La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) describe en su 8º Informe de farmacovigilancia de vacunas contra la COVID -19, los avances en el seguimiento de la seguridad de las vacunas COVID-19, y los cambios en las fichas técnicas de estas vacunas COVID-19 tras la revisión de los datos de seguridad disponibles, que ha evaluado el PRAC, comité europeo de evaluación de riesgos en farmacovigilancia.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha publicado en su 8º Informe de Farmacovigilancia de las Vacunas COVID-19 las novedades y los cambios que, tras la revisión de los datos de seguridad disponibles por parte del Comité europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC), se han incorporado en la información de las fichas técnicas y de los prospectos de las vacunas COVID-19 correspondientes (AEMPS, 2021a).
En España, desde el inicio de la pandemia hasta el 10 de septiembre de 2021, se han confirmado 4.907.461 de casos de COVID-19 y 85.290 fallecimientos de infectados confirmados mediante test antes de fallecer (Ministerio de Sanidad, 2021a).
En España se han administrado, hasta el día 5 de septiembre de 2021, un total de 66.835.878 dosis de vacunas frente a la COVID-19, que corresponden a un total de 36.877.329 personas. Las dosis administradas según el Registro de Vacunación, del Ministerio de Sanidad, correspondieron:
- Comirnaty®: el 70% de las dosis.
- Vaxzevria® (antes COVID-19 Vaccine AstraZeneca): el 15% de dosis.
- Spikevax® (antes COVID-19 Vaccine Moderna): el 12% de dosis.
- COVID-19 Vaccine Janssen®: el 3% restante.
La Estrategia de Vacunación y sus ocho actualizaciones posteriores, hasta el 6 de junio de 2021, pueden consultarse en la web del Ministerio de Sanidad (Ministerio de Sanidad, 2021b).
Hasta el 5 de septiembre, se han registrado en la base de datos FEDRA, del Sistema Español de Farmacovigilancia Humana (SEFV-H) un total de 41.751 notificaciones de acontecimientos adversos (Figura 1), con un total de 115.993 acontecimientos (una notificación puede contener una media de 3 acontecimientos o síntomas clínicos), lo que correspondería a 62 notificaciones por cada 100.000 dosis administradas. El número de notificaciones es proporcional al número de dosis administradas de cada una de las 4 vacunas COVID-19 utilizadas (Figura 1).
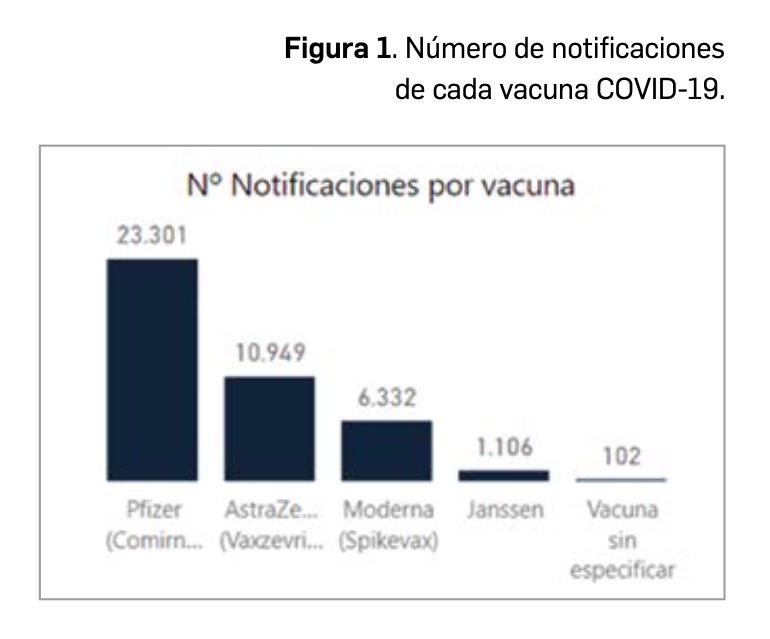
Es de destacar que ha habido 102 notificaciones en las que se ha notificado como fármaco sospechoso “una vacuna COVID-19”. Se deben hacer los esfuerzos necesarios para identificar qué vacuna ha sido la relacionada con el acontecimiento. Si no se sabe la marca, es como si no se supiera nada. Solo representa el 0,2% del total de notificaciones, pero es inútil notificar así una sospecha de relación causal si no se identifica qué vacuna concreta es la sospechosa de causar el acontecimiento adverso.
