, la enfermedad meningocócica afecta sobre todo a los niños, aunque puede aparecer en cualquier edad. En general predomina en los niños entre 3 meses y 1 año de edad. En España, el 50 % de los casos se dan en niños menores de 5 años, y en un porcentaje muy significativo (15-25 %) en menores de 1 año. A medida que la edad del niño aumenta, se incrementa la posibilidad de que su infección se limite la nasofaringe, quedando únicamente como portador. También suele presentarse en epidemias en poblaciones cerradas (cuarteles, residencias, etc.).
Los antibacterianos modernos y las medidas de soporte iniciados precozmente han reducido la tasa de mortalidad de la meningitis bacteriana aguda a menos del 10%; sin embargo, la meningitis tratada tardíamente y la neonatal o del anciano suelen ser mortales y los pacientes que sobreviven suelen presentar signos de lesión de los pares craneales o de infarto cerebral, convulsiones recurrentes o deterioro cognitivo.
Hace años, la vacunación antimeningocócica sólo era empleada durante las epidemias y en las poblaciones cerradas cuando se sospechaba una extensión epidémica; además, los miembros de la familia, el personal sanitario y otros contactos cercanos con un paciente infectado deben recibir quimioprofilaxis con rifampicina durante 48 h. La cuestión era que hasta hace poca más de una década no había vacunas que cubriesen suficientemente el amplio espectro de serotipos del meningococo; por otro lado, la tecnología de las vacunas distaba de ser la más eficiente desde el punto de vista de la respuesta inmune. En el año 2000 se registró en España una vacuna antimeningocócica C de tipo conjugado, que mejoraba notablemente la capacidad preventiva, incorporándose al calendario oficial de vacunaciones. En este sentido, la mejora de la farmacotecnia de las vacunas conjugadas con proteínas (que tan espectaculares resultados ya había producido con la vacuna del Haemophillus influenzae de tipo b) permitió el desarrollo de una vacuna antimeningocócica C conjugada mucho más inmunogénica que las anteriores.
Junto con la vacuna frente al serotipo C, se dispone de otras frente al resto de los principales serotipos implicados en la mayoría de los cuadros invasivos meningocócicos, salvo para el grupo B que, lamentablemente es el más común (tanto por ser el más prevalente de forma natural, como por el hecho de que la población se haya ido seleccionando paulatinamente – al menos en los países desarrollados – al vacunarse frente al resto de los serotipos).
Hasta ahora, no había sido posible el desarrollo de una vacuna frente al serotipo B siguiendo las líneas marcadas por el resto de serotipos de meningococos. El motivo de ello es que su lipooligosacárido capsular es antigénicamente similar al de algunas moléculas fisiológicas humanas, particularmente la molécula de adhesión celular neural fetal, lo que limita notablemente su capacidad inmunogénica y facilita la formación de autoanticuerpos. Por este motivo, los investigadores han centrado sus esfuerzos en el desarrollo de vacunas de origen no capsular o, al menos, no enteramente capsular.
En este sentido, uno de los candidatos inmunogénicos alternativos investigados han sido las vesículas de la membrana externa (outer membrane vesicles, OMV); sin embargo, la protección conseguida con vacunas monovalentes es específica para cada cepa bacteriana, ya que la respuesta inmune va dirigida fundamentalmente frente a la proteína inmunodominante (PorA), muy variable entre los diferentes clones (Findlow, 2014). Por este motivo, las vacunas OMV monovalentes son poco eficaces en la prevención de la enfermedad meningocócica B en zonas donde coexistan diversas variantes antigénicas, lo cual es relativamente común; de ahí que se hayan ensayado formulaciones multivalentes (hasta 9 subserotipos OMV diferentes). Tras haber conseguido la secuenciación completa del genoma del serotipo B de Neisseria meningitidis, se estudiaron cerca de 600 proteínas con potencial antigénico producidas por esta bacteria, susceptibles de convertirse en vacunas efectivas.
ACCIÓN Y MECANISMO
Se trata de una vacuna combinada de varias proteínas procedentes del serotipo B de la Neisseria meningitidis indicada para la inmunización activa de individuos a partir de 2 meses de edad frente a la enfermedad meningocócica invasiva causada por Neisseria meningitidis grupo B.
ASPECTOS MOLECULARES
La vacuna está formada por la combinación de cuatro ingredientes inmunológicamente activos. El primero es extraído mediante la utilización de detergentes sobre las vesículas de la membrana externa (outer membrane vesicles, OMV) de la cepa NZ98/254 (PorA P1.4), y los tres restantes corresponden a sendos antígenos procedentes de la superficie celular, las proteínas 961c, 287-953 y 936-741 de Neisseria meningitidis grupo B, producidas por tecnología de ADN recombinante. Se trata de la proteína de unión al factor H (factor H binding protein, fHbp), antígeno de unión a heparina (Neisseria heparin binding antigen, NHBA) y adhesina A (Neisseria adhesión A, NadA). Adicionalmente, se han utilizado otras proteínas procedentes del genoma de Neisseria (GNA 2091 y 1030) como proteínas de fusión accesorias de fHbp y NHBA para incrementar la inmunogenicidad y la estabilidad de estas últimas.
Las principales proteínas procedentes de la membrana externa (outer membrane proteins, OMP) de la Neisseria meningitidis se clasifican en cinco clases diferentes. Las de las clases 1 (PorA), 2 (PorB2) y 3 (PorB3) son porinas específicas para determinados aniones y presentan una elevada variabilidad antigénica, aunque todas las cepas de meningococo presentan PorB2 y PorB3. Las de clase 4 (reduction-modifiable proteins, RMP) están estrechamente relacionadas con ciertas proteínas (PIII) de la Neisseria gonorrhoeae, son antigénicamente invariables y suelen asociarse a las porinas, actuando como estabilizadoras de éstas. Finalmente, la de clase 5 (Opc) es una proteína presente en el exterior de la superficie celular, donde forma trímeros o tetrámeros.
EFICACIA Y SEGURIDAD CLÍNICAS
La capacidad inmunogénica y la seguridad de la vacuna frente a la Neisseria meningitidis grupo B han sido adecuadamente contrastadas mediante dos ensayos clínicos de fase 3, multicéntricos, aleatorizados, parcialmente ciegos (para el observador) y controlados con placebo, en lactantes y niños desde dos meses de edad y en adolescentes. Las pautas posológicas utilizadas en lactantes fueron de tres dosis, siendo la primera a los dos meses de edad, seguida de otras dos a los 4 y 6 meses de edad, o a los 3 y 4 meses, más otra de recuerdo entre los 12 y 24 meses; para los lactantes no vacunados de 6 a 11 meses se utilizaron dos dosis separadas por 2 meses, con una dosis de recuerdo en el segundo año de vida; los niños de 12 a 23 meses recibieron dos dosis separadas por 2 meses, con una dosis de recuerdo 12-23 meses después de la última; los niños de 2 a 10 años recibieron dos dosis separados por dos meses y los adolescentes y adultos (≥11 años), dos dosis separadas por un mes.
El ensayo clínico principal realizado en lactantes y niños pequeños (V72P13; Vesikari, 2013) es un estudio de fase 3, parcialmente ciego, aleatorizado, multicéntrico (70 localizaciones) y controlado con placebo para evaluar la inmunogenicidad, seguridad y homogeneidad de los diferentes lotes de la vacuna recombinante frente a meningococo B, administrada sola o conjuntamente con el régimen convencional de vacunación infantil. Este estudio tuvo una extensión abierta, en la que los pacientes que finalizaron el anterior recibieron una dosis de recuerdo durante el segunda año de vida, y aquellos mayores de 6 meses no vacunados anterioremente recibieron dos dosis durante su segunda año de vida.
Los 2.481 niños incluidos en el estudio tenían entre 55 y 89 días de vida (mediana de 74,3 días), una edad gestacional de al menos 37 semanas (mediana de 39,5) y un peso al nacer de al menos 2,5 kg (mediana 3,5 kg); un 52% eran varones, 99% de raza caucásica, con una mediana de peso de 5,8 kg y de longitud de 59,5 cm al inicio del estudio. En la fase de refuerzo/recuerdo, el número de niños mayores de 12 meses fue de 2.249, con una mediana de edad de 12,3 meses, 51% varones, 10,0 kg de peso y 76,5 cm de altura.
El objetivo del estudio fue demostrar que todos los lotes ensayados eran homogéneos desde el punto de vista de la inducción de anticuerpos específicos une mes después de la tercera dosis, mediante un ensayo bactericida sérico con complemento humano (human serum bactericidal assay, hSBA) frente a las cepas vacunales ensayadas, considerándose como seroconversión a partir de títulos de 1:5 (≥1:5) para cada antígeno.
Los resultados obtenidos mostraron que la relación de los títulos de anticuerpos entre los diferentes lotes ensayados osciló entre 0,74 y 1,33 (IC95%), demostrando la homogeneidad inmunogénica. El 100% de los niños vacunados (IC95% 99 a 100) mostraron una titulación hSBA ≥1:5 para la adesina A (NadA) y proteína de unión al factor (fHbp), del 84% (IC95% 82 a 86) para la OMV de la cepa NZ98/254 (PorA P1.4) y del 84% (IC95% 75 a 91) para el antígeno de unión a heparina (NHBA).
A partir de los doce meses de edad, tras la administración de la cuarta dosis (la de recuerdo) los respectivos porcentajes fueron un mes después de su administración del 100% (NadA y fHbp), 94% para PorA P1.4 y 98% para NHBA. Doce meses después de la dosis de recuerdo, estos porcentajes habían descendido al 62% (IC95% 56 a 67) para fHbp; 97% (IC95% 95 a 99) para NadA; 17%% (IC95% 13 a 22) para PorA P1.4 y 36% (IC95% 31 a 42) para NHBA.
Los resultados de los esquemas de dos dosis de vacuna a los 13 y 15 meses o 12 y 14 meses de edad en niños no vacunados previamente mostraron títulos ≥1:5 en el 100% para todas los antígenos, salvo para PorA P1.4 en el grupo 12+14 (96%).
Por su parte, en el ensayo clínico realizado sobre 1.631 adolescentes entre 11 y 17 años (mediana de 13,8 años, 13,2 kg de peso, 158,4 cm de altura; 48% varones y 100% hispanoamericanos), fue un estudio de fase 2b/3, multicéntrico (12 localizaciones de Chile), aleatorizado, parcialmente ciego y controlado con placebo, se les administró 1 (N= 375), 2 (628) o 3 (628) dosis de vacuna, con intervalos, en su caso, de 1, 2 o 6 meses (V72P10; Santolaya, 2012). Como criterios de eficacia inmunogénica se utilizaron los mismos que en el anterior estudio, salvo por el hecho de establecer el límite de título de anticuerpos como seroconversión en ≥1:4.
Después de 2 o 3 dosis, el 99-100% de los sujetos tenían un título de anticuerpos por encima del límite de seroconversión, frente al 92-95% de aquellos que recibieron una única dosis (p< 0,0145) y del 29-50% con placebo. A los seis meses entre el 91% y el 100% presentaban títulos ≥1:4 para cada uno de los cuatro antígenos después de dos o tres dosis, frente a solo el 73-76% con una sola.
Desde el punto de vista de la seguridad, los eventos adversos más comúnmente observados (>10%) en niños pequeños y lactantes fueron pérdida de apetito, somnolencia, diarrea, vómitos, erupciones cutáneas, fiebre e irritabilidad, así como induración y enrojecimiento en el punto de inyección. La fiebre (≥38,5º C) se manifestó en el 69-79% de los lactantes y niños pequeños cuando la vacuna se administró en conjunto con el resto de las vacunas infantiles habituales, frente al 44-59% cuando se administró sola; no obstante, menos del 1% experimentó reacciones febriles intensas (≥ 40ºC). Por su parte, en adolescentes y adultos los eventos adversos más comunes fueron cefalea, náuseas, mialgia, artralgia y dolor e induración en el punto de inyección.
ASPECTOS INNOVADORES
Se trata de una vacuna combinada de varias proteínas procedentes del serotipo B de la Neisseria meningitidis indicada para la inmunización activa de individuos a partir de 2 meses de edad frente a la enfermedad meningocócica invasiva causada por Neisseria meningitidis grupo B.
Los resultados serológicos sobre la capacidad inmunogénica de la nueva vacuna son claros y contundentes, con porcentajes de sujetos con títulos preventivos de anticuerpos por encima del 98% medidos un mes después de la última dosis. Los estudios clínicos son de actividad bactericida de los anticuerpos generados y de la persistencia de estos, ya que es prácticamente realizar un estudio clínico convencional de eficacia, dada la baja incidencia de la enfermedad invasiva por meningococo. Por ello, el beneficio real de esta nueva vacuna solo podrá evaluarse realmente tras su implantación en los programas oficiales de vacunación sistemática. En cualquier caso, la utilización del ensayo bactericida ha demostrado anteriormente una estrecha correlación con la protección frente a la enfermedad invasiva meningocócica. La duración de la protección todavía no es conocida, aunque se ha podido observar que los títulos de anticuerpos para dos de los antígenos (PorA P1.4 y NHBA) declinan rápidamente (6 meses después de la primovacunación y 12 después de la dosis de recuerdo.
La dosificación finalmente aceptada por la Agencia Europea de Medicamentos (EMA) es en lactantes de 2 a 5 meses, 3 dosis separadas por un mes, a partir de los dos meses de edad, con una dosis de recuerdo entre los 12 y 15 meses de edad; en lactantes no vacunados de 6 a 11 meses de edad, dos dosis separadas por dos meses, con una dosis de recuerdo durante el segundo año de vida, al menos dos meses después de la última dosis; en niños de 12 a 23 meses no vacunados, dos dosis separadas por dos meses, con una dosis de recuerdo 12-23 meses después de la última; en niños de 2 a 10 años, dos dosis separadas por dos meses, sin que se haya establecido la conveniencia de una dosis de recuerdo; en adolescentes y adultos (≥11 años), dos dosis separadas por un mes, sin dosis de recuerdo.
El perfil de seguridad de la vacuna es similar al de otras vacunas convencionales, aunque la incidencia de fiebre parece ser mayor cuando la vacuna frente al meningococo B se administra conjuntamente con otras vacunas que cuando se administra sola (Carter, 2013). Por otro lado, no se dispone de datos relativos a pacientes inmunodeprimidos, niños prematuros o ancianos.
Con todo, debe valorarse en su justo término el hecho de que la enfermedad invasiva por meningococo B es un cuadro grave, con una mortalidad asociada superior al 8%, que afecta fundamentalmente a los niños pequeños, con un segundo pico de incidencia durante la adolescencia, y hasta ahora no existía ninguna vacuna eficaz para su prevención. Por ello, la vacuna está claramente dirigida para estos grupos de edad; no obstante, en caso de focos epidémicos es deseable vacunar a toda la población.
