Hasta la fecha de 5 de febrero, en el año 2021 la AEMPS no ha publicado ninguna Nota Informativa de Seguridad.
Archive
Revista PAM: 440
Número 440, Enero – Febrero 2021
Patología de las glándulas salivares
Resumen
Las glándulas salivales son un elemento fundamental para mantener el equilibrio fisiológico de la cavidad oral, jugando un importante papel en diversas funciones desarrolladas en dicha cavidad, como la masticación, la gustación, la deglución y la fonación. Anatómicamente, se dividen en tres pares de glándulas salivales principales –parótida, submandibular y sublingual–, si bien existe también un amplio número de glándulas salivales menores.
El conjunto de estas glándulas puede verse implicado en una gran cantidad de afecciones relacionadas con la propia estructura de estos elementos anatómicos (acinos y conductos excretores) o con su estructura celular. Las enfermedades de las glándulas salivales abarcan desde lesiones inflamatorias e infecciosas por gérmenes comunes o específicos hasta afecciones tumorales y pseudotumorales, alteraciones funcionales y enfermedades sistémicas. En el presente artículo se resumen las principales características fisiopatológicas y aspectos clínicos de algunas de las más comunes.
- Vallejo Hernández R, Arana Zumaquero M, Ortega Polar E, Gómez González del Tánago P, Panadero Carlavilla FJ. Patología de las glándulas salivales. Panorama Actual Med. 2021; 45(440): 21-27
Anatomía
Las glándulas salivales se dividen en mayores, constituidas por tres pares de glándulas (parótida, submaxilar y sublingual), y alrededor de 800 glándulas salivales menores, situadas fundamentalmente en la cavidad oral y la orofaringe.
Desde un punto de vista estructural, las glándulas salivales mayores son de tipo tubuloacinar. La glándula parótida es la de mayor tamaño, está contenida en la fosa retromandibular y recubierta por una cápsula que es un desdoblamiento de la fascia cervical superficial. Limita profundamente con la rama ascendente de la mandíbula y el músculo masetero, por detrás con el conducto auditivo externo, cranealmente con el cigoma, e inferiormente limita con el músculo esternocleidomastoideo. Funcionalmente compuesta de acinos serosos, drena en la boca a través del conducto de Stenon a la altura del segundo molar superior, y tiene una relación muy importante con el nervio facial, que se divide en su espesor en sus cinco ramas distales.
La glándula submaxilar o submandibular se sitúa en la celda submaxilar, en la parte alta y central del cuello, bajo la rama horizontal de la mandíbula. Su límite inferior es el músculo digástrico, el superior la mandíbula, y se apoya en el músculo milohioideo. Atravesada por la arteria facial y con relaciones nerviosas de vecindad importantes (nervio hipogloso y nervio lingual), desemboca en el suelo de la boca a través del conducto de Wharton, donde segrega saliva seromucosa.
Por su parte, la glándula sublingual está situada en los dos tercios anteriores del suelo de la boca. Tiene acinos mucosos y drena en el suelo de la boca a través de los múltiples conductos de Rivinus.
Fisiología
En general, la función de todas las glándulas salivares es la producción de saliva. La secreción salival sirve para el mantenimiento de la higiene bucodental, para la preparación del alimento en la masticación, gusto y deglución, para la iniciación de la fase precoz de digestión de los hidratos de carbono y para la regulación indirecta de la hidratación del organismo. La saliva también sirve como vehículo excretor de antibióticos, distintos metales e incluso algunos virus.
Con una producción diaria que oscila entre litro y litro y medio de saliva al día, dicha secreción consta de dos tipos: una fracción serosa, que contiene ptialina (una amilasa alfa que contribuye a la digestión de almidones), y una fracción mucosa que se encarga de la lubricación. Las glándulas parótidas secretan fracción serosa, las submaxilares serosa y mucosa, las sublinguales fundamentalmente mucosa, y las bucales solo fracción mucosa. Existe una inversión iónica con respecto al plasma sanguíneo, siendo la concentración de iones potasio en la saliva siete veces mayor que en el plasma.
Exploración y diagnóstico
La parte más definitoria de la orientación clínica en afecciones de las glándulas salivales es la anamnesis, que debe recoger los síntomas, su inicio, la periodicidad, la duración y la patología asociada. La exploración física incluye la inspección y la palpación. Además, debe precisarse si existe tumoración y/o tumefacción, así como el aspecto de la piel y de los conductos excretores. En ciertas patologías inflamatorias es muy útil realizar una palpación bimanual con expresión glandular.
El estudio por imagen es útil para confirmar la impresión clínica. La radiografía simple tiene poco interés pues gran parte de la patología podría pasar desapercibida. En cambio, la sialografía, que se realiza introduciendo contraste a través del conducto de Wharton o Stenon, permite obtener imágenes sucesivas de manera indirecta, observando cómo se modifica la morfología canalicular. Las técnicas utilizadas han cambiado en los últimos años y cada vez cobra más fuerza la tomografía axial computerizada (TAC) y la resonancia magnética nuclear (RMN).