El 72% de las notificaciones han sido comunicadas por profesionales sanitarios y el 28% por ciudadanos. La mayoría de las notificaciones corresponden a personas de entre 18 y 65 años (88%) y mayoritariamente a mujeres (76%).
Recordemos que un “acontecimiento adverso” es “cualquier problema de salud que ocurre después de la vacunación, sin que necesariamente tenga que estar ocasionado por la vacuna”. Estos acontecimientos adversos pueden ser simplemente coincidentes en el tiempo, relacionados con el propio acto de vacunarse (ansiedad, desmayo, error en la administración), relacionados con un potencial problema de calidad de la vacuna, o realmente relacionados con la propia vacuna (como por ejemplo dolor en el lugar de la inyección o fiebre). Por lo tanto, hay que recordar que los “acontecimientos adversos” que se notifican no significa que estén relacionados con la vacunación, como causa-efecto. Por tanto, no son reacciones adversas ni sirven para comparar el perfil de reacciones adversas de las diferentes vacunas.
Estas son las conclusiones de la evaluación periódica de los datos de farmacovigilancia de cada una de las vacunas COVID-19:
Comirnaty® (BioNTech/Pfizer)
Desde que se autorizó su comercialización en la Unión Europea hasta el 2 de septiembre de 2021, se han administrado casi 392 millones de dosis de Comirnaty® en el Espacio Económico Europeo (EEE) de 30 países, los 27 de la UE más Islandia, Noruega y Liechtenstein:
- Trastornos menstruales: el PRAC está analizando los casos de trastornos menstruales notificados tras la vacunación frente a la COVID-19. Hasta el momento actual, no se ha establecido una relación causal entre estos trastornos y las vacunas COVID-19, que son muy frecuentes y a veces pueden suceder sin que exista ninguna enfermedad subyacente en la mujer, como en situaciones de estrés o cansancio. Se han solicitado datos adicionales a los laboratorios titulares de la autorización de comercialización (TAC) de todas estas vacunas, y el PRAC revisará toda la evidencia disponible.
- Glomerulonefritis y síndrome nefrótico: ha comenzado también la evaluación de los casos de glomerulonefritis (inflamación de los pequeños filtros que existen en los riñones) y síndrome nefrótico (un trastorno del riñón que causa que estos filtren demasiada cantidad de proteínas a la orina), notificados tras recibir alguna de las vacunas basadas en ARNm (Comirnaty® y Spikevax®). Los pacientes con estos trastornos pueden presentar orina sanguinolenta o espumosa, edema (inflamación de los párpados, pies o abdomen), o fatiga. El motivo para iniciar esta evaluación ha sido la publicación en la literatura médica de un pequeño número de casos, incluyendo algunos en los que los pacientes presentaban recaídas de enfermedades renales preexistentes.
- Eritema multiforme: con motivo de la notificación de algunos casos de eritema multiforme después de haber recibido alguna de las vacunas basadas en ARNm (Comirnaty® y Spikevax®), el PRAC ha iniciado la evaluación de esta señal, con el fin de establecer si puede ser una reacción adversa (RAM) asociada a alguna de ellas. El eritema multiforme es una reacción de hipersensibilidad (alérgica) que se caracteriza por la aparición de lesiones en la piel redondeadas, y que también pueden afectar a las mucosas en las cavidades internas del organismo. Los casos notificados son acontecimientos que se han observado tras la vacunación, pero no significa que estén relacionados o causados directamente por la vacuna. Se han solicitado más datos y análisis adicionales a los laboratorios TAC para continuar con la evaluación.
- Síndrome inflamatorio multisistémico (SIM): con motivo de la notificación de un caso de SIM tras la vacunación con Comirnaty® en un hombre de 17 años, en Dinamarca, que se recuperó, el PRAC está evaluando si existe un riesgo aumentado de sufrir este síndrome con las vacunas frente a la COVID-19. Se han notificado otros casos de SIM con esta y las demás vacunas frente a la COVID-19, y también en adultos, dentro y fuera del EEE, pero se debe tener en cuenta que los casos notificados son acontecimientos adversos que se han observado tras la vacunación, y que no significa necesariamente que estén relacionados o causados por la vacuna. Este síndrome es una condición inflamatoria grave, que afecta a varias partes del organismo. Sus síntomas pueden incluir cansancio, fiebre intensa y persistente, diarrea, vómitos, dolor de estómago, cefalea, dolor torácico y dificultad para respirar. El SIM se presenta raramente, siendo su tasa de incidencia global de 0,51 casos por 100.000 pacientes-año (datos de BIFAP en 2019). El SIM se ha asociado también a la propia enfermedad COVID-19.