Al hecho de ser la primera vacuna capaz de inmunizar frente al serotipo B del meningococo, que es el más prevalente en España y en el resto de la Unión Europea, hay que agregar que se trata de la primera vacuna eficaz que no utiliza el sistema de conjugación de proteínas y polisacáridos capsulares empleado con tanto éxito para prevenir meningitis y septicemias de otros serotipos de meningococos (A, C, W, Y), de neumococos (13 serotipos) y de Heamophillus influenzae tipo b. Esto último también supone un avance farmacotécnico nada desdeñable, que abre nuevas líneas de investigación para el desarrollo de vacunas frente a bacterias y otros microorganismos patológicamente relevantes.
|
VALORACIÓN |
|
|---|---|
|
Vacuna meningocócica del grupo B ► BEXSERO® (Novartis) |
|
|
Grupo Terapéutico (ATC): J07AH. TERAPIA ANTIINFECCIOSA SISTÉMICA. Vacunas antibacterianas: meningococos. |
|
|
Indicaciones autorizadas: Inmunización activa de individuos a partir de 2 meses de edad frente a la enfermedad meningocócica invasiva causada por Neisseria meningitidis grupo B. |
|
|
Valoración global: INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar. |
♣ ♣ ♣ |
|
Novedad clínica: Supone la incorporación de una nueva vía preventiva en ausencia de alternativas, con una gran relevancia clínica |
⇑ |
|
Novedad económico-técnica: Mejora aspectos farmacotécnicos. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Meningococo C |
Neisvac C |
Baxter |
2001 |
|
Meningococo C |
Menjugate |
Novartis |
2005 |
|
Meningococo C |
Meningitec |
Pfizer |
2008 |
|
Meningogoco A+C+Y |
Menveo |
Novartis |
2010 |
|
Meningococo A+C |
Nimenrix |
GlaxoSmithKline |
2013 |
|
Meningococo B |
Bexsero |
Novartis |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
Bibliografía
Decitabina DACOGEN® (Janssen Cilag)
LEUCEMIA MIELOIDE AGUDA (LMA)
La leucemia mieloide aguda como una proliferación neoplásica de células inmaduras de estirpe mieloide que proceden de un progenitor hematopoyético lesionado con capacidad de maduración alterada y que, debido a su expansión, desplaza a las células normales de la médula ósea. Se trata del tipo más común de leucemia aguda en adultos y representa un 40% de las leucemias en el mundo occidental. En la Unión Europea cada año se diagnostican alrededor de 18.000 casos, con una mediana de edad en el momento del diagnóstico entre 64 y 67 años.
La decitabina es un agente antitumoral de tipo antimetabolito (es un análogo de la citidina), estructural y farmacológicamente relacionado con la azacitidina; ha sido autorizado para el tratamiento de pacientes adultos de 65 o más años de edad con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional. Dada la muy baja prevalencia de esta condición, el medicamento ha sido designado como huérfano.
El único ensayo clínico de fase III (confirmatorio de eficacia y seguridad) disponible, comparativo con citarabina en bajas dosis, ha mostrado consistentemente una mayor eficacia en términos de supervivencia global, con una mejora de 2,7 meses, así como en otras variables clínicamente significativas. Dichas diferencias favorables a la decitabina en relación a la citarabina son consideradas como clínicamente relevantes, habida cuenta de que los pacientes sobre los que se utilizaron ambas eran de alto riesgo (un 40% tenían ≥75 años). Por otro lado, su perfil toxicológico hace que sea relativamente bien tolerado, aunque con las obvias manifestaciones hematológicas adversas (trombocitopenia, anemia, neutropenia, etc.).
La médula ósea produce inicialmente células madre sanguíneas indiferenciadas o pluripotenciales que, posteriormente, se transforman en células sanguíneas maduras, tanto de tipo mieloide como linfoide. Mientras que las células linfoides inmaduras acaban produciendo diferentes estirpes de linfocitos, las mieloides se pueden transformar en eritrocitos, ciertos leucocitos y megacariocitos (precursores de las plaquetas).
La leucemia puede ser aguda, progresando rápidamente y con muchas células inmaduras neoplásicas, o crónica, que progresa lentamente con células leucémicas de apariencia más madura. Las leucemias agudas son cuadros neoplásicos específicos de células hemopoyéticas de tipo pluripotencial; es decir, se trata de diferentes formas de expansión de un clon maligno específico de células sanguíneas. Se pueden clasificar en dos grandes grupos: las leucemias linfoides agudas (LLA) y las leucemias mieloides agudas (LMA), también conocidas, por contraposición a las anteriores, como leucemias no linfoides agudas (LNLA).
En el caso de la leucemia mieloide aguda, habitualmente las células madre mieloides se transforman en un tipo de glóbulo blanco inmaduro (mieloblasto o blastocito mieloide) y anómalo que no se transforman en glóbulos blancos sanos. En otras formas de LMA, se produce un excesivo número de células madre que se transforman en glóbulos rojos o plaquetas anormales o leucémicos, que acaban desplazando de la médula ósea y de la sangre a sus congéneres sanos, provocando infecciones, anemia o hemorragias graves. Además, las células leucémicas se pueden diseminar fuera del torrente sanguíneo y de los órganos hematopoyéticos a otras partes del organismo, particularmente al sistema nervioso central (cerebro y médula espinal), piel y encías.
Así pues, se puede definir a la leucemia mieloide aguda como una proliferación neoplásica de células inmaduras de estirpe mieloide que proceden de un progenitor hematopoyético lesionado con capacidad de maduración alterada y que, debido a su expansión, desplaza a las células normales de la médula ósea. La LMA se conoce también como leucemia mielógena aguda, leucemia mieloblástica aguda, leucemia granulocítica aguda o leucemia no linfocítica aguda.
La LMA es el tipo más común de leucemia aguda en adultos y representa un 40% de las leucemias en el mundo occidental. En la Unión Europea cada año se diagnostican alrededor de 18.000 casos, con una mediana de edad en el momento del diagnóstico entre 64 y 67 años. En Estados Unidos se registran 14.500 casos anualmente, mayoritariamente en personas de más de 60 años; de hecho, un tercio tiene más de 75 años (Malik, 2014). Su incidencia en este país es de 0,59:100.000 en menores de 20 años, pero asciende a 1,54 entre los 20 y los 49, a 5,02 entre los 50 y 64, a 14,80 entre los 65 y 74 y 24,40 a partir de los 75 (SEER, 2014).
Entre los posibles factores de riesgo de LMA están el ser varón (proporción 1,3:1 frente a mujeres), fumar (especialmente después de los 60 años), haber recibido un tratamiento con quimioterapia (un 10-20% de todos los casos) o radioterapia en el pasado, haber padecido leucemia linfoblástica aguda (LLA) infantil en el pasado, estar expuesto a la radiactividad o al benceno y tener antecedentes de trastornos hematológicos proliferativos, como el síndrome mielodisplásico. Por otro lado, el riesgo de desarrollar LMA es tres veces mayor entre personas con parentesco en primer grado de pacientes.
El inicio de la enfermedad es relativamente rápido y raramente se superan los tres meses entre la aparición de los primeros síntomas y el diagnóstico de la LMA. Los síntomas son consecuencia de la anemia producida por el déficit de glóbulos rojos (cansancio, mareos, palidez), de plaquetas (hematomas, sangrado de encías, nariz u otras mucosas) y de granulocitos (fiebre e infecciones). En ocasiones puede observarse el crecimiento de los ganglios linfáticos, así como hepato y/o esplenomegalia. Puedes observarse síntomas neurológicos, que denotan una infiltración del sistema nervioso central (dolor de cabeza, vómitos, somnolencia, etc.), aunque también pueden afectar a piel (nódulos diseminados o zonas de piel engrosada), mucosas (inflamación de las encías), ocular (visión borrosa, ceguera), etc.
El origen de la leucemia mieloide aguda (LMA) está en el proceso de malignificación de algún clon de mieloblasto, como consecuencia de la acumulación de una serie de mutaciones en su ADN, lo que le impide proseguir en su proceso de diferenciación celular normal, por lo que queda en un estado más o menos indiferenciado (inmaduro); cuando a ello se le agregan otras mutaciones que le confieren la capacidad para proliferar rápidamente, se produce una multiplicación incontrolada del clon que acaba por arrinconar literalmente a las células sanguíneas normales de la médula ósea y del torrente circulatorio, desencadenante los síntomas de la enfermedad.
La gran diversidad y heterogeneidad de tipos celulares que pueden observarse en la LMA se debe a que la transformación leucémica puede ocurrir en diferentes estadios a lo largo del proceso de diferenciación, lo que condiciona el tipo de células cancerígenas que se podrán encontrar en un determinado paciente. Esto es determinante para su clasificación y pronóstico. Las anomalías cromosómicas consisten básicamente en translocaciones (t) e inversiones (inv) de trozos de cromosomas, que provocan la formación nuevo material genético susceptible de generar nuevas proteínas de fusión que pueden perder su función original o ejercerla pero de forma incontrolada, escapando a los sistemas de regulación de la expresión génica. En el caso de que estas estas proteínas anómalas actúen como factores de transcripción, se produce una interrupción de la diferenciación y la posterior proliferación incontrolada. La OMS ha establecido (2008) la siguiente clasificación:
- Leucemia mieloide aguda (LMA) con anomalías genéticas recurrentes
- LMA con t(8;21)(q22;q22); RUNX1-RUNX1T1
- LMA con inv(16)(p13.1q22) o t(16;16)(p13.1;q22); CBFB-MYH11
- LMA con t(9;11)(p22;q23); MLLT3 MLL
- LMA con t(6;9)(p23;q34); DEK-NUP214
- LMA con inv(3)(q21q26.2) o t(3;3)(q21;q26.2); RPN1-EVI1
- LMA (megacarioblástica) con t(1;22)(p13;q13); RBM15-MKL1
- Leucemia promielocítica aguda (LPA) con t(15;17)(q22;q12); PML-RARA
- LMA con mutación NPM1
- LMA con mutación CEBPA
- LMA con mielodisplasia
- Neoplasias mieloides relacionadas con el tratamiento
- LMA sin otra especificación (NOS)
- LMA con diferenciación mínima
- LMA sin maduración
- LMA con maduración
- Leucemia mielomonocítica aguda
- Leucemia aguda monoblástica/monocítica
- Leucemia eritroide aguda
- Leucemia eritroide pura
- Eritroleucemia, eritroide/mieloide
- Leucemia aguda megacarioblástica
- Leucemia aguda basófila
- Panmielosis aguda con mielofibrosis (mielofibrosis aguda, mieloesclerosis aguda)
- Sarcoma mieloide (tumor mieloide extramedular, sarcoma granulocítico, cloroma)
- Proliferaciones mieloides relacionadas con el síndrome de Down
- Mielopoyesis transitoria anormal (trastorno mieloproliferativo transitorio)
- Leucemia mieloide asociada con síndrome de Down
- Neoplasias de células dendríticas plasmocitoides
- Leucemias agudas de linaje ambiguo
- Leucemia aguda indiferenciada
- Leucemia aguda fenotipo mezclado con t(9;22)(q34;q11.2); BCR-ABL1
- Leucemia aguda fenotipo mezclado con t(v;11q23); MLL reordenado
- Leucemias agudas de fenotipo mixto, B/mieloide, NOS
- Leucemias agudas de fenotipo mixto, T/mieloide, NOS
- Entidad provisional: natural killer (NK) de células de leucemia linfoblástica/linfoma
El tratamiento de la leucemia mieloide aguda en adultos habitualmente tiene dos fases.
- Terapia de inducción de la remisión: Tiene como objetivo destruir las células leucémicas en la sangre y en la médula ósea. Esto induce la remisión de la leucemia.
- Terapia de posremisión (continuación de la remisión): Comienza después de que la leucemia está en remisión. Su objetivo es destruir cualquier célula leucémica aislada que quede, que tal vez no esté activa pero que pueda comenzar a volver a crecer y producir una recaída.
En general, y salvo los casos de leucemia promielocítica aguda, los tratamientos requeridos para inducir la remisión de la LMA son muy agresivos, lo que les contraindica en caso de edad avanzada o en caso de graves patologías concomitantes. De hecho el 70% de los pacientes mayores de 65 años no reciben tratamiento intensivo y su supervivencia es de dos meses. En estos casos, las opciones terapéuticas son escasas y con resultados insatisfactorios. Las actuales guías clínicas recomiendan tratamiento de soporte (no intensivo) con hidroxiurea y transfusiones sanguíneas, o tratamiento activo sistémico con citarabina a dosis bajas, o agentes hipometilantes como la azacitidina y la decitabina (AEMPS, 2014).
Pacientes hasta 60 años
Se utiliza como tratamiento de inducción el esquema 3 + 7, que consiste en administrar 3 días de antraciclinas (daunorubicina, idarubicina o mitoxantrona) y 7 días con citarabina, sigue siendo en la actualidad el estándar para el tratamiento de inducción. Con dicho esquema se logra la respuesta completa en el 60-80% de los adultos jóvenes. Ninguna otra intervención ha demostrado de manera convincente ser mejor. La quimioterapia de inducción se debe iniciar después de que el trabajo de diagnóstico se ha completado, con la mínima demora. Los datos retrospectivos indican que el resultado del tratamiento puede verse afectado cuando el tiempo desde el diagnóstico hasta el inicio del tratamiento aumenta más allá de 5 días (Novelli, 2011).
Existen varias estrategias posremisión:
- Tratamiento intensivo: cuatro ciclos de altas dosis de citarabina (HiDAC): 3 g/m2 cada 12 horas los días 1, 3 y 5 de cada mes.
- Tratamiento de mantenimiento: sin beneficio en cuanto a la duración de la remisión o la supervivencia global a los 3 años con el mantenimiento intensivo en comparación con el trasplante hematopoyético como terapia posterior a la remisión. Sí parece ser superior a la consolidación con HiDAC secuencial y mitoxantrona (HiDAC 1 g/m2 cada 12 horas los días 1, 2, 8 y 9; mitoxantrona 10 mg/m2 en los días 3, 4, 10 y 11). La quimioterapia de mantenimiento generalmente no se administra sistemáticamente fuera de ensayos clínicos para los pacientes con LMA (no promielocítica).
- TASPE (trasplante autólogo de progenitores hematopoyéticos de sangre periférica): es considerado como una opción alternativa para pacientes con una citogenética favorable y riesgo intermedio, mientras que no puede ser recomendado en pacientes con alto riesgo. El resultado después del TASPE es al menos tan bueno como la quimioterapia posremisión; hasta la fecha no ha habido ninguna evidencia de una mejora en los resultados.
- Alo-TPH (trasplante alogénico de progenitores hematopoyéticos): se asocia con menores tasas de recaída. Este beneficio es atribuible tanto a la terapia con altas dosis de los regímenes de acondicionamiento estándar como a un potente efecto injerto contra leucemia (graft versus leukemia, GVL). Sin embargo, los beneficios del alo-TPH se han visto limitados por una mortalidad relacionada con el trasplante. Se suele practicar en pacientes con LMA con pronóstico desfavorable y en aquéllos refractarios.