No obstante, el estudio histológico constituye la clave diagnóstica de toda la patología de las glándulas salivales. El estudio citológico con punción-aspiración con aguja fina (PAAF) puede ser de utilidad en manos de un patólogo experimentado. La realización de biopsias labiales tiene su importancia en el estudio del síndrome seco o de Sjögren o para el diagnóstico de enfermedades sistémicas.
Patología no tumoral
1. MALFORMACIONES DE LAS GLÁNDULAS SALIVARES
La ausencia total de glándulas salivales es excepcional. Pueden existir aplasias unilaterales o hipoplasias, generalmente asociado a síndromes malformativos. También se pueden encontrar distopías, ectopías o heterotopías de glándulas salivales: en definitiva, tejido glandular en zonas no habituales, como en la hipófisis, la región mastoidea o el cuello.
Como una patología relativamente frecuente, debemos señalar la existencia de ránula (literalmente significa “rana pequeña”): se trata de un quiste de retención situado debajo de la lengua, de carácter congénito o adquirido por obliteración de los pequeños conductos excretores de la glándula sublingual. Su aspecto es de tumoración de contenido líquido, traslúcida y de coloración azulada, que se visualiza al levantar la punta de la lengua. La palpación bimanual nos orienta hacia el diagnóstico, siendo su tratamiento la exéresis o extirpación completa del quiste con preservación de mucosa. Se debe realizar un diagnóstico diferencial con el quiste dermoide, el cual a la palpación es duro (contenido sólido).
2. INFLAMACIONES AGUDAS
Aunque todas las infecciones de las glándulas salivales pueden denominarse de forma general sialitis, cuando la infección es inicialmente parenquimatosa se denomina sialoadenitis y cuando afectan a los conductos hablamos de sialodoquitis.
Inflamaciones agudas bacterianas
La parotiditis aguda bacteriana es la infección bacteriana de una o ambas glándulas parótidas. Hoy en día es menos frecuente que hace unos años y se presenta mayoritariamente en ancianos, por lo general después de una cirugía abdominal o cardiotorácica, donde la disminución de la secreción salival y la ectasia de saliva favorecen la infección. El agente causal más frecuente es el Staphylococcus aureus, seguido de Streptococcus pneumoniae.
En términos de histopatología, la lesión suele ser mixta (parenquimatosa y canalicular), con tres posibles formas: catarral, que es una inflamación banal, supurativa, por la existencia de pequeños abscesos miliares, y gangrenosa, por infiltración necrótica de toda la glándula. El cuadro clínico produce una tumefacción inflamatoria de toda la glándula con dolor intenso, fiebre y mal estado general. Aparece secreción purulenta por el conducto de Stenon y, con frecuencia, trismus.
El diagnóstico se basa en el cuadro clínico y en los hallazgos exploratorios, mientras que el tratamiento consiste en la administración de antibióticos parenterales de amplio espectro, junto con abundante hidratación y medidas locales (por ejemplo, calor y masajes). En caso de absceso, se precisará drenaje quirúrgico, para lo cual es importante tener en cuenta el recorrido de las ramas del facial.
En la infancia, el proceso infeccioso suele ser recurrente, a diferencia de lo que sucede en el adulto, y constituye el proceso de agudización de una parotiditis crónica bacteriana. Los síntomas corresponden a una inflamación aguda con fiebre elevada, dolores y malestar general. La sialografía como en todo proceso agudo está contraindicada, pero no demuestra alteraciones después de la resolución del mismo. En esta fase aguda debe realizarse un tratamiento conservador y sintomático en el que tiene gran importancia los antibióticos y la hidratación del niño.
Inflamaciones víricas
La parotiditis aguda epidémica o paperas supone una infección vírica de ambas parótidas por un virus del grupo paramixovirus (familia Paramyxoviridae, género Rubulavirus), si bien también existen cuadros clínicos similares producidos por otros agentes víricos, entre los que destacan los virus coxsackie, los echovirus y el citomegalovirus. Se trata de una enfermedad típica de la infancia, con un pico de incidencia entre los 4 y 8 años, que suele aparecer en forma de pequeñas epidemias locales en guarderías y colegios, de forma preferente en invierno o primavera. El periodo de incubación es de 2 a 3 semanas.
En cuanto a su patogenia, predominan las lesiones a nivel del tejido conjuntivo intersticial, pero también se producen alteraciones necróticas de las células acinosas. El cuadro clínico se caracteriza por la aparición súbita de una tumefacción parotídea uni o bilateral con dolor asociado, sin supuración que, por lo general, está precedida por fiebre y malestar. La saliva es clara. Debido al carácter neurotropo del virus causal, pueden producirse lesiones irreversibles en el nervio auditivo con sordera unilateral o bilateral. De forma simultánea a la infección de las glándulas salivales o bien posteriormente, pueden afectarse también el páncreas, los testículos o los ovarios (puede provocar esterilidad) y el sistema nervioso central (encefalitis).