Spikevax® (antes COVID-19 Vaccine Moderna®)
Desde que se autorizó su comercialización en la UE hasta el 2 de septiembre de 2021, se han administrado más de 54,2 millones de dosis de Spikevax® en el EEE. Los aspectos de seguridad, evaluados o en evaluación por el PRAC desde el último informe, son los siguientes:
- Trastornos menstruales: ver apartado para vacuna Comirnaty®.
- Eritema multiforme: ver apartado para la vacuna Comirnaty®.
- Glomerulonefritis y síndrome nefrótico: ver apartado para la vacuna Comirnaty®.
- Síndrome inflamatorio multisistémico (SIM): ver apartado para la vacuna Comirnaty®.
Vaxzevria® (antes COVID-19 Vaccine AstraZeneca)
Desde que se autorizó su comercialización en la UE hasta el 29 de julio de 2021, se han administrado más de 68,4 millones de dosis de Vaxzevria® en el EEE. Los aspectos de seguridad en evaluación por el PRAC desde el último informe son los siguientes:
- Trastornos menstruales: para esta vacuna de AstraZeneca ya se han revisado los casos notificados de trastornos menstruales tras la administración, junto con una revisión de la literatura científica e incluyendo información de estudios preclínicos. Hasta el 31 de julio de 2021, se habían notificado en total 12.410 casos en todo el mundo en el contexto de alrededor de 592 millones de dosis administradas hasta el 25 de julio. Los casos notificados son acontecimientos que se han observado tras la vacunación y no significa necesariamente que estén relacionados o causados por la vacuna. La evaluación de estos casos, llevada a cabo con el asesoramiento de especialistas en ginecología e incluyendo el análisis del tipo de síntomas, que son transitorios, no pudo identificar un patrón común, ni un potencial mecanismo de acción de la vacuna. Se ha tenido en cuenta el tiempo transcurrido entre la administración de la vacuna y el inicio de los trastornos, la duración de estos y su desenlace, así como la administración de otros tratamientos y la historia médica previa de las pacientes. Basándose en la evaluación de toda la información disponible hasta el momento, el PRAC ha concluido que no hay evidencia que sugiera una relación causal entre los trastornos menstruales notificados y la administración de la vacuna Vaxzevria®. Estos trastornos son muy habituales y a veces pueden suceder sin que exista ninguna enfermedad subyacente en la mujer, como en situaciones de estrés o cansancio.
- Síndrome inflamatorio multisistémico (SIM): ver apartado para la vacuna Comirnaty®.
- Trombosis de senos venosos cerebrales sin trombocitopenia: el PRAC está evaluando, en el proceso de revisiones mensuales, los casos de trombosis de los senos venosos cerebrales (una forma de ictus poco frecuente en la que el coágulo de sangre se forma en los senos venosos del cerebro) sin trombocitopenia (es decir, sin presentar niveles bajos de plaquetas en sangre), que se han notificado tras la vacunación con Vaxzevria®. Se han solicitado al laboratorio TAC datos adicionales para el próximo informe mensual.
- Síndrome de fuga capilar: en junio de 2021, se identificó este síndrome como posible reacción adversa de Vaxzevria® y se actualizaron la ficha técnica y el prospecto (ver nota de seguridad de la AEMPS) (AEMPS, 2021b). El síndrome de fuga capilar es un trastorno muy raro, pero grave y potencialmente mortal, que causa extravasación de los fluidos desde los capilares sanguíneos hacia el exterior. En España, hasta el 8 de agosto de 2021, no se ha notificado ningún caso de síndrome de fuga capilar tras la administración de Vaxzevria®. En la reunión de septiembre de 2021, el PRAC ha evaluado diferentes hipótesis mecanicistas para el desarrollo de este síndrome tras la vacunación, sin que se pueda identificar un mecanismo definitivo. Se recuerda a las personas que han tenido este síndrome con anterioridad que no deben ser vacunadas con Vaxzevria® (esta vacuna está contraindicada en este caso), y a las personas que experimenten una rápida inflamación de brazos y piernas, ganancia de peso repentina, y sensación de desmayo (baja presión arterial) en los días siguientes a la vacunación con Vaxzevria®, que deben buscar atención médica inmediata.