Pacientes de 60 a 75 años
El aumento de la edad se asocia con factores predictivos de muerte prematura, comorbilidades diversas (diabetes, hipertensión arterial, cardiopatía, etc.) y a la resistencia al tratamiento más frecuentemente observada en LMA con citogenética adversa y en LMA secundarias. Independientemente de estas asociaciones, la edad sigue siendo un importante predictor de mal pronóstico. Los estudios sugieren que la quimioterapia de inducción de remisión proporciona una mejor calidad de vida y una mayor supervivencia que sólo el tratamiento de soporte.
Para los pacientes con una puntuación en la escala ECOG (Eastern Cooperative Oncology Group) menor de 2, la terapia estándar de inducción de la remisión es a menudo una opción plausible que resulta en tasas de remisión del 50%. La elección de la antraciclina (daunorubicina, idarubicina o mitoxantrona) es de poca importancia, partiendo de dosis de toxicidad equivalentes; además, en casos individuales puede considerarse la reducción de la dosis. El grado de respuesta de la terapia de inducción estándar depende en gran medida de la citogenética, ya que una citogenética adversa es un fuerte factor pronóstico independiente del fracaso para alcanzar una respuesta completa. Para este subgrupo de pacientes de mayor edad, las tasas de respuesta completa son del 30% o menos y la supervivencia global es menor del 5%.
Como terapia de posremisión, no parece haber diferencias entre la utilización de dos ciclos de quimioterapia relativamente intensiva (citarabina 500 mg/m2 cada 12 horas con mitoxantrona 5 mg/m2 cada 12 horas por 6 dosis) con ciclos menos intensivos (citarabina 100 mg/m2, días 1-5). Para los pacientes sin citogenética adversa, buen estado general y sin comorbilidades significativas el estándar es el 3 + 7 de inducción seguido de ciclos repetitivos de consolidación de dosis quimioterápica de intensidad moderada (Novelli, 2011).
Finalmente, el alo-TPH (trasplante alogénico de progenitores hematopoyéticos) en pacientes de edad avanzada se ha convertido en un campo activo y prometedor de investigación. En particular, los trasplantes de intensidad reducida (TIR) o minialotrasplantes se han desarrollado para reducir la mortalidad asociada al trasplante en los pacientes ancianos o en aquéllos más jóvenes pero con comorbilidades.
Pacientes mayores de 75 años
El empleo de dosis bajas de citarabina (20 mg SC, 2 veces al día, durante 10 días) se asocia con una mayor supervivencia que con la hidroxiurea, aunque solo marginalmente.
ACCIÓN Y MECANISMO
La decitabina es un agente antitumoral de tipo antimetabolito (es un análogo de la citidina), autorizado para el tratamiento de pacientes adultos de 65 o más años de edad con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional.
Las células indiferenciadas de la leucemia mieloide aguda se caracterizan por presentar intensos procesos de metilación del ADN, que se relacionan con el control de la expresión genética. Una vez en el interior de la célula, la decitabina es fosforilada mediante el enzima desoxicitidina cinasa hasta producir el derivado trifosfatado, el cual compite con la citosina, reemplazándola, en la pareja CpG (citosina-guanosina dinucleótido) del ADN. Una vez intercalada en la cadena de ADN, se une de forma estable (mediante un enlace covalente) al enzima ADN metiltransferasa (DNMT, DNA methyltransferase), la cual queda bloqueada y no es capaz de catalizar las reacciones de metilación (incorporación de grupos metilo: -CH3) a las bases nucleicas de varios genes promotores; como consecuencia de la reducción de la capacidad metiladora, algunos de los genes silenciados por las mutaciones propias de la leucemia mieloide – incluyendo a varios genes supresores tumorales: p15/INK4b, p16, p14, SHP-1, p21WAF1, p73, C/EBPd – vuelven a expresarse y, con ello, pueden prevenir la transformación tumoral de las células hematopoyéticas precursoras e inducir su diferenciación normal. Asimismo, la decitabina puede facilitar también la recuperación de la expresión de genes implicados en la inmunogenicidad y en el reconocimiento inmune – antes suprimidos – de las células tumorales.
El efecto inactivante del enzima DNMT no solo parece ser debido a su unión covalente con el ADN, previniendo su síntesis, sino que también podría facilitar la degradación de aquella ya sintetizada, al potenciar los procesos de ubiquitinación y degradación del enzima en el proteasoma.
Sin embargo, la decitabinina es capaz de ejercer efectos citotóxicos no relacionados con una reducción de la metilación de los genes supresores tumorales y e inmunológicos, sino que también puede dar lugar a la formación de complejos – aductos – de DNMT y el ADN que interfieren con la síntesis de nuevo ADN durante la división celular, poniendo en marcha los mecanismos de apoptosis (muerte celular programada). En este sentido, parece que la inducción de la apoptosis celular por decitabina estaría ligada a la recuperación de la expresión del gen supresor tumoral p73.
Finalmente, según se ha constatado en diversos modelos tumorales experimentales en animales, la decitabina también ejerce un efecto antiangiogénico, reduciendo la formación de vasos sanguíneos en el interior tumoral.
ASPECTOS MOLECULARES
{kind=link}
Se trata de un análogo molecular de la base nucleica citidina, de núcleo pirimidínico. La decitabina está estrechamente relacionada estructural y farmacológicamente con otros antimetabolitos citotóxicos análogos de la citidina, como citarabina, gemcitabina, capecitabina y, en particular, con la azacitidina. De hecho, en términos químicos, la decitabina es la 4-amino-1-[(2R,4S,5R)-4-hidroxi-5-(hidroximetil)oxolan-2-il]-1,3,5-triazin-2-ona2-desoxiazacitidina, es decir, la 2-desoxiazacitidina.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas han sido adecuadamente contrastadas mediante un ensayo clínico de fase III multinacional (15 países), abierto y aleatorizado (DACO-016; Kantarjian, 2012), en el que se comparó la administración de dacitabina (20 mg/m2, infusión IV en 1 h cada 24 h) durante los 5 primeros días de cada ciclo de 4 semanas, frente a citarabina en dosis bajas (20 mg/m2, subcutánea cada 24 h) durante los 10 primeros días de cada ciclo de 4 semanas, en un conjunto de 485 pacientes con leucemia mieloide aguda primaria (64%) o secundaria (34%), que fueron valorados como de alto riesgo global, aunque con riesgo citogenético bajo (36%) o intermedio (64%); la proporción mediana de elementos blásticos en médula era del 46% (un 43% de los pacientes tenían ≥50%). La mediana de edad de los pacientes era de 73 años (un 40% tenían ≥75 años) y un 59% eran varones. El tratamiento se mantuvo hasta la muerte del paciente o la pérdida del beneficio clínico (progresión).
Como variable primaria de eficacia se utilizó la mediana de la supervivencia global (periodo desde el inicio del tratamiento hasta la muerte del paciente, con independencia de su causa); como variable secundaria se determinó la suma de las tasas de remisión completa morfológica y de remisión completa sin recuperación de plaquetas (CR+CRp). Como las variables terciarias se calcularon la remisión completa citogenética (CRt; sobre pacientes con anomalías citogenéticas previas), la supervivencia libre de eventos adversos, la supervivencia libre de enfermedad y la supervivencia libre de recaídas (para pacientes con respuesta CR).
Los resultados mostraron que la supervivencia global fue de 7,7 (IC95% 6,2 a 9,2) meses con dacitabina vs. 5,0 (IC95% 4,7 a 6,3) meses con citarabina, con una reducción del 18% en la tasa de riesgo de muerte en el grupo de la decitabina (HR= 0,82; IC95% 0,68 a 0,99; p= 0,0373)1. Asimismo, excluyendo a los pacientes que habían recibido terapia antileucémica adicional (38% en el grupor de dacitabina y 44% en el de citarabina), la mediana de la supervivencia global fue de 8,5 vs. 5,3 meses (p= 0,044)
En cuanto al resto de variables, las tasas de remisión completa CR+CRp fueron del 17,8% con dacitabina vs. 7,8% con citarabina, siendo la probabilidad 2,5 veces mayor con dacitabina (odds ratio, OR= 2,5; IC95% 1,40 a 4,78; p= 0,0011); asimismo, las tasas de remisión completa citogenética (CRc) fueron del 10,0% vs. 7,3%. En cuanto a las medianas de supervivencia libre de eventos fueron 3,5 meses (IC95% 2,5 a 4,1) vs. 2,1 (IC95% 1,9 a 2,8), de supervivencia libre de enfermedad 3,7 meses (IC95% 2,7 a 4,6) vs. 2,1 (IC95% 1,9 a 3,1) y de supervivencia libre de recaídas fueron 6,7 (IC95% 2,9 a 13,4) vs. 8,3 (IC95% 4,6 a 11,4).
El perfil de seguridad de la decitabina es similar al de la citarabina en dosis bajas. La toxicidad fundamental fue de tipo hematológico, con una mayor incidencia de trombocitopenia (40 vs. 32%), anemia (34 vs. 25%) y neutropenia (32 vs. 18%) en el grupo tratado con decitabina. Las complicaciones de tipo infeccioso fueron también más comunes con decitabina, con episodios febriles asociados a neutropenia (32 vs. 22%) y neumonía (21 vs. 18%). No obstante, es preciso indicar que los pacientes tratados con decitabina estuvieron expuestos al tratamiento durante más tiempo que aquellos tratados con citarabina (4,4 vs. 2,4 meses), lo que es probable que facilitase un reporte mayor de eventos adversos entre los tratados con decitabina. Por otro lado, la tasa de descontinuación del tratamiento por eventos adversos fue ligeramente inferior con decitabina (6%) que con citarabina (8%).
ASPECTOS INNOVADORES
La decitabina es un agente antitumoral de tipo antimetabolito (es un análogo de la citidina), estructural y farmacológicamente relacionado con la azacitidina; ha sido autorizado para el tratamiento de pacientes adultos de 65 o más años de edad con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional. Dada la muy baja prevalencia de esta condición, el medicamento ha sido designado como huérfano.
La capacidad de la decitabina para limitar sustancialmente los procesos de metilación del ADN, parece ser determinante para permitir la recuperación de la expresión de ciertos genes supresores tumorales y para dificultar la conversión tumoral de los precursores celulares hematológicos.
El único ensayo clínico de fase III (confirmatorio de eficacia y seguridad) disponible, comparativo con citarabina en bajas dosis, ha mostrado consistentemente una mayor eficacia en términos de supervivencia global, con una mejora de 2,7 meses, así como en otras variables clínicamente significativas. Dichas diferencias favorables a la decitabina en relación a la citarabina son consideradas como clínicamente relevantes, habida cuenta de que los pacientes sobre los que se utilizaron ambas eran de alto riesgo (un 40% tenían ≥75 años). Por otro lado, su perfil toxicológico hace que sea relativamente bien tolerado, aunque con las obvias manifestaciones hematológicas adversas (trombocitopenia, anemia, neutropenia, etc.).
Es difícil establecer el papel de la decitabina en el tratamiento, aunque dada la notable escasez de alternativas existente en los pacientes con peor pronóstico, no cabe duda que supone una opción plenamente válida; asimismo, su perfil toxicológico no excesivamente problemático la convierte en una atractiva alternativa en terapias de combinación con otros agentes antileucémicos (Malik, 2014).
Por otro lado, tal como señala el Informe de Posicionamiento Terapéutico de la AEMPS , los datos no permiten comparar clínicamente a la decitabina con la azacitidina, aunque el nivel de evidencia de la primera es mayor en pacientes mayores de 65 años con leucemia mieloide aguda, tanto primaria como secundaria.
|
VALORACIÓN |
|
|---|---|
|
Decitabina ► DACOGEN® (Janssen Cilag) |
|
|
Grupo Terapéutico (ATC): L01BC. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Citostáticos: antimetabolitos análogos de la pirimidina. |
|
|
Indicaciones autorizadas: Tratamiento de pacientes adultos de 65 o más años de edad con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional. |
|
|
Valoración global: INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar. |
♣ ♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Fluorouracilo |
Fluoro Uracil |
Meda |
1964 |
|
Tegafur |
Utefos** |
Almirall |
1978 |
|
Citarabina |
Citarabina Pfizer |
Pfizer |
1984 |
|
Gemcitabina |
Gemzar |
Lilly |
1985 |
|
Capecitabina |
Xeloda |
Roche |
2001 |
|
Azacitidina |
Vidaza |
Celgene |
2009 |
|
Decitabina |
Dacogen |
Janssen Cilag |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
** Retirado del mercado
Bibliografía
Sofosbuvir SOVALDI® (Gilead)
HEPATITIS C
En España, la prevalencia de la infección por VHC C se sitúa en torno al 1,2-1,7% de la población general, lo que supone un número de personas infectadas de entre 500.000 y 1.000.000; más del 50% de los pacientes que han necesitado un trasplante de hígado son pacientes con hepatitis C que han evolucionado a una enfermedad hepática terminal.
El sofosbuvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos. Presenta unos excelentes resultados clínicos, bien avalados por ensayos clínicos amplios y rigurosos metodológicamente, con un amplio espectro de actividad frente a los diversos genotipos de VHC circulantes, a lo cual hay que añadir su elevada barrera frente a la generación de resistencias por mutaciones de la proteína NS5B.
El virus de la hepatitis C (VHC) se clasifica dentro de la familia Flaviviridae por ser un virus envuelto, de aproximadamente 50 nm de diámetro, con una sola cadena de ARN en sentido positivo (5′-3′), formando parte del género Hepacivirus. Su prevalencia global se estima entre 150 y 180 millones (2-3 % de la población general), con una incidencia anual de 3-4 millones de nuevos casos. En España, la prevalencia de la infección por VHC C se sitúa en torno al 1,2-1,7% de la población general, lo que supone un número de personas infectadas de entre 500.000 y 1.000.000; más del 50% de los pacientes que han necesitado un trasplante de hígado son pacientes con hepatitis C que han evolucionado a una enfermedad hepática terminal.
Existen varios genotipos del VHC. En Europa y EEUU, el más frecuente es el 1 (70% de los casos), seguido por el 2 y 3 (25%). El 2 (varios subtipos) y 4 (b, c, e y m) en África del Norte y Central, y el 6p y 2i en Asia. En concreto, en Europa predomina el genotipo 1b, seguido de 2a, 2b, 2c y 3ª; por su parte, en Norte América predomina el genotipo 1a, seguido de 1b, 2a, 2b y 3a.
Una vez en el interior del hepatocito, el virus de la hepatitis C (VHC) utiliza la maquinaria celular para replicarse. La expresión del ARN viral conduce a la síntesis de una única proteína (poliproteína) de gran tamaño (3.011 aminoácidos), que requiere de la acción de varias proteasas para dar lugar a las formas activas de las proteínas virales, que incluyen a tres proteínas estructurales (E) y a siete no estructurales (NS). Esta poliproteína (C(F)-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B) genera al menos diez proteínas maduras al ser procesada por la señal peptidasa del retículo endoplásmico, la cual libera las proteínas C, E1, E2 y p7 que forman la cápside o núcleo viral (C), la envoltura de la partícula (E1 y E2) y p7 que se parece intervenir en la liberación del genoma al formar canales iónicos en la envoltura viral; la región de las proteínas no estructurales (NS2-NS5B) que participan en la replicación son procesadas por dos enzimas virales: NS2 y NS3 que generan un corte autocatalítico en las regiones NS2-NS3 y NS3-NS4A respectivamente y permiten la formación del complejo proteasa de serina NS3/4A que procesa el resto de la poliproteína viral, provocando la maduración de las proteínas NS4B, NS5A y NS5B con función de polimerasa de ARN dependiente de ARN (NS5B) y de fosfoproteína (NS5A); por el momento, la función de la proteína NS4B no está plenamente comprendida.