El diagnóstico se establece por la historia clínica y los datos de la exploración física. El tratamiento es sintomático e incluye reposo, analgésicos y calor local, así como forzar una buena hidratación. En la mayoría de los casos la enfermedad confiere inmunidad para toda la vida, aunque se debe recordar que los recién nacidos y los niños hasta los 9 meses no suelen padecerla por la inmunidad pasiva adquirida de la madre. Hoy en día, la incidencia de esta infección en España está disminuyendo gracias a la vacunación universal, que por lo general se administra junto con las vacunas de la rubeola y el sarampión a los 15 meses de vida, con una dosis de recuerdo a los 4 años.
Submaxilitis aguda supurativa no litiasica
Es una afección rara, más frecuente en el recién nacido y en el lactante. El tratamiento se lleva a cabo mediante antibioterapia.
Inflamación secundaria a fármacos y alérgenos
La atropina, la fenilbutazona y los derivados de la fenotiacina pueden producir una tumefacción de la glándula salival, y también una inflamación súbita de la glándula parótida, posiblemente mediado por un mecanismo de tipo alérgico.
3. INFLAMACIONES CRÓNICAS
Parotiditis crónica recidivante
La existencia de inflamaciones recidivantes con sobreinfección de una parótida es una enfermedad de patogenia desconocida. Aparece preferentemente en la infancia y se supone que existen ectasias congénitas del conducto excretor como factor predisponente, que posteriormente se sobreinfecta. El cuadro clínico se caracteriza por presentar episodios repetidos de inflamación unilateral de la glándula parótida. Ésta aparece indurada y dolorosa y segrega una saliva espesa y lechosa, a veces muy purulenta. Los ataques se repiten a intervalos irregulares, si bien durante los periodos intercrisis el paciente se halla asintomático.
El diagnóstico se realiza por la anamnesis y la evolución clínica. En la sialografía es frecuente encontrar ectasias ductales arrosariadas. Se objetiva una reducción y alteración de la formación de saliva (disquilia) y se evidencia una activación intraglandular excesiva del sistema calicreína-cinina. En la sialadenitis electrolítica se produce un aumento desmesurado de la viscosidad y eventualmente la obstrucción del sistema excretor (el sodio y la actividad de la fosfohexosa isomerasa están incrementados; ocasionalmente, se producen sialolitiasis o sialoadenitis obstructivas). Histopatológicamente se evidencia un infiltrado inflamatorio linfoplasmocitario en el intersticio y una metaplasia del conducto.
No se dispone de tratamiento etiológico alguno. Se suelen tratar con antibióticos de amplio espectro (que suelen acortar la duración de los brotes) y aprotinina, garantizando también el flujo salival (mediante la estimulación de la salivación); en muchos casos, se aconseja el masaje de la glándula por el propio paciente. En casos raros, la falta de respuesta a estas medidas obliga a la parotidectomía parcial con conservación del facial. En las formas infantiles existe la tendencia a que la enfermedad mejore de forma espontánea llegando al periodo puberal.
Sialoadenitis crónica esclerosante de la glándula submaxilar
Antiguamente llamada tumor de Küttner, suele presentarse en mujeres de 50 años o más, quienes refieren una tumefacción unilateral o bilateral, moderadamente dolorosa y con evolución a la cronicidad. La patogenia está aún poco esclarecida, aunque se le atribuye una influencia hormonal en la mujer menopáusica. Aunque se trata de una afección benigna, dado el aspecto pseudotumoral de la glándula y el inconcluyente resultado de la PAAF (punción aspirativa del tiroides con aguja fina), se recomienda la exéresis de la glándula.
Tuberculosis
Afecta principalmente a la parótida y puede sobrevenir a cualquier edad. Se caracteriza por la aparición de uno o varios nódulos en el interior de la glándula. Suele ser unilateral y siempre se trata de una tuberculosis secundaria. El diagnóstico se lleva a cabo mediante la realización de PAAF y de las reacciones cutáneas. El tratamiento debe ser médico, aunque es rara la desaparición total de los nódulos, lo que llevaría a la realización de una parotidectomía superficial con conservación del nervio facial.
Sífilis
Es una afección muy rara, que afecta generalmente a la glándula parótida y casi siempre es bilateral.
Sialadenitis por radioterapia
En este cuadro se produce una disminución de la secreción salival por aparición de trastornos funcionales de las glándulas salivales, debidos a fibrosis en los conductos mayores y menores, pudiendo llegar a una xerostomía completa acompañada de ageusia. Suele aparecer tras irradiación de tumores de cabeza y cuello con niveles superiores a 10-15 Gy. Las manifestaciones clínicas pueden durar meses o años: se debe iniciar una terapia sustitutiva con saliva artificial para evitar la formación de caries dental y otras patologías bucodentales.
Litiasis salival (sialolitiasis)
Consiste en la aparición de cálculos en los conductos de excreción de una glándula salival, que provocan obstrucción y un proceso inflamatorio secundario. Se trata de una enfermedad del adulto que se presenta, sobre todo, entre las décadas 5ª y 8ª de la vida. Es más frecuente en la glándula submaxilar, por diversas razones: la saliva es seromucosa, el drenaje es contra la gravedad y, además, el conducto de Wharton es muy estrecho. Porcentualmente, la glándula submaxilar presenta aproximadamente el 