- Síndrome de trombosis con trombocitopenia (STT): en mayo de 2021, la información de Vaxzevria® se actualizó para incluir el riesgo (muy raro) de síndrome de trombosis (formación de coágulos de sangre en los vasos sanguíneos) con trombocitopenia (niveles bajos de plaquetas). Este síndrome continúa vigilándose estrechamente con la finalidad de caracterizar posibles factores de riesgo. En la reunión de septiembre de 2021, el PRAC ha recomendado actualizar la ficha técnica y el prospecto de este medicamento para eliminar la referencia que especifica que los casos notificados ocurrían sobre todo en mujeres de hasta los 60 años, debido a que la magnitud de las diferencias por edad y sexo que se observaban al principio actualmente son menores. El último análisis de estos casos de STT notificados espontáneamente incluye un 43% de casos en varones y un 37% en personas mayores de 60 años. El análisis de los datos que ofrece la literatura científica no identifica grandes diferencias por sexo (Arepally et al., 2021). Hasta el 31 de julio de 2021, se habían notificado 1.503 casos en todo el mundo, habiéndose administrado alrededor de 592 millones de dosis de Vaxzevria® hasta el 25 de julio. En España, hasta el 8 de agosto se han registrado 31 casos sugerentes o confirmados de STT, de los cuales 7 tuvieron desenlace mortal. La mayoría de las trombosis se presentaron en localizaciones inusuales (senos venosos o venas esplácnicas). De los 31 casos, 30 se produjeron tras la primera dosis; hasta esa fecha se habían administrado cerca de 9,6 millones de dosis. Se recuerda a las personas que experimenten cefalea intensa o persistente, visión borrosa, confusión, convulsiones, dificultad para respirar, dolor en el pecho, inflamación o dolor en las piernas, dolor abdominal persistente, o hematomas o manchas en la piel en forma de puntos pequeños redondos, que no se sitúan en el lugar de la vacunación, hasta tres semanas después de la administración de Vaxzevria®, que busquen atención médica inmediata.
- Síndrome de Guillain-Barré (SGB): el SGB es un trastorno del sistema inmune, muy poco frecuente, que causa inflamación de los nervios periféricos y puede resultar en dolor y/o adormecimiento, inicialmente de las extremidades, debilidad muscular y dificultad para la deambulación. En casos muy severos puede progresar a parálisis. La mayoría de los pacientes se recuperan de los síntomas. El pasado julio de 2021 ya se incluyó en la ficha técnica y el prospecto de esta vacuna información a este respecto con una advertencia para profesionales sanitarios y personas a las que se les administre Vaxzevria®, recomendando buscar atención médica inmediata en caso de debilidad y parálisis en las extremidades que puede progresar al pecho y a la cara. El PRAC también solicitó al TAC en el contexto del informe mensual algunos datos adicionales, para aclarar si se necesita actualizar de nuevo la información del producto y el plan de gestión de riesgos de esta vacuna. Una vez terminada la evaluación de estos datos, se ha concluido que el SGB es una posible reacción adversa de esta vacuna, cuya frecuencia de aparición es muy rara, por lo que se incluirá como tal en su ficha técnica y prospecto. Asimismo, se actualizará la advertencia actual del prospecto indicando que los pacientes diagnosticados de este síndrome tras recibir la primera dosis de la vacuna consulten con el médico antes de recibir una segunda dosis. Hasta el 31 de julio de 2021, se habían notificado 833 casos de SGB tras la vacunación con Vaxzevria® en todo el mundo, habiéndose administrado alrededor de 592 millones de dosis. En España, hasta el 8 de agosto de 2021 se habían registrado 32 casos de SGB confirmados, ninguno con desenlace mortal. Hasta esa fecha se habían administrado cerca de 9,6 millones de dosis.
COVID-19 Vaccine Janssen®
Desde que se autorizó su comercialización en la UE hasta el 2 de septiembre de 2021, se han administrado más de 13,8 millones de dosis de COVID-19 Vaccine Janssen en el EEE. Las conclusiones de la evaluación periódica de los datos de farmacovigilancia son las siguientes:
- Trastornos menstruales: ver apartado para vacuna Comirnaty®.
- Síndrome inflamatorio multisistémico (SIM): ver apartado para la vacuna Comirnaty®.
