Presentación inicial del caso
Mujer de 79 años, que acaba de sufrir un evento cardiovascular agudo, acude a la Farmacia Comunitaria con la bolsa de los medicamentos que está tomando y el informe con los tratamientos activos. La paciente presenta un elevado grado de confusión y desesperación porque no sabe para qué son los medicamentos ni cómo debe tomarlos.
En el informe aparecen los siguientes diagnósticos activos:
- Diabetes mellitus tipo 2
- Carencias de componentes del complejo B
- Hipertrigliceridemia
- Hiperlipidemia mixta
- Ansiedad
- Depresión
- Insuficiencia de la película lagrimal no especificada
- Hipertensión arterial
- Infarto agudo de miocardio
- Otras formas agudas y subagudas de cardiopatía isquémica
- Ardor de estómago
- Los medicamentos que utiliza son:
- Trajenta® comp. recubiertos 5 mg
- Levemir® plumas precargadas 100 U/ml
- Ferplex® viales bebibles 40 mg
- Nervobion® caps.
- Secalip® comp. recubiertos 145 mg
- Atorvastatina comp. recubiertos 80 mg
- Omacor® caps. 1.000 mg
- Tepazepan caps.
- Heipram comp. recubiertos 10 mg
- Acuolens® colirio
- Ixia® comp. recubiertos 40 mg
- Ramipril comp. 5 mg
- Isosorbida mononitrato comp. 40 mg
- Bisoprolol comp. recubiertos 5 mg
- Duoplavin® comp. recubiertos 75/100 mg
- Nexium® Mups comp. gastrorr. 40 mg
Descripción del caso y evaluación
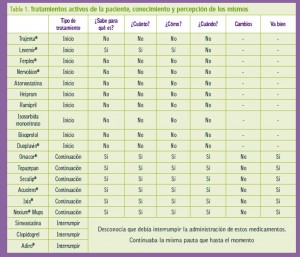
Se realiza una evaluación de los tratamientos activos y del grado de conocimiento de los mismos por parte de la paciente. Los resultados se recogen en la Tabla 1.
Intervención
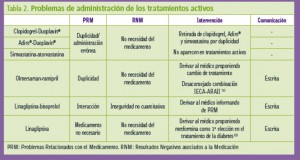
Tras realizar el análisis de la información obtenida, se detectan problemas en la administración de los tratamientos activos, recogidos en la Tabla 2.
A partir de este punto, se elabora un Sistema Personalizado de Dosificación (SPD).
Resultados
Se corrigió una falta de entendimiento entre médico y paciente, ya que cuando la paciente acude a la Farmacia Comunitaria desconoce cómo tomar la nueva medicación.
Con el SPD se consiguió que solo se administrara la medicación correcta, siguiendo la pauta marcada por el facultativo. Además, se facilitó la toma de medicamentos, aumentando la adherencia al tratamiento.
De las tres propuestas enviadas al médico, este decidió retirar olmesartan. Mantuvo el tratamiento con bisoprolol y linagliptina porque la paciente los necesitaba. No se aceptó metformina como primera línea para la diabetes por intolerancia.
Comentarios
El grado de saturación de los centros y el poco tiempo del que dispone el médico en consulta hace difícil asegurarse de que el paciente conozca su medicación y cumpla su tratamiento.
La Farmacia Comunitaria es un lugar idóneo donde revisar la medicación del paciente y resolver sus dudas, además de ser una labor para la cual el profesional farmacéutico está totalmente preparado.
Es necesario el desarrollo de Servicios Profesionales en la Farmacia Comunitaria enfocados a solucionar este problema, pero no de forma independiente, sino colaborando con instituciones, ambulatorios, hospitales, etc., y con una remuneración adecuada. Un ejemplo de esto sería el SPD.
BIBLIOGRAFIA
- Mancía G, Fagarol R, Narkiewicz K, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension. J Hypertens. 2013;31:1281-357.
- Menéndez-Torre E, Lafita Tejedor J, Artola Menéndez S, et al. Recomendaciones para el tratamiento farmacológico de la hiperglucemia en la diabetes tipo 2. Av Diabetol. 2010;26:331-8.