Las proteínas NS toman el ARN viral formando un complejo de replicación – sistema replicón – que es asociado a membranas ciplasmáticas modificadas. Concretamente, la proteínas NS5B es una ARN polimerasa ARN dependiente que es responsable de producir la hebra de ARN complementaria – ARN(-) – que servirá de molde para fabricar las auténticas hebras de ARN viral – ARN(+) –, que a su vez podrán ser de nuevo replicados y traducidas, o bien empaquetados en las proteínas estructurales para formar nuevas partículas virales de VHC, que son liberadas mediante un proceso de exocitosis. La capacidad de replicación del VHC es enorme, calculándose en un billón (1012) el número de nuevos virus que son capaces de ser producidos durante un día en una persona infectada.
La proteína NS3 del VHC es una enzima multifuncional ya que presenta en el primer tercio de su estructura, una actividad de proteasa de serina y en el resto función de ARN helicasa DexH/D que revierte el enrollamiento de cadenas dobles de ARN formadas durante la replicación viral. El extremo NS4A es una proteína anfipática de 54 aminoácidos que se asocia no covalentemente con las cadenas A0 y A1 de NS3 y provoca una reorganización en su estructura, optimizando así la actividad de proteasa de NS3; además NS4A promueve la localización del complejo NS3/4A a la membrana del retículo endoplásmico donde es procesada la poliproteína viral.
Por su parte la proteína NS5A es una proteína intensamente fosforilada que, una vez escindida de la poliproteína viral, localiza a las membranas donde se une a la fracción 3′-terminal del ARN viral recién sintetizado y participa en la replicación del genoma viral, en parte a través de interacciones con la ARN polimerasa dependiente del ARN viral (NS5B). Finalmente, la NSB5 es la ARN polimerasa dependiente del ARN viral que desarrolla un papel esencial en la replicación del ARN viral, utilizando la cadena de este última como molde para nuevas cadenas y catalizando el proceso de polimerización de los ribonucleótido-trifosfato (rNTP) durante este proceso de replicación.
En el año 2011 se produjo un cambio notable en el panorama terapéutico de la hepatitis C, al comercializarse el boceprevir y el telaprevir – la primera generación de inhibidores selectivos y reversibles de la proteasa NS3 – para el tratamiento de pacientes infectados por VHC de tipo 1, tanto no tratados previamente (naïve) como tratados, en combinación de con peginterferón alfa y ribavirina (terapia triple). En estas circunstancias, las tasas de respuesta tanto en pacientes naïve como en pretratados llegaban a doblarse prácticamente, permitiendo acortar la duración del tratamiento en muchos de los pacientes (60%) de 48 a 24 semanas. Sin embargo, boceprevir y telaprevir presentaban perfiles toxicológicos importantes que obligaban a suspender el tratamiento en un porcentaje de pacientes netamente superior a los tratados solo con peginterferín y ribavirina, amén de un amplio abanico de interacciones farmacológicas, lo cual, asociado con la propia complejidad del tratamiento, dejaba un amplio margen para la mejora.
Tras esta primera generación de inhibidores de la proteasa del VHC ha llegado una nueva oleada de agentes con propiedades farmacodinámicas, farmacocinéticas y toxicológicas más satisfactorias, que está en la fase final del proceso de autorización por las agencias reguladoras (EMA, en el caso de la Unión Europea) o han recibido dicha autorización en los últimos meses. Se trata del asunaprevir, inhibidor de la NS3); simeprevir, faldaprevir y vaniprevir, inhibidores duales de NS3 y NS4A; daclatasvir y ledipasvir, de la NS5A; y sofosbuvir y deleobuvir, de la NS5B (Cuéllar, 2014).
ACCIÓN Y MECANISMO
El sofosbuvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos.
El sofosbuvir es un profármaco que requiere ser fosforilado hasta formar el trifosfato, la forma biológicamente activa. Como tal compite con el uridilato por la ARN polimesa, provocando la finalización prematura del proceso de replicación de la nueva cadena de ARN viral en formación. El sofosbubir es activo sobre los genotipos 1, 2, 3, 4, 5 y 6 del VHC, con valores de la concentración inhibitoria para el 50% (CI50%) que van desde 0,01 a 2,6 µM.
Las proteínas NS toman el ARN viral formando un complejo de replicación – sistema replicón – que es asociado a membranas ciplasmáticas modificadas. Concretamente, la proteínas NS5B es una ARN polimerasa dependiente del ARN viral que es responsable de producir la hebra de ARN complementaria – ARN(-) – que servirá de molde para fabricar las auténticas hebras de ARN viral – ARN(+) –, que a su vez podrán ser de nuevo replicados y traducidas, o bien empaquetados en las proteínas estructurales para formar nuevas partículas virales de VHC, que son liberadas mediante un proceso de exocitosis. La capacidad de replicación del VHC es enorme, calculándose en un billón (1012) el número de nuevos virus que son capaces de ser producidos durante un día en una persona infectada.
La resistencia del VHC a sofosbuvir está directamente relacionada con mutaciones que conducen a determinadas variaciones en la secuencia peptídica de la ARN polimerasa NSB5, siendo la más comúnmente descrita como causante de una reducida susceptibilidad al fármaco por los VHC la mutación S282T, por sustitución de serina por treonina en la posición 282 de la cadena peptídica. Esta mutación no parece afectar a la susceptibilidad del VHC a otros tipos de antivirales de acción directa.
ASPECTOS MOLECULARES
{kind=link}
El sofosbuvir es un análogo estructural del uridilato (uridina monofosfato, UMP) y del uracilo (U), una de las cuatro bases nucleicas que constituyen el ARN (aunque en el ADN, el uracilo es reemplazado por la timina). El sofosbuvir incluye también en su molécula al aminoácido L-alanina. Químicamente es el (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopirimidin-1-il)-4-fluoro-3-hydroxy-4-metiloxolan-2-il]metoxi-fenoxifosforil]amino]propanoato de propan-2-ilo.
Tras un proceso de fosforilación intracelular, el sofosbuvir es transformado en un derivado trifosfatado, que es la forma biológicamente activa del fármaco, actuando como una falsa base nucleica (un falso uridilato) que es capaz de bloquear a la ARN polimerasa dependiente del ARN viral, provocando la finalización prematura del proceso de replicación y, con ello, deteniendo la reproducción del VHC.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad del sofosbuvir en la indicación autorizada han sido contrastadas mediante varios ensayos clínicos principales de fase 3, tanto en pacientes naïve (no tratados previamente) como en aquellos pretratados. En todos los casos, la variable primaria de eficacia utilizada fue la tasa de pacientes con respuesta virológica sostenida, es decir, aquellos con niveles séricos de menos de 25 copias/ml de ARN de VHC, medidos 12 semanas después de haber finalizado el tratamiento completo antiviral (RVS12). La dosis empleada de sofosbuvir fue de 400 mg/24 h por vía oral, la de ribabvirina fue de 500 o 600 mg/12 h (según peso) por vía oral, y de la peginterferón alfa-2a fue de 180 µg/semana, por vía subcutánea.
Eficacia en infección por VHC de genotipos 1, 4, 5 y 6
El estudio NEUTRINO (Lawitz, 2013) es un ensayo clínico abierto y multicéntrico realizado sobre 327 pacientes naïve con hepatitis C, que recibieron un tratamiento de 12 semanas con sofosbuvir (SOF), ribavirina (RBV) y peginterferón alfa-2a (IFN). La mediana de edad era de 54 años, un 64% eran varones, un 79% de raza blanca, con un índice de masa corporal (IMC) medio de 29 y un 17% tenían cirrosis hepática; la mediana de la carga viral era de 6,6 log10 (4.000.000) copias/ml (el 78% tenía más de 6 log10, es decir, 1.000.000 copias/ml). Un 89% presentaban VHC de genotipo 1, 9% genotipo 4, 0,3% genotipo 5 y 1,8% genotipo 6.
La tasa global de respuesta (RVS12) fue del 90,5% (IC95% 86,8 a 93,5), siendo del 92,7% entre los no cirróticos y del 79,6% entre los cirróticos; las tasas de recaídas y de fallo virológico fue del 8,6%. En los pacientes con VHC de genotipo 1 fue del 89,7% (91,6% en 1a y 83,3% en 1b), mientras que considerando conjuntamente a los genotipos 4, 5 y 6 fue del 97,1%.
Eficacia en infección por VHC de genotipos 2 y 3
El estudio FISSION (Lawitz, 2013) es un ensayo clínico abierto, aleatorizado y multicéntrico realizado sobre 499 pacientes naïve, que recibieron un tratamiento de 12 semanas con sofosbuvir (SOF) y ribavirina (RBV) de 12 semanas, o de peginterferón alfa-2a (IFN) más ribavirina (RBV) de 24 semanas. La mediana de edad era de 50 años, un 66% eran varones, un 87% de raza blanca, con un IMC medio de 28 y un 20% tenían cirrosis hepática; la mediana de la carga viral era de 6,3 log10 (2.000.000) copias/ml (el 70% tenía más de 6 log10). Un 51% presentaban VHC de genotipo 2 y el 49% de genotipo 3.
La tasa global de respuesta (RVS12) fue del 67% (IC95% 60,7 a 72,5) con SOF/RBV12 vs. del 67% (IC95% 60,4 a 72,6) con IFN/RBV24, con una diferencia ajustada de 0,3 puntos porcentuales (IC95% -7,5 a 8,0), siendo del 97% vs. 81% (genotipo 2) y del 61% vs. 71% (genotipo 3) entre los no cirróticos y del 83% vs. 62% (genotipo 2) y del 34% vs. 30% (genotipo 3) entre los cirróticos; la tasa de recaídas fue del 30,1% (SOF/RBV) vs. 21,2% (IFN/RBV) y la de fallo virológico del 29,6% (SOF/RBV) vs. 26,3% (IFN/RBV).
El estudio POSITRON (Jacobson, 2013) es un ensayo clínico multicéntrico, aleatorizado, doblemente ciego y controlado con placebo, realizado sobre 278 pacientes intolerantes, que no eran candidatos o que habían rechazado ser tratados con interferón y que recibieron un tratamiento de 12 semanas con sofosbuvir (SOF) y ribavirina (RBV), o con placebo. La mediana de edad era de 54 años, un 54% eran varones, un 91% de raza blanca, con un IMC medio de 28; el 70% tenía una carga viral igual o mayor de 6 log10. Un 51% presentaban VHC de genotipo 2 y el 49% de genotipo 3.
La tasa global de respuesta (RVS12) fue del 78% (IC95% 71,5 a 83,2) vs. 0% (IC95% 0 a 5,1) con placebo, siendo del 92% vs. 0% (genotipo 2) y del 98% vs. 0% (genotipo 3) entre los no cirróticos y del 94% vs. 0% (genotipo 2) y del 21% vs. 0% (genotipo 3) entre los cirróticos; la tasa global de recaídas fue del 20,5% (SOF/RBV) vs. 0% (placebo) y la de fallo virológico del 20,3% (SOF/RBV) vs. 97,2% (placebo).
El estudio FUSION (Jacobson, 2013) es un ensayo clínico aleatorizado y doblemente ciego, realizado sobre 201 pacientes previamente tratados con interferón (con recaída o sin respuesta) y que recibieron un tratamiento de 12 o de 16 semanas con sofosbuvir (SOF) y ribavirina (RBV). La mediana de edad era de 56 años, un 70% eran varones, un 87% de raza blanca, con un IMC medio de 29; el 73% tenía una carga viral igual o mayor de 6 log10. Un 37% presentaban VHC de genotipo 2 y el 63% de genotipo 3; el 34% presentaba cirrosis.
La tasa global de respuesta (RVS12) fue del 49,5% (IC95% 39,5 a 59,5) con 12 semanas vs. 74,4% (IC95% 61,4 a 80,1) con 16 semanas de tratamiento, siendo del 87% vs. 62% (genotipo 2) y del 60% vs. 78% (genotipo 3) entre los no cirróticos y del 37% vs. 63% (genotipo 2) y del 19% vs. 71% (genotipo 3) entre los cirróticos; las tasas globales de recaídas y de fallo virológico fueron del 46,6% (12 sem.) vs. 28,5% (16 sem.).
En el estudio VALENCE (Zeuzem, 2014), un ensayo clínico aleatorizado, doblemente ciego y controlado, se comparó el tratamiento de de sofosbuvir más ribavirina vs. placebo durante 12 semanas, en un conjunto de 419 pacientes tanto naïve como tratados anteriormente; sin embargo, a mitad del estudio se modificaron las condiciones y todos los pacientes con VHC de genotipo 3 fueron tratados durante 24 semanas con sofosbuvir y ribavirina, finalizándose el grupo placebo. La mediana de edad era de 51 años, un 60% eran varones, con un IMC medio de 25; la mediana de la carga viral era de 6,4 log10 (2.500.000) copias/ml. Un 22% presentaban VHC de genotipo 2 y el 78% de genotipo 3; y el 21% presentaba cirrosis.
Las tasas globales de respuesta (RVS12) fueron del 93% (genotipo 2, 12 semanas) y del 98% (genotipo 3, 24 semanas). En los pacientes sin tratamiento previo (naïve) fue del 97% (genotipo 2, 12 semanas) y del 93% (genotipo 3, 24 semanas) entre los no cirróticos y del 100% (genotipo 2, 12 semanas) y del 92% (genotipo 3, 24 semanas) entre los cirróticos. Por su parte, en los pacientes con tratamiento previo fue del 91% (genotipo 2, 12 semanas) y del 85% (genotipo 3, 24 semanas) entre los no cirróticos y del 88% (genotipo 2, 12 semanas) y del 60% (genotipo 3, 24 semanas) entre los cirróticos.
Por último, el estudio PHOTON-1 (Sulkowski, 2014) es un ensayo clínico abierto y multicéntrico, realizado sobre 223 pacientes, de los que 182 eran naïve (114 de genotipo 1 y 68 de genotipo 2 o 3) y los restantes 41 habían sido tratados anteriormente con interferón y ribavirina (todos eran de genotipo 2 o 3). Los pacientes naïve recibieron 12 semanas de tratamiento con sofosbuvir y ribavirina, y previamente tratados 24 semanas.
La tasa global de respuesta (RVS12) fue del 78% (genotipo 1) y 75% (genotipos 2 o 3) entre los pacientes naïve y del 93% entre los tratados anteriormente (genotipos 2 o 3). Considerando específicamente el genotipo, entre los pacientes con VHC de genotipo 2, la tasa de eficacia fue del 88% en pacientes naïve (88% sin cirrosis y 100% con cirrosis) y del 93% en aquellos con tratamiento previo (92% sin cirrosis y 100% con cirrosis). Por su parte, entre los pacientes con VHC de genotipo 3, la tasa de eficacia fue del 67% en pacientes naïve (67% con o sin cirrosis) y del 92% en aquellos con tratamiento previo (100% sin cirrosis y 80% con cirrosis).