- Tromboembolismo venoso (TEV): se están revisando los datos de los casos de tromboembolismo venoso (coágulos de sangre en las venas) que se han recibido con COVID-19 Vaccine Janssen. El TEV se incluyó en el Plan de Gestión de Riesgos de esta vacuna en el momento de su autorización como un asunto de seguridad que debía ser investigado, ya que en los ensayos clínicos se observó una proporción más alta de casos en el grupo vacunado que en el grupo no vacunado. Recientemente, el PRAC ha revisado los datos procedentes de dos ensayos clínicos aún en marcha como parte de la evaluación de este riesgo, concretamente los estudios denominados COV3001 y COV3009. Los resultados de la evaluación ya se han publicado (AEMPS, 2021c). En el ensayo COV3001 se observó un mayor riesgo de TEV en personas que habían recibido la vacuna: 26 (0,1%) personas de las 21.894 que han recibido la vacuna, en comparación con 9 (0,04%) en las 21.882 personas que han recibido placebo (mediana de seguimiento de 123 días). La mayoría de los casos fueron trombosis venosas profundas o embolismos pulmonares, y se observaron en personas que tenían al menos un factor de riesgo para TEV. En el ensayo COV3009 no se observó una mayor frecuencia de TEV en las personas vacunadas respecto a las que recibieron placebo (15.708 y 15.592 personas en cada grupo respectivamente, con una mediana de seguimiento de 70 días). Los datos de notificación de acontecimientos adversos registrados durante las campañas de vacunación indican que es posible la aparición de este efecto adverso, ya que se han notificado casos en personas sin factores de riesgo para TEV, y en algunos grupos de edad los casos notificados son mayores a los esperados en la población general no vacunada del mismo grupo etario. En España, hasta el 26 de septiembre, el SEFV-H ha recibido 29 notificaciones que incluyen TEV. Hasta esta misma fecha, se habían vacunado en España cerca de 2 millones de personas con esta vacuna.
- Trombocitopenia inmune (TPI): el PRAC recomendó en su reunión del mes de agosto de 2021, actualizar la ficha técnica y prospecto de la vacuna COVID-19 Vaccine Janssen para incluir la información sobre la TPI como posible reacción adversa. La TPI es una condición médica en la que el sistema inmune ataca y destruye, por error a las células de la sangre llamadas plaquetas, que son necesarias para la coagulación normal de la sangre. En todo el mundo, hasta el 18 de junio de 2021, se habían notificado 120 casos de sospechas de TPI, 27 en los ensayos clínicos y 93 a través de las campañas de vacunación; de todos ellos, 4 tuvieron un desenlace mortal. Hasta el 30 de junio de 2021, más de 21 millones de personas habían recibido esta vacuna en todo el mundo. El análisis6 de los casos notificados indica que las personas con antecedentes de TPI pueden presentar un mayor riesgo de reducción de plaquetas y de TPI sintomática tras la administración de la vacuna COVID-19 Vaccine Janssen. En España, hasta el 26 de septiembre, el SEFV-H ha registrado dos notificaciones que incluyen TPI; ninguno de estos casos tuvo desenlace mortal. Hasta esta misma fecha, se habían vacunado en España cerca de dos millones de personas con esta vacuna.
- Síndrome de Trombosis con Trombocitopenia (STT): hasta el 8 de agosto de 2021, en España, se han registrado 5 casos, confirmados o probables, de STT en personas vacunadas con COVID-19 Vaccine Janssen, con cerca de 1,8 millones de dosis administradas. Las trombosis se presentaron en localizaciones inusuales (trombosis cerebrales o venas esplácnicas); dos de los pacientes fallecieron.
- Síndrome de Guillain-Barré (SGB): el SGB es un trastorno del sistema inmune muy poco frecuente que causa inflamación de los nervios periféricos y puede resultar en dolor y/o adormecimiento, inicialmente de las extremidades, debilidad muscular y dificultad para la deambulación. En casos muy severos puede progresar a parálisis. La mayoría de los pacientes se recuperan de los síntomas. El SGB se ha incluido en la información del producto de la vacuna COVID-19 Vaccine Janssen como una posible reacción adversa, junto con una advertencia para informar a los profesionales sanitarios y a las personas que reciban esta vacuna (ver nota de seguridad de la AEMPS) (AEMPS, 2021d). En todo el mundo, hasta el 30 de junio de 2021, se han notificado 108 casos de SGB, habiendo recibido esta vacuna más de 21 millones de personas. Uno de los casos tuvo desenlace mortal. En España, hasta el 8 de agosto de 2021 se han notificado 5 casos de SGB confirmados, habiendo recibido esta vacuna cerca de 1,8 millones de dosis. Ninguno de los casos tuvo desenlace mortal. Aunque los casos se han notificado muy raramente, en vista de la gravedad de este síndrome, los profesionales sanitarios deben estar atentos a los signos y síntomas de SGB para permitir un diagnóstico temprano, y los cuidados y el tratamiento adecuados. Se aconseja a las personas que han recibido esta vacuna buscar atención médica inmediata si desarrollan signos y síntomas que puedan sugerir SGB (dolor y/o adormecimiento, inicialmente de las extremidades, debilidad muscular y dificultad para la deambulación).