Seguridad
La incidencia de eventos adversos de gravedad de grado 2 o superior en los ensayos clínicos pivotales fue del 30% con placebo (PLA), 42% sofosbuvir y ribavirina en 12 semanas (SOF+RBV/12), 42% sofosbuvir y ribavirina en 16 semanas (SOF+RBV/16), 69% con peginterferón alfa-2a y ribavirina en 24 semanas (IFN+RBV/24) y 59% con sofosbuvir, peginterferón alfa-2a y ribavirina en 12 semanas (SO+IFN+RBV/12).
- Eventos adversos graves: 2,8% (P); 3,9% (SOF+RBV/12); 3,1% (SOF+RBV/16); 1,2% (IFN+RBV/24) y 1,2% (SOF+IFN+RBV/12).
- Suspensión del tratamiento por adversos: 4,2% (P); 1,4% (SOF+RBV/12); 0% (SOF+RBV/16); 10,7% (IFN+RBV/24) y 1,5% (SOF+IFN+RBV/12).
-
Eventos adversos más comunes:
- Fatiga: 24% (P); 41% (SOF+RBV/12); 47% (SOF+RBV/16); 55% (IFN+RBV/24) y 59% (SOF+IFN+RBV/12).
- Cefalea: 20% (P); 23% (SOF+RBV/12); 33% (SOF+RBV/16); 44% (IFN+RBV/24) y 36% (SOF+IFN+RBV/12).
- Náusea: 18% (P); 20% (SOF+RBV/12); 20% (SOF+RBV/16); 29% (IFN+RBV/24) y 34% (SOF+IFN+RBV/12).
- Insomnio: 4,2% (P); 16% (SOF+RBV/12); 29% (SOF+RBV/16); 29% (IFN+RBV/24) y 25% (SOF+IFN+RBV/12).
- Irritabilidad: 1,4% (P); 10% (SOF+RBV/12); 11% (SOF+RBV/16); 17% (IFN+RBV/24) y 13% (SOF+IFN+RBV/12).
- Anemia: 0% (P); 10% (SOF+RBV/12); 4% (SOF+RBV/16); 12% (IFN+RBV/24) y 21% (SOF+IFN+RBV/12).
ASPECTOS INNOVADORES
El sofosbuvir es un agente antiviral activo frente a virus de la hepatitis C (VHC), que actúa inhibiendo específicamente la proteína NS5B, una ARN polimerasa dependiente del ARN viral, lo que impide la replicación del material genético viral. Ha sido autorizado para el tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos.
Las tasas globales de respuesta virológica sostenida en pacientes con VHC de genotipo 1 son, en combinación con peginterferón alfa-2a y ribavirina son muy elevadas (90% y 80% en pacientes sin y con cirrosis, respectivamente). Por otro lado, la utilización de pautas de 24 semanas de duración y sin interferón obtiene tasas del 50-70%, superiores a las obtenidas con pautas de interferón y ribavirina de 48 semanas. Por otro lado, se obtienen tasas cercanas al 80% en pacientes coinfectados con VIH utilizando pautas de 12 o 24 semanas de sofosbuvir y ribavirina (con o sin tratamiento previo para el VIH).
En infecciones por VHC de genotipo 2, las tasas de respuesta están en torno al 95% en pacientes naïve y del 93% en aquellos inelegibles o intolerantes al interferón, con pautas de sofosbuvir y ribavirina de 12 semanas. Por su parte, en los casos ligados al genotipo 3 se requieren tratamientos de sofosbuvir y ribavirina de 24 semanas de duración, ya que las de 12 semanas producen resultados notablemente inferiores. Por último, los datos correspondientes a los genotipos 4, 5 y 6 – que son raros en la Unión Europea – son escasos, aunque no parecen diferir sustancialmente de los obtenidos para el genotipo 1.
Algunos datos (Lawitz, 2014) sugieren resultados muy convincentes asociando simeprevir y sofosbuvir, tanto en pacientes pretratados no respondedores al peginterferón alfa como en naïve y con independencia del nivel de fibrosis hepática. Precisamente, la eficacia de esta combinación, en ausencia de interferón, es muy apreciable al añadir el ahorro de la toxicidad del interferón y mejorar así la tolerabilidad del tratamiento (Gaetano, 2014).
Igualmente, se han obtenido excelentes resultados en pacientes con VHC de genotipo 1, con la combinación de ledipasvir y sofosbuvir, solos o en combinación con ribavirina, tanto en pacientes naïve con tasas de respuesta virológica sostenida por encima del 95% con tratamientos de 12 y 24 semanas (ION-1; Afdhal, 2014a), como en pacientes previamente tratados (ION-2; Afdhal, 2014b), e incluso en tratamientos abreviados de 8 semanas de duración (ION-3; Kowdley, 2014).
Asimismo, también hay datos consistentes que apuntan la utilidad del sofosbuvir en pacientes sometidos a trasplante hepático, algo especialmente relevante; a lo que hay que agregar la utilidad del fármaco en el retratamiento de pacientes no curados anteriormente.
El sofosbuvir tiene presenta una buena tolerabilidad en combinación con ribavirina, con o sin interferón, sin que se hayan observado efectos adversos específicos del fármaco, mostrando mejor tolerabilidad que el interferón.
Es importante tener en cuenta la reducción de la duración del tratamiento que supone la incorporación del sofosbuvir a los regímenes, con 12 semanas de pacientes naïve, frente a las 24-48 semanas con interferón, con la ventaja adicional de requerir una única dosis oral diaria (Kholi, 2014) y la práctica ausencia de interacciones farmacológicas clínicamente relevantes (Degasperi, 2014).
Sin duda, el sofosbuvir presenta unos excelentes resultados clínicos, bien avalados por ensayos clínicos amplios y rigurosos metodológicamente, con un amplio espectro de actividad frente a los diversos genotipos de VHC circulantes, a lo cual hay que añadir su elevada barrera frente a la generación de resistencias por mutaciones de la proteína NS5B.
Tras la primera oleada de antivirales específicos frente al VHC, con boceprevir y telaprevir, que ciertamente modificaron el pronóstico y las expectativas terapéuticas en este campo, el sofosbuvir forma parte de una segunda oleada, junto con simeprevir, daclastavir, ledipasvir y algunos otros, que aportan elevadas tasas de curación en todos los subtipos de pacientes, a través de regímenes más sencillos y mejor tolerados (Jiménez Galán, 2014), con la particularidad de que van eliminando la otrora imprescindible presencia del interferón alfa en el tratamiento.
Estos nuevos fármacos, autorizados hace apenas unos meses, han tenido tal impacto que han dejado obsoleta rápidamente a la guía de práctica clínica de la Asociación Europea para el Estudio del Hígado (European Association for the Study of the Liber, EASL) publicada en diciembre de 2013, obligando a una nueva edición (EASL, 2014) en la que incluye las nuevas recomendaciones.
En concreto, en el caso del tratamiento de la hepatitis C de genotipo 1, la guía establece como opciones preferentes de tratamiento las combinaciones de sofosbuvir o de simeprevir con peginterferon + ribavirina. Como alternativas recomienda daclatasvir junto con peginterferon + ribavirina, sofosbuvir + simeprevir durante 12 semanas o sofosbuvir + daclatasvir durante 12 o 24 semanas.
Para el genotipo 2 se recomienda una combinación de ribavirina y sofosbuvir durante 12 semanas (16-20 en casos de pacientes cirróticos). Para el genotipo 3, la combinación es de sofosbuvir con peginterferon alfa-2a y ribavirina durante 12 semanas, y como alternativas ribavirina y sofosbuvir durante 12 semanas (no aconsejable en pacientes cirróticos) o sofosbuvir y daclastavir durante 12 (naïve) o 24 semanas (pretratados). Para los cuadros asociados al genotipo 4, se aconseja el simeprevir durante 12 semanas asociados a peginterferón y ribavirina durante 24 o 48 semanas (naïve o pretratados), citándose como alternativas peginterferón más ribavirina y daclastavir durante 24 semanas o sofobuvir y ribavirina durante 24 semanas. Finalmente, para las infecciones por el genotipo 5 o 6 se recomienda peginterferón más ribavirina y sofosbuvir durante 12 semanas o, como alternativa, ribavirina y sofosbuvir durante 24 semanas.
Es importante no olvidar que la prevalencia de la hepatitis C en España se sitúa en entorno al 1-2% de la población, lo que supone que el número de personas infectadas está entre 500.000 y 1.000.000. Habida cuenta de que un 60-80% desarrollan la infección crónica y que un 20% de estos evoluciona a cirrosis hepática al cabo de 20-25 años, de los cuales un 80% acabará por padecer un cáncer de hígado, es evidente el interés sanitario y económico de un tratamiento curativo eficaz.
|
VALORACIÓN |
|
|---|---|
|
Sofosbuvir ► SOVALDI® (Gilead) |
|
|
Grupo Terapéutico (ATC): J05AE. ANTIINFECCIOSOS SISTÉMICOS. Antivirales de acción directa: inhibidores de la proteasa. |
|
|
Indicaciones autorizadas: Tratamiento, en combinación con otros medicamentos, de la hepatitis C crónica en adultos. |
|
|
Valoración global: INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar. |
♣ ♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar |
⇑ |
|
Novedad molecular: Incorpora variaciones significativas en el mecanismo de acción: |
⇑ |
|
Novedad toxicológica: Mejora el perfil toxicológico con relación a la terapia farmacológica estándar. |
⇑ |
|
Novedad físico-química: Mejora las características farmacocinéticas, con incidencia en las condiciones de uso. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Boceprevir |
Victrelis |
Merck Sharp Dohme |
2011 |
|
Telaprevir |
Incivo |
Janssen Cilag |
2011 |
|
Simeprevir |
Olysio |
Janssen Cilag |
2014 |
|
Sofosbuvir |
Solvaldi |
Gilead |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
Bibliografía
Medicamentos con nuevos principios activos autorizados por EMA/AEMPS
|
MEDICAMENTOS DE USO HUMANO AUTORIZADOS (EMA/AEMPS) DURANTE LOS ÚLTIMOS DOCE MESES, CON NUEVOS PRINCIPIOS ACTIVOS QUE AÚN NO ESTÁN COMERCIALIZADOS EN ESPAÑA |
|||||
|---|---|---|---|---|---|
|
Principio activo |
Medicamento® |
Laboratorio |
Autorización |
GT |
Indicación autorizada1 |
|
Daclatasvir |
Daklinza |
Bristol Myers Squibb |
2014/08/22 |
J05AX |
Hepatitis C |
|
Ataluren |
Translarna |
PTC Therapeutics |
2017/07/31 |
– |
Distrofia muscular de Duchenne (O) (C) |
|
Obinotuzumab |
Gazyvaro |
Roche |
2014/07/23 |
L01XC |
Leucemia linfocítica crónica (O) |
|
Peginterferón beta-1a |
Plegridy |
Biogen |
2014/07/18 |
L03AB |
Esclerosis múltiple |
|
Trametinib |
Mekinist |
Glaxo |
2017/06/30 |
L01XE |
Melanoma |
|
Vedolizumab |
Entyvio |
Takeda |
2014/05/22 |
L04AA |
Enfermedad Inflamatoria Intestinal |
|
Siltuximab |
Sylvant |
Janssen Cilag |
2014/05/22 |
– |
Hiperplasia nódulo linfático (O) |
|
Empagliflozina |
Jardiance |
Boehringer Ingelheim |
2014/05/22 |
A10BX |
Diabetes tipo 2 |
|
Elosulfasa alfa |
Vimizim |
BioMarin |
2014/04/28 |
A16AB |
Mucopolisacaridosis IV (O) |
|
Umeclidinio, bromuro |
Incruse |
Glaxo |
2014/04/28 |
R03BB |
EPOC |
|
Delamanid |
Deltyba |
Otsuka |
2014/04/28 |
J04AK |
Tuberculosis multirresistente (O) (C) |
|
Riociguat |
Adempas |
Bayer |
2014/03/27 |
C02KX |
Hipertensión pulmonar (O) |
|
Albiglutida |
Eperzan |
GlaxoSmithKline |
2014/03/21 |
A10BX |
Diabetes mellitus de tipo 2 |
|
Lurasidona |
Latuda |
Takeda |
2014/03/21 |
N05AE |
Esquizofrenia |
|
Cabozantinib |
Cometriq |
TMC |
2014/03/21 |
L01XE |
Cáncer de tiroides (O) (C) |
|
Bedaquilina |
Sirturo |
Janssen Cilag |
2014/03/05 |
J04AX |
Tuberculosis multirresistente (O) (C) |
|
Florbetaben (18F) |
Neuraceq |
Piramidal Imaging |
2014/02/20 |
V09AX |
Diagnóstico Alzheimer |
|
Fumarato de dimetilo |
Tecfidera |
Biogen Idec |
2014/01/30 |
N07XX |
Esclerosis múltiple |
|
Sofosbuvir |
Sovaldi |
Gilead |
2014/01/16 |
J05AX |
Hepatitis C |
|
Macitentam |
Opsumit |
Actelion |
2013/12/20 |
C02KX |
Hipertensión pulmonar (O) |
|
Vortioxetina |
Brintellix |
Lundbeck |
2013/12/18 |
N06AX |
Depresión |
|
Virus gripales inactivados |
Fluenz Tetra |
Med Immune |
2013/12/04 |
J07BB |
Gripe, profilaxis |
|
Elvitegravir2 |
Vitekta |
Gilead |
2013/11/13 |
J05AX |
VIH |
|
Turoctocog alfa |
NovoEight |
Novo Nordisk |
2013/11/13 |
B02BD |
Hemofilia A |
|
Canagliflozina |
Invokana |
Janssen Cilag |
2013/11/15 |
A10BX |
Diabetes mellitus de tipo 2 |
|
Radio (223Ra), cloruro |
Xofigo |
Bayer |
2013/11/13 |
V10XX |
Cáncer de próstata |
|
Defibrótido |
Defitelio |
Gentium |
2013/10/18 |
B01AX |
Enfermedad hepática venooclusiva (O) (E) |
1 O= Medicamento huérfano; C= Autorizado condicionalmente; E= Autorizado en condiciones excepcionales
2 Elvitegravir ya está comercializado en España (Stribild®), asociado a cobicistat, emtricitabina y tenofovir (2013/06/26)
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA EN LOS ÚLTIMOS DOCE MESES
|
PAM |
Principio activo |
Medicamento |
INNOVACIÓN |
Indicación principal |
Laboratorio |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
|
A |
B |
C |
D |
E |
F |
|||||
|
377 |
Sofosbuvir |
Sovaldi |
⇑ |
⇑ |
⇑ |
⇑ |
|
♣♣♣ |
Hepatitis C |
Gilead |
|
377 |
Decitabina |
Dacogen |
⇑ |
|
|
|
|
♣♣♣ |
Leucemia mieloide aguda |
Janssen Cilag |
|
377 |
Vilanterol/Fluticasona |
Relvar Ellipta |
|
|
|
⇑ |
|
♣♣ |
Asma y EPOC |
Glaxo SmithKline |
|
377 |
Vacuna meningococo B |
Bexsero |
⇑ |
|
|
|
⇑ |
♣♣♣ |
Prevención meningitis meningocócica B |
Novartis |
|
377 |
Dexmedetomidina |
Dexdor |
|
⇑ |
|
|
|
♣♣ |
Sedoanalgesia |
Orion |
|
377 |
Dolutegravir |
Tivicay |
⇑ |
|
|
⇑ |
|
♣♣ |
Infección por VIH |
Glaxo SmithKline |
|
376 |
Brentuximab vedotina |
Adcetris |
⇑ |
⇑ |
|
|
|
♣♣ |
Linfomas |
Takeda |
|
376 |
Ocriplasmina |
Jetrea |
⇑ |
|
⇑ |
|
|
♣♣ |
Tracción vitreomacular |
Alcon Cusi |
|
376 |
Ofatumumab |
Arzerra |
⇑ |
|
|
|
|
♣♣ |
Leucemia linfocítica crónica |
Glaxo SmithKline |
|
376 |
Pirfenidona |
Esbriet |
⇑ |
⇑ |
|
|
|
♣♣ |
Fibrosis pulmonar idiopática |
InterMune |
|
376 |
Simeprevir |
Olysio |
⇑ |
⇑ |
⇑ |
⇑ |
|
♣♣♣ |
Hepatitis C |
Janssen Cilag |
|
376 |
Tetradecilsulfato sódico |
Veinfibro |
|
|
|
|
|
♣ |
Escleroterapia de venas varicosas |
STD |
|
375 |
Afatinib |
Giotrif |
⇑ |
|
|
|
|
♣♣ |
Cáncer de pulmón |
Boehringer Ingelheim |
|
375 |
Linaclotida |
Constella |
⇑ |
⇑ |
|
|
|
♣♣ |
Síndrome del intestino irritable |
Almirall |
|
375 |
Pertuzumab |
Perjeta |
⇑ |
⇑ |
|
|
|
♣♣♣ |
Cáncer de mama |
Roche |
|
375 |
Pomalidomida |
Imnovid |
⇑ |
|
⇑ |
|
|
♣♣ |
Mieloma múltiple |
Celgene |
|
374 |
Axitinib |
Inlyta |
⇑ |
|
|
|
|
♣♣ |
Cáncer renal |
Pfizer |
|
374 |
Brimonidina3 |
Mirvaso |
⇑ |
|
|
|
|
♣♣ |
Rosácea |
Galderma |
|
374 |
Dabrafenib |
Tafinlar |
⇑ |
|
|
⇑ |
|
♣♣ |
Melanoma |
Pfizer |
|
374 |
Vemurafenib |
Zelboraf |
⇑ |
|
|
⇑ |
|
♣♣ |
Melanoma |
Roche |
|
373 |
Lisdexanfetamina |
Elvanse |
⇑ |
|
|
|
|
♣♣ |
Trastorno por déficit de atención-hiperactividad |
Shire |
|
372 |
Avanafilo |
Spedra |
|
|
|
|
|
♣ |
Disfunción eréctil |
Menarini |
|
372 |
Mirabegrón |
Betmiga |
|
⇑ |
⇑ |
|
|
♣♣ |
Incontinencia urinaria |
Astellas |
|
372 |
Regadenosón |
Rapiscan |
|
|
|
⇑ |
|
♣♣ |
Diagnóstico en cardiología |
Mallincrockrodt |
|
371 |
Catridecacog |
Novothirteen |
|
|
|
|
⇑ |
♣♣ |
Déficit congénito Factor XIII |
Novo Nordisk |
|
371 |
Florbetapir [18F] |
Amyvid |
⇑ |
|
|
|
|
♣♣ |
Diagnóstico Alzheimer |
Lilly |
|
370 |
Clevidipino |
Cleviprex |
⇑ |
|
|
⇑ |
|
♣♣ |
Hipertensión perioperatoria |
Ferrer |
|
370 |
Crizotinib |
Xalkori |
|
⇑ |
|
|
|
♣♣ |
Cáncer de pulmón |
Pfizer |
|
370 |
Elvitegravir (+ Cobicistat, Emtricitabina y Tenofovir) |
Stribild |
|
|
|
⇑ |
|
♣♣ |
Infección VIH |
Gilead |
|
370 |
Perampanel |
Fycompa |
⇑ |
⇑ |
|
|
|
♣♣ |
Epilepsia inicio parcial |
Eisai |
|
370 |
Tafamidis |
Vyndaqel |
⇑ |
⇑ |
|
|
|
♣♣ |
Amiloidosis transtiretina |
Pfizer |
|
369 |
Dapagliflozina |
Forxiga |
⇑ |
⇑ |
|
|
|
♣♣ |
Diabetes mellitus tipo 2 |
Bristol Myers Squibb |
|
368 |
Ulipristal |
Esmya |
|
|
|
|
|
♣ |
Miomas uterinos |
Gedeon Richter |
A. Clínica: Mejora la eficacia clínica con relación al tratamiento estándar.
B. Molecular: Incorpora variaciones significativas en la estructura molecular
y/o en el mecanismo de acción.
C. Seguridad: Mejora el perfil toxicológico con relación a la terapia estándar.
D. Farmacocinética: Mejora las características farmacocinéticas, con incidencia potencial o demostrada en las condiciones de uso y en la respuesta del paciente.
E. Técnico-Económica: Mejora algún aspecto farmacoeconómico y/o farmacotécnico.
F. Valoración Global
3 La brimonidina estaba previamente comercializada para el tratamiento tópico oftálmico del glaucoma.
VALORACIÓN DE LA INNOVACIÓN TERAPÉUTICA EN PANORAMA ACTUAL DEL MEDICAMENTO
Es importante indicar que se valora el grado de innovación. Todos los medicamentos no innovadores tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas. Asimismo, debe considerarse que ésta es una evolución que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta ese momento, lo que no prejuzga, en ningún caso, el potencial desarrollo de nuevas indicaciones clínicas ni la posible aparición de aspectos desfavorables (efectos adversos graves previamente desconocidos, interacciones, etc.). Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
– SIN INNOVACIÓN. No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas (♣)
– INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar (♣ ♣)
– INNOVACIÓN IMPORTANTE. Aportación sustancial a la terapéutica estándar (♣ ♣ ♣).
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
– Evidencia clínica: mediante estudios controlados específicamente diseñados y desarrollados para demostrar lo que pretende ser un avance o mejora sobre la terapia estándar.
– Potencialidad: existencia de aspectos en el medicamento que racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostradas mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA DURANTE LOS ÚLTIMOS DOCE MESES, POR GRUPOS TERAPÉUTICOS
|
GRUPO TERAPËUTICO |
Principio activo |
Medicamento |
Laboratorio |
PAM |
|---|---|---|---|---|
|
A. Tracto alimentario y metabolismo |
Dapagliflozina |
Forxiga |
Bristol Myers Squibb |
369 |
|
Linaclotida |
Constella |
Almirall |
375 |
|
|
B. Sangre y órganos hematopoyéticos |
Catridecacog |
Novothirteen |
Novo Nordisk |
371 |
|
C. Aparato cardiovascular |
Clevidipino |
Cleviprex |
Ferrer |
370 |
|
Regadenosón |
Rapiscan |
Mallinckrodt |
372 |
|
|
Tetradecilsulfato sódico |
Veinfibro |
STD |
376 |
|
|
D. Terapia dermatológica |
Brimonidina |
Mirvaso |
Galderma |
374 |
|
G. Terapia genitourinaria |
Avanafilo |
Spedra |
Menarini |
372 |
|
Mirabegrón |
Betmiga |
Astellas |
372 |
|
|
Ulipristal |
Esmya |
Gedeon Richter |
368 |
|
|
J. Antiinfecciosos, uso sistémico |
Dolutegravir |
Tivicay |
Glaxo SmithKline |
377 |
|
Elvitegravir (+ Cobicistat, Emtricitabina y Tenofovir) |
Stribild |
Gilead |
370 |
|
|
Simeprevir |
Olysio |
Janssen Cilag |
376 |
|
|
Sofosbuvir |
Sovaldi |
Gilead |
377 |
|
|
Vacuna meningococo B |
Bexsero |
Novartis |
377 |
|
|
L. Antineoplásicos y terapia inmunomoduladora |
Afatinib |
Giotrif |
Boehringer Ingelheim |
375 |
|
Axitinib |
Inlyte |
Pfizer |
374 |
|
|
Brentuximab vedotina |
Adcetris |
Takeda |
376 |
|
|
Crizotinib |
Xalkori |
Pfizer |
370 |
|
|
Dabrafenib |
Tafinlar |
Pfizer |
374 |
|
|
Decitabina |
Dacogen |
Janssen Cilag |
377 |
|
|
Ofatumumab |
Arzerra |
Glaxo SmithKline |
376 |
|
|
Pertuzumab |
Perjeta |
Roche |
375 |
|
|
Pirfenidona |
Esbriet |
InterMune |
376 |
|
|
Pomalidomida |
Immovid |
Celgene |
375 |
|
|
Vemurafenib |
Zelboraf |
Roche |
375 |
|
|
N. Sistema nervioso |
Dexmedetomidina |
Dexdor |
Orion |
377 |
|
Lisdexanfetamina |
Elvanse |
Shire |
373 |
|
|
Perampanel |
Fycompa |
Eisai |
370 |
|
|
Tafamidis |
Vyndaqel |
Pfizer |
370 |
|
|
R. Aparato respiratorio |
Vilanterol/Fluticasona |
Relvar Ellipta |
Glaxo SmithKline |
377 |
|
S. Órganos de los sentidos |
Ocriplasmina |
Jetrea |
Alcon Cusi |
376 |
|
V. Varios |
Florbetapir [18F] |
Amyvid |
Lilly |
371 |
MEDICAMENTOS CON NUEVOS PRINCIPIOS ACTIVOS COMERCIALIZADOS EN ESPAÑA DURANTE LOS ÚLTIMOS DOCE MESES, POR LABORATORIOS
|
Laboratorio |
Principio activo |
Medicamento |
PAM |
|---|---|---|---|
|
Alcon Cusi |
Ocriplasmina |
Jetrea |
376 |
|
Almirall |
Linaclotida |
Constella |
375 |
|
Astellas |
Mirabegrón |
Betmiga |
372 |
|
Boehringer Ingelheim |
Afatinib |
Giotrif |
375 |
|
Bristol Myers Squibb |
Dapagliflozina |
Forxiga |
369 |
|
Celgene |
Pomalodomida |
Imnovid |
375 |
|
Eisai |
Perampanel |
Fycompa |
370 |
|
Ferrer |
Clevidipino |
Cleviprex |
370 |
|
Galderma |
Brimonidina |
Mirvaso |
374 |
|
Gedeon Richter |
Ulipristal |
Esmya |
368 |
|
Gilead |
Elvitegravir (+ Cobicistat, Emtricitabina yTenofovir) |
Stribild |
370 |
|
Sofosbuvir |
Sovaldi |
377 |
|
|
Glaxo SmtihKline |
Dolutegravir |
Tivicay |
377 |
|
Ofatumumab |
Arzerra |
376 |
|
|
Vilanterol/Fluticasona |
Relvar Ellipta |
377 |
|
|
InterMune |
Pirfenidona |
Esbriet |
376 |
|
Janssen Cilag |
Decitabina |
Dacogen |
377 |
|
Simeprevir |
Olysio |
376 |
|
|
Lilly |
Florbetapir [18F] |
Amyvid |
371 |
|
Mallinckrodt |
Regadenosón |
Rapiscan |
372 |
|
Menarini |
Avanafilo |
Spedra |
372 |
|
Novartis |
Vacuna meningococo B |
Bexsero |
377 |
|
Novo Nordisk |
Catridecacog |
Novothirteen |
371 |
|
Orion |
Dexmedetomidina |
Dexdor |
377 |
|
Pfizer |
Axitinib |
Inlyta |
374 |
|
Crizotinib |
Xalkori |
370 |
|
|
Dabrafenib |
Tafinlar |
374 |
|
|
Tafamidis |
Vyndaqel |
370 |
|
|
Roche |
Pertuzumab |
Perjeta |
375 |
|
Vemurafenib |
Zelboraf |
374 |
|
|
Shire |
Lisdexanfetamina |
Elvanse |
373 |
|
STD |
Tetradecilsulfato sódico |
Veinfibro |
376 |
|
Takeda |
Brentuximab vedotina |
Adcetris |
376 |
Patología Vascular Urgente
Resumen
La isquemia arterial aguda es la patología vascular urgente más grave. Produce una alta tasa de morbimortalidad si no se trata a tiempo. Una adecuada anamnesis y una exploración exhaustiva siguen siendo los pilares fundamentales para el diagnóstico de todas las patologías vasculares. La anticoagulación con heparinas de bajo peso molecular (HBPM) y Antagonistas de la vitamina K sigue siento un tratamiento eficaz en la trombosis venosa profunda pero requiere un adecuado seguimiento clínico y analítico.
INTRODUCCION
Las consecuencias de las enfermedades vasculares periféricas son importantes tanto para el paciente como para el sistema sanitario, por la invalidez que producen, la morbimortalidad asociada y el coste socio económico que suponen.
Es importante resaltar que en el caso de la patología aguda de EEII (extremidades inferiores), pese a la proliferación de pruebas complementarias utilizadas para la realización de un diagnóstico adecuado que ha tenido lugar en las últimas décadas, siguen siendo la anamnesis y la exploración física la base que sustenta el propio diagnóstico de sospecha a partir del cual debemos tomar las decisiones apropiadas.
Clasificación
A continuación vamos a dividir la patología vascular urgente en dos grupos. Primero la patología arterial aguda y segundo grupo la patología venosa aguda.
-
Patología arterial aguda: (Isquemia Aguda)
- Embolia arterial (30%)
- Trombosis arterial aguda (70%)
-
Patología venosa aguda:
- Trombosis venosa profunda.
- Trombosis venosa superficial
ISQUEMIA AGUDA
Es una manifestación clínica producida por la interrupción brusca de aporte sanguíneo a un territorio determinado de una extremidad como consecuencia de la oclusión súbita de la arteria que la irriga.
Fisiopatología
La isquemia aguda está condicionada por varios hechos de relevancia:
- El obstáculo mecánico que significa la obliteración brusca de la arteria. Es el factor fundamental y el más grave y de él dependen en gran parte los demás.
La isquemia no sería tan grave si al obstáculo mecánico no se añadiera habitualmente una trombosis secundaria, que da lugar a dos consecuencias importantes:
- Mayor extensión del territorio obstruido.
- Oclusión de la circulación colateral, es este sentido, el estado de la circulación colateral es un factor condicionante ya que una buena circulación colateral puede evitar en si misma la muerte del tejido cuando se produce la oclusión del tronco arterial principal.
- Espasmo: Sobre todo en los casos de isquemia aguda por embolia existe un importante espasmo, tanto en el tronco principal por debajo del obstáculo como a nivel de la circulación colateral. Por lo tanto, éste sería un factor agravante de la isquemia, al menos en su fase inicial.
- Resistencia de los tejidos a la isquemia: La gravedad de la isquemia va a estar también condicionada en función de los tejidos afectados. Así los nervios y fibras musculares son capaces de soportar una situación de déficit de aporte arterial entre 4 y 6 horas (a partir de ese tiempo las lesiones que se produzcan serán irreversibles). Sin embargo la piel es capaz de soportar la isquemia durante 12-24 horas.
Los tejidos sometidos a isquemia no mueren inmediatamente, pero su función si que cesa total o parcialmente. Al principio la falta de aporte de sangre arterial en presencia de un buen drenaje venoso origina palidez (por disminución de la cantidad de sangre en el tejido isquémico) y frialdad (por la pérdida de la capacidad termorreguladora del sistema capilar). Posteriormente, al ocluirse la circulación colateral disminuye aún mas el aporte y los tejidos agotan el poco oxígeno que les llega, siendo además incapaces de liberarse de esa sangre que se habrá hecho rica en anhidrido carbónico. Esto justifica la cianosis en la isquemia grave avanzada.
Etiología
Las causas más habituales de isquemia aguda son:
- Embolia
- Trombosis arterial aguda.
La embolia supone una obliteración brusca de una arteria sana o patológica por material procedente de otro territorio proximal (émbolo), el cual se desprende y es arrastrado por la corriente sanguínea yendo a ocluir una arteria de menor calibre. El origen de los émbolos puede ser diverso: trombos de procedencia cardíaca e incluso material embolígeno de naturaleza no trombótica (placas de ateromas, cristales de colesterol).
- Cardíacas: Arritmias, Valvulopatías , Fallo cardiaco, Infarto de miocardio, endocarditis, tumores cardíacos, aneurismas ventriculares.
- Arteriales: Ateroembolias, Aneurismas arteriales.
- Otras: Embolia paradójica (a partir de Trombosis venosa profunda en caso de coexistir Comunicación interauricular o interventricular). Iatrogénicas: Catéteres, vávulas cardíacas, cirugía de aneurismas.
Por su parte, la trombosis arterial aguda es una obliteración brusca de una arteria, habitualmente patológica, por material originado en el mismo lugar donde se produce la obliteración. Los principales factores concurrentes son:
- Factor de la pared vascular: ateroesclerosis, aneurismas, displasias, necrosis quística, vasculitis, tromboangeitis obliterante, traumatismos.
- Factor hemodinámico: aneurismas, anomalías anatómicas, hipotensión, bajo gasto, deshidratación.
- Factor hematológico: sindromes mieloproliferativos, trombocitosis esencial, crioglobulinemias, coagulopatía intravascular, neoplasias, estadíos de hipercoagulabilidad.
Clínica
La isquemia aguda se caracteriza por ser un cuadro agudo y aparatoso, que se manifiesta por una serie de síntomas y signos: (tabla 1)
- DOLOR de aparición brusca, agudo, intenso y persistente en la extremidad afecta que no se modifica con la postura y que existe tanto en decúbito como en bipedestación.
- PALIDEZ inicialmente. A medida que transcurre el tiempo de isquemia se traduce en cianosis.
- FRIALDAD intensa y persistente.
- ANESTESIA Y PARÁLISIS: Por afectación sensitiva y motora, al cabo de las 4-8 horas de isquemia.
- IMPOTENCIA FUNCIONAL, al cabo del tiempo se transforma en paresia, anestesia y parálisis.
- AUSENCIA DE PULSOS.
|
Tabla 1. Diagnóstico diferencial entre embolia y trombosis. |
||
|---|---|---|
|
|
Embolia |
Trombosis |
|
Inicio |
Súbito |
Mas lento |
|
Edad |
Cualquier edad |
Más de 45 años |
|
Severidad |
Severo |
Menos severo |
|
Localización |
Preciso |
Poco preciso |
|
Fuente embólica |
Identificable |
No |
|
Antecedente de claudicación |
No |
Habitual |
|
Pulsos |
Ausente solo en sitio ocluido |
Ausencia o disminución del pulso en otros territorios. |
|
Arteriografía |
Stop súbito, no circulación colateral |
Ateroesclerosis en varios territorios, circulación colateral, adelgazamiento |
Diagnóstico
Con los síntomas y signos descritos anteriormente tenemos suficiente información para llegar a un diagnóstico de certeza. Por lo tanto, salvo en casos de duda, no debemos insistir en utilizar otras exploraciones, ya que además de no aportar datos que modifiquen la actitud terapéutica van a demorar la instauración del tratamiento.
También es importante establecer el diagnóstico diferencial con dolor agudo en una extremidad
Tratamiento
- Derivación urgente al hospital en las primeras 6 horas.
- Mientras se gestiona el traslado al hospital, aplicar las medidas generales:
- Declive moderado
- Protección
- Evitar calor
- Medicación inmediata: Sueroterapia, analgesia, heparinización (Bolo endovenoso de 1-1.5 mg de heparina sódica/kg de peso.
Evolución
La isquemia aguda puede evolucionar de tres formas:
- GANGRENA SECA: La cianosis se hace fija (no desaparece con la digitopresión) y da lugar a la formación de una escara o a la momificación de las partes acras.
- GANGRENA HÚMEDA: La cianosis da lugar a la formación de flictenas con intenso edema y licuefacción tisular, provocando la salida de líquido serohemático.
- ESTABILIZACIÓN Y CRONIFICACIÓN: Recuperación parcial en el caso de compensación hemodinámica de la isquemia y estabilización clínica, dando lugar a un síndrome de isquemia crónica.
Pronóstico
Va a depender del nivel de la obliteración, siendo más graves cuanto más proximales.
Factores pronósticos:
- Extensión proximal y distal de la trombosis secundaria.
- Estado de la circulación colateral.
- Tiempo de evolución y sensibilidad tisular a la isquemia. El tiempo de evolución es un factor pronóstico definitivo. Así, las isquemias agudas tratadas correctamente en las primeras 6-8 horas tienen habitualmente un excelente pronóstico, mientras que a partir de las 10-12 horas el pronóstico es malo y conlleva frecuentemente a la amputación de la extremidad o secuelas neurológicas definitivas.
- La coexistencia de otras enfermedades: cardiopatías neoplasias, etc.
PATOLOGIA VENOSA
TROMBOSIS VENOSA PROFUNDA
La trombosis venosa profunda (TVP) hace referencia a la presencia de un trombo en el sistema venoso profundo de las extremidades inferiores, extremidades superiores, intracraneal o en las venas abdominales. La TVP afecta a una de cada mil personas a lo largo de un año; en muchas ocasiones es asintomática. Las complicaciones más importantes son el tromboembolismo pulmonar (TEP) y el síndrome postflebítico. El TEP y la TVP son enfermedades cardiovasculares muy habituales, hasta tal punto que ocupan el tercer lugar en frecuencia tras la enfermedad coronaria y el ictus.
Factores de riesgo
- Anticoncepción hormonal y tratamiento hormonal sustitutivo, sobre todo en fumadoras.
- Hipercoagulabilidad hereditaria o adquirida.
- Inmovilización.
- Traumatismo en extremidades inferiores.
- Embarazo o puerperio (máximo riesgo seis semanas postparto).
- Hospitalización.
- Comorbilidad, una o más enfermedades (cardíaca, metabólica, endocrina, respiratoria, inflamatoria o infecciones agudas).
- Cáncer activo.
- Cirugía reciente.
- Edad > 60 años.
- Deshidratación.
- Antecedentes personales o familiares de TVP.
- Viajes en avión de más de 8 horas de duración.
- Venas varicosas con flebitis.
- Catéter venoso central.
- Obesidad (IMC > 30 kg/m2), hipertensión, diabetes, hipercolesterolemia, síndrome metabólico y hábito tabáquico, todos ellos factores de riesgo de la enfermedad cardiovascular y de TVP.
A través de una historia clínica y de un examen físico detallados es posible identificar los factores de riesgo en más de la mitad de los casos. No hay suficientes datos para saber si la suma de dos factores juega un papel mayor que la mera adición de ambos; se sabe el papel multiplicador de la interacción entre factor V de Leiden y el uso de la píldora anticonceptiva combinada (Sign, 2010). El cribado de trombofilias hereditarias no está indicado, aunque la existencia de una trombofilia hereditaria identificada previamente podría influir en la evaluación del riesgo (Baglin, 2010).
Diagnóstico
La TVP se puede manifestar con cualquiera de los síntomas y signos que se reseñan a continuación
- Edema en el tobillo, en la pantorrilla o en toda la pierna.
- Sensibilidad o dolor en reposo, al caminar o al presionar la pantorrilla.
- Cordón venoso palpable.
- Aumento de la temperatura cutánea.
- Cianosis, dilatación o tensión en el sistema venoso superficial.
- Signo de Homans positivo (dolor en la pantorrilla con la dorsiflexión del pie).
- Signo de Lowenberg positivo (dolor intenso en la pantorrilla a la presión del esfingomanómetro por debajo de 180 mmHg).
Se debe sospechar TVP en los pacientes con uno o varios de los síntomas y signos sugestivos. Muchos de los casos de TEP no han tenido una TVP clínicamente evidente. Alrededor del 30% de los pacientes con TVP sintomática tienen un TEP.
Los síntomas y los signos son poco sensibles y poco específicos. Se tendrán en cuenta otras posibilidades en el diagnóstico diferencial (tabla 2).
|
Tabla 2. Diagnóstico diferencial de dolor y edema en extremidad inferior (Muñoz, 2004) |
|
|---|---|
|
Patología venosa |
Otras |
|
Tromboflebitis superficial |
Celulitis |
|
Síndrome posflebítico |
Quiste de Baker |
|
Insuficiencia venosa crónica |
Linfedema |
|
Obstrucción venosa |
Edema por ICC, hepática o renal |
|
|
Rotura fibrilar |
|
|
Inflamación o rotura tendón de Aquiles |
En los pacientes ambulatorios, ante la sospecha de TVP o TEP, se recomienda usar los criterios de Wells para iniciar la estrategia diagnóstica (tabla 3). Este modelo clínico está validado, ayuda a establecer la probabilidad de la TVP además de ser una de las reglas de predicción clínica de TVP más utilizadas.
|
Tabla 3. Criterios de Wells |
|
|---|---|
|
Características clínicas |
Valor |
|
Cáncer activo (tratamiento quimioterápico actual o en los 6 meses previos o en tratamiento paliativo) |
1 |
|
Parálisis, paresia o inmovilización reciente de una extremidad |
1 |
|
Reposo reciente en cama de más de 3 días, o cirugía mayor en el último mes |
1 |
|
Dolor en trayecto de sistema venoso profundo |
1 |
|
Tumefacción de toda la extremidad |
1 |
|
Aumento del perímetro mayor de 3 cm respecto a extremidad asintomática, medido 10 cm por debajo de la tuberosidad tibial |
1 |
|
Edema con fóvea (mayor en la extremidad sintomática) |
1 |
|
Presencia de circulación colateral superficial no varicosa |
1 |
|
Diagnóstico alternativo al menos tan probable como la TVP |
-2 |
|
Valores: < 1: probabilidad baja; 1-2: probabilidad moderada; ≥ 3: probabilidad alta. |
|
Es importante que se usen sólo en los casos indicados: pacientes ambulatorios y con síntomas o signos sugestivos. Esta regla no está validada para casos en los que haya existido una TVP previa, en pacientes hospitalizados y en mujeres embarazadas.
- Probabilidad baja y dímero D negativo, excluye diagnóstico.
- Probabilidad baja o moderada y dímero D positivo, se debe hacer prueba de imagen para confirmar o excluir TVP.
-
Probabilidad moderada/alta, no es preciso solicitar dímero D, se hará ecografía doppler.
- Si ecografía doppler negativo se solicitará dímero D.
-
Si ecografía doppler negativa y dímero D positivo se valoran varias alternativas:
- Iniciar tratamiento
- Repetir la ecografía a los 3 y a los 7 días
- Realizar otra prueba de imagen
- Si ambos resultados son negativos se puede retrasar el inicio de la anticoagulación
Se realizarán una historia clínica y un examen físico exhaustivos con la intención de detectar factores subyacentes que faciliten la presentación de trombosis y valorar la adecuación del tratamiento antitrombótico.
No está indicado el despistaje de cáncer tras un episodio de TVP o TEP.
No está indicado realizar estudio rutinario de trombofilias en los pacientes con un primer episodio de TVP. No existen recomendaciones validadas sobre cómo realizar la selección, ni de la actuación adecuada ante los resultados, sean estos positivos o negativos; tampoco se sabe la influencia de los mismos en el riesgo. Sin embargo, las recomendaciones actuales sobre duración del tratamiento tras un primer episodio de TVP incluyen esta variable.
En los pacientes en los que se desarrolla necrosis cutánea asociada al uso de antagonistas de la vitamina K (AVK) puede ser adecuado realizar un estudio de deficiencia de proteína C y S tras la suspensión del tratamiento.
Pruebas complementarias
Dímero-D
Producto de degradación de la fibrina que se detecta en sangre en la fase aguda de la TVP. Es una prueba muy sensible y poco específica y se puede presentar en otros procesos además de en la TVP. Se debe solicitar en los casos de probabilidad baja o moderada según los criterios de Wells. Hay otros problemas de salud que producen elevación de dímero D (tabla 4). No se debe solicitar en pacientes asintomáticos.
|
Tabla 4. Procesos que cursan |
|
|---|---|
|
1 |
Infecciones. |
|
2 |
Neoplasias |
|
3 |
Insuficiencia cardíaca y renal |
|
4 |
IAM |
|
5 |
ACV |
|
6 |
Coagulación intravascular diseminada |
|
7 |
Cirugía reciente |
|
8 |
Enfermedad inflamatoria |
|
9 |
Embarazo |
|
10 |
Situaciones graves |
Técnicas de imagen
- Ecografía-Doppler. Es la prueba de elección para el diagnóstico de TVP sintomática proximal de extremidades inferiores. Se valora la presencia de trombo, la posibilidad de comprimir la vena y el flujo sanguíneo a través de una oclusión incompleta. Tiene una sensibilidad alta (94-99%) y una especificidad de 89-96% comparadas con el patrón oro, que es la flebografía. La sensibilidad y la especificidad son más bajas en las TVP asintomáticas, tanto en el muslo como en la parte inferior de la rodilla. El valor predictivo negativo de una ecografía simple normal para excluir TVP en un paciente sintomático es alta. Sin embargo, ante una alta sospecha clínica, una ecografía normal no excluye el diagnóstico, motivo por el que se debe repetir la exploración ecográfica en un período de tres a siete días.
Está indicado realizar ecografía doppler con técnica de compresión en los casos de probabilidad moderada-alta y en los casos de probabilidad baja con dímero D elevado.
- Flebografía. Como se ha dicho, se considera el patrón oro para el diagnóstico de la TVP; permite distinguir entre trombos nuevos y antiguos. Se realiza en casos muy seleccionados por sus inconvenientes: precisa el uso de contraste, es costosa, incómoda para el paciente y tiene el riesgo de trombosis secundaria.
- Otras técnicas que se utilizan son el TAC y la RMN.
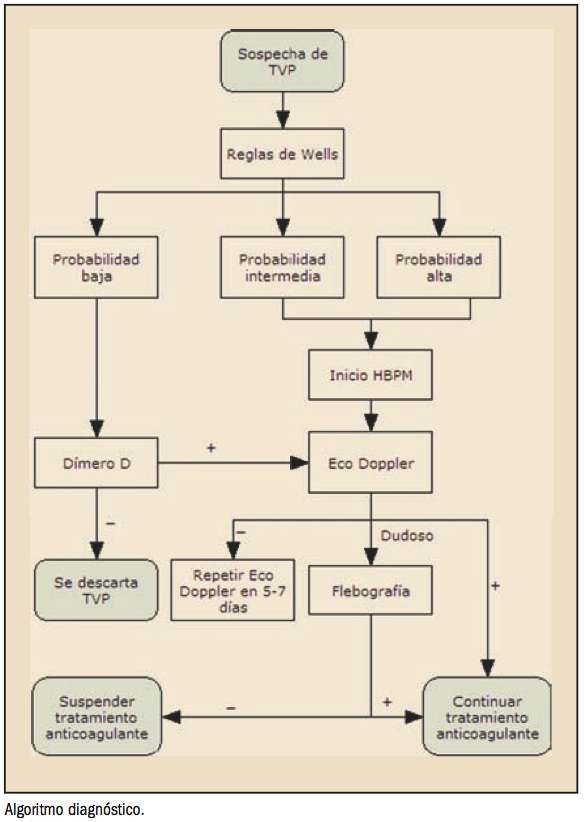{kind=link}
Tratamiento
Todos los pacientes diagnosticados de TVP proximal deben iniciar tratamiento con anticoagulantes, salvo que existan contraindicaciones. Los pacientes diagnosticados de TVP distal también son tributarios de tratamiento anticoagulante. Aunque se puede considerar como opción aceptable la vigilancia de la evolución del trombo, no existen estudios acerca de la seguridad de esta alternativa.
- Heparinas de bajo peso molecular (HBPM). Tras el diagnóstico se iniciará tratamiento con HBPM preferible a la utilización de heparina no fraccionada. En los pocos estudios en los que se han comparado HBPM entre sí no se ha podido demostrar la superioridad de una sobre las otras. Salvo situaciones especiales, se puede realizar el tratamiento de manera ambulatoria. La mayoría de los pacientes pueden cambiar de HBPM a antagonistas de la vitamina K a los 6-10 días de iniciado el tratamiento. Este cambio no es aconsejable en pacientes oncológicos, embarazadas o con mala tolerabilidad a los antagonistas de la vitamina K, que deben seguir el tratamiento con HBPM durante tres o seis meses.
- Heparina no fraccionada (HNF). La utilización de HNF de administración únicamente intravenosa se reserva para determinadas situaciones: cuando se considera la necesidad de realizar trombolisis, en el período postoperatorio inmediato o si existe riesgo de sangrado.
- Antagonistas de la vitamina K (AVK). Se dispone de dicumarina o acenocumarol, sintrom® y warfarina, aldocumar®, ambos de uso oral. Cuando esté indicado el cambio se realizará a los 5 días del inicio de la administración de HBPM. El objetivo para la TVP es un INR de 2,5 (rango 2,0-3,0).
- Inhibidor directo del factor Xa (Fondaparinux). Se aplica por vía subcutánea en dosis diaria, tiene una vida media de 17-21 horas. No se conoce ningún antídoto que contrarreste su efecto. Se recomienda un uso cauteloso en personas con riesgo de sangrado, insuficiencia renal y con peso menor de 50 kg. Para monitorizar su efecto se utiliza el anti-factor-Xa, el recuento de plaquetas previo y en las fases iniciales del tratamiento, se necesitan calibradores nuevos de su efecto. Al igual que las HBPM, se recomienda mantener durante un mínimo de 5 días antes de introducir la anticoagulación oral.
- Inhibidores orales directos de la trombina (Dabigatran). Su única indicación actual es la prevención primaria de episodios tromboembólicos venosos en pacientes adultos sometidos a cirugía programada de reemplazo total de cadera o de rodilla. No necesita monitorización. Interacciona con amiodarona, claritromicina y verapamilo, precisan reducción de dosis. En asociación con rifampicina y hierba de san Juan, precisan aumento de dosis. No existen interacciones con el Diclofenaco; no hay estudios con el resto de AINEs. No se recomienda el uso concomitante con anticoagulantes o antiagregantes plaquetarios, medicamentos trombolíticos y antagonistas de los receptores GPIIb/IIIa.
- 4. Trombolisis. El tratamiento trombolítico iv o trombolisis sólo se recomienda para casos individualizados de pacientes con TVP iliofemoral masiva, en los que está amenazada la viabilidad de la extremidad y con riesgo bajo de sangrado. No hay datos sobre su incidencia en la disminución de la morbilidad y mortalidad de los pacientes hemodinámicamente estables.
- 5. Trombectomía quirúrgica. Se ha utilizado en trombosis venosa extensa y con contraindicaciones para otro tipo de tratamiento. La decisión debe ser individualizada y tomada como último recurso por su morbilidad y mortalidad. Antes del inicio del tratamiento anticoagulante se evaluará:
- Si existen procesos subyacentes que faciliten la aparición de TVP o TEP.
- La seguridad de la anticoagulación en cada paciente.
- Si la anticoagulación puede controlarse de manera adecuada.
- La posibilidad de monitorizar los efectos secundarios del tratamiento.
- Los riesgos de la anticoagulación y las contraindicaciones a la misma (riesgo de hemorragia, cirugía reciente o inminente, enfermedad hepática, sangrado gastrointestinal o anemia).
- Es necesario disponer de un estudio de coagulación, de plaquetas y de la situación de la función renal. La HBPM se metaboliza en el riñón; es necesario ajustar la dosis en pacientes con un filtrado glomerular < 30 ml/m, ya que se pueden dar casos de hemorragias fatales por dosis mal ajustadas, tanto de HBPM como de dicumarínicos.
El índice de riesgo de sangrado informa sobre la posibilidad de tener un sangrado mayor en los pacientes a tratamiento con dicumarínicos; esto implica una evaluación clínica simple en la que incluye: recuento sanguíneo, evaluación de la creatinina, edad del paciente, historia de sangrado gastrointestinal, de ictus hemorrágico o isquémico y la presencia de enfermedad asociada (infarto reciente, afectación renal, anemia con hematocrito < 30 o diabetes).
- 6. Filtros en vena cava (IVC). La implantación de un filtro en la vena cava se indica en situaciones de riesgo de mortalidad: casos en los que la anticoagulación está contraindicada, tromboembolismo progresivo a pesar de que la anticoagulación sea adecuada e hipertensión pulmonar subyacente, casos en los que un TEP puede ser fatal.
- 7. Medidas físicas. Elevación de MMII en sedestación, deambulación precoz y medias de compresión gradual. Tras un episodio de TVP en MMII es importante insistir en la utilización de medias de compresión gradual (30-40 mmHg) en la pierna afectada durante un período de un año. Su uso reduce el riesgo de síndrome posflebítico, el dolor y la tumefacción. Esta medida está contraindicada en caso de arteriopatía periférica o edemas por insuficiencia cardíaca (ICSI, 2010).
TROMBOSIS VENOSA SUPERFICIAL (TVS)
La trombosis venosa superficial (TVS) es la aparición de dolor, eritema, sensibilidad e induración debido a la presencia de trombosis y la reacción inflamatoria que la acompaña en alguna de las venas que se localizan bajo la piel. La localización más frecuente es en miembros inferiores, asociada a varices, aunque también puede localizarse en brazos, generalmente relacionada con cateterización o punciones venosas repetidas (fármacos endovenosos, drogadicción parenteral). Habitualmente es un proceso benigno y autolimitado; aunque en ocasiones puede complicarse con una trombosis venosa profunda e incluso un tromboembolismo pulmonar (Menezes NS, 2012). Presenta una incidencia estimada en población general entre el 3% y 11%.
Los factores de riesgo de la trombosis venosa superficial son: estasis venoso (varices, inmovilización, obesidad), embarazo, anticonceptivos orales, neoplasia, trombofilias hereditarias, e inyección intravenosa de sustancias irritantes (fármacos, estupefacientes). Se puede clasificar en:
- Tromboflebitis aséptica: se suele asociar a varices o traumatismos. Es más frecuente en miembros inferiores, y no existe evidencia de infección. Habitualmente se resuelve en unos 10 días, aunque el pronóstico depende del desarrollo de complicaciones.
- Tromboflebitis séptica o supurativa: se relaciona con canalizaciones venosas o heridas; es más frecuente en los brazos, y presenta mal pronóstico si no se trata.
Los factores de riesgo de la TVS son: embarazo, anticonceptivos orales, neoplasia, trombofilias hereditarias, varices e inyección intravenosa de sustancias irritantes (fármacos, estupefacientes)
Diagnóstico
La presentación clínica se caracteriza por edema, eritema y dolor a lo largo del trayecto venoso afectado, en pacientes con factores de riesgo conocidos. En la mayoría de los casos la vena trombosada es palpable(cordón venoso). Puede haber fiebre y afectación del estado general en el caso de la tromboflebitis séptica.
Las complicaciones más frecuentes son:
- Trombosis Venosa Profunda (TVP): aparece en menos del 10% de los casos, más si existen factores de riesgo, como cuando la trombosis afecta a la vena safena mayor cerca de la unión safeno-femoral; en sujetos con antecedentes de trombosis venosa profunda, trombofilias, neoplasia o terapia estrogénica. Es mas frecuente en varones, mayores de 60 años, cuando coexiste trombosis bilateral de venas superficiales, en presencia de infección sistémica y si aparece en personas que no tenían varices previamente (Menezes NS, 2012).
- Tromboembolismo (infrecuente)
- Hiperpigmentación cutánea.
- Persistencia de nódulo subcutáneo en el lugar de la trombosis.
- Abscesificación, sepsis, etc. en el caso de la tromboflebitis séptica.
En la mayoría de los casos el diagnóstico es clínico. El empleo del Eco-Doppler depende de la situación clínica y de si la información obtenida determinará un cambio en la actitud terapéutica. El Eco-Doppler identifica la presencia, localización y extensión de la trombosis venosa. Esta indicado si afecta al tercio proximal de la vena safena interna del muslo, si existe evidencia clínica de extensión de la flebitis, si la hinchazón de la extremidad es superior a la esperada de una flebitis superficial o en caso de duda diagnóstica.
Tratamiento
El tratamiento de la trombosis venosa superficial no complicada (segmento venoso pequeño y lejos de la unión safeno-femoral) es ambulatorio y sintomático, basado en la elevación de la extremidad, compresas frías o tibias y antiinflamatorios no esteroideos (AINE). Ningún antiinflamatorio ha demostrado superioridad respecto a otros, recomendándose los de mejor perfil de seguridad: naproxeno (500 mg/12 horas), ibuprofeno (400 mg/8 horas), diclofenaco (50 mg/8 horas). En caso de embarazo, se puede usar Paracetamol (1.000 mg/6 horas). No están indicados los antibióticos, salvo en el caso de la tromboflebitis séptica.
En ausencia de contraindicación (arteriopatía periférica) las medias elásticas de compresión de clase II (20-30 mmHg) con una longitud que abarque toda la extensión de la vena afectada pueden mejorar el dolor y el edema. Los tratamientos tópicos producen un alivio sintomático, pero no afectan a la evolución de la trombosis. Si la trombosis es extensa, afecta a la vena safena interna en la unión safeno-femoral o el paciente presenta factores de riesgo para la trombosis venosa profunda está indicada la anticoagulación durante 4 semanas.
Comparados con placebo tanto los AINE como la heparina no fraccionada y las heparinas de bajo peso molecular han demostrado disminuir la extensión del trombo y/o la recurrencia de la flebitis. Ninguno ha demostrado una disminución significativa en la incidencia de embolismos pulmonares. Dado el riesgo de hemorragia asociado a la anticoagulación, se recomiendan los AINE como terapia inicial en pacientes con bajo riesgo de tromboembolismo, reservando la anticoagulación para los de alto riesgo. La American College of Chest Physicians (ACCP) recomienda dosis intermedias de heparinas de bajo peso molecular (dalteparina 5.000 UI cada 12 horas o enoxaparina 40 mg/día vía subcutánea) durante un mes (Mearon C, 2008).
Bibliografía
El trastorno obsesivo-compulsivo (TOC) como factor de riesgo para la esquizofrenia
El diagnóstico de trastorno obsesivo-compulsivo (TOC) parece asociarse con mayores tasas de esquizofrenia y de trastornos del espectro esquizofrénico, sugiriendo que todos ellos presentan elementos etiológicos comunes.
A pesar de una notable concurrencia del trastorno obsesivo-compulsivo y la esquizofrenia, se sabe poco acerca de la relación clínica y etiológica ambos trastornos. Por ello, existe un especial interés en explorar el grado en que estos trastornos comparten factores etiológicos.
El estudio utilizó los datos individuales de los registros longitudinales daneses en todo el país, lo que implica un estudio de cohorte prospectivo con 45 millones de personas-año de seguimiento. Todos los análisis de supervivencia se ajustaron por sexo, edad, año natural, edad de los padres, y lugar de residencia en el momento del nacimiento, realizándose un seguimiento a partir del 1 de enero de 1995 hasta el 31 de diciembre de 2012 sobre un total de 3 millones de personas nacidas entre el 1 de enero de 1955 y el 30 de noviembre de 2006. Durante este periodo, 30.556 personas desarrollaron esquizofrenia o trastornos de espectro esquizofrénico.
Los resultados mostraron que la presencia de un diagnóstico previo de TOC se asoció con un mayor riesgo de desarrollar esquizofrenia (RR= 6,90; IC95% 6,25 a 7,60) o un trastorno del espectro esquizofrénico (RR= 5,77; IC95% 5,33 a 6,22) a lo largo de la vida. Del mismo modo, los hijos de padres con diagnóstico de TOC tenían un mayor riesgo de esquizofrenia (RR= 4,31; IC95% 2,72 a 6,43) y de trastornos del espectro esquizofrénico (RR= 3,10; IC95% 2,17 a 4,27). Los resultados siguieron siendo estadísticamente significativos después de ajustar por antecedentes familiares de trastornos psiquiátricos y la historia psiquiátrica del paciente.
Meier SM, Petersen L, Pedersen MG, Arendt MC, Nielsen PR, Mattheisen M, Mors O, Mortensen PB. Obsessive-Compulsive Disorder as a Risk Factor for Schizophrenia: A Nationwide Study. JAMA Psychiatry. 2014 Sep 3. doi: 10.1001/jamapsychiatry.2014.1011. [Epub ahead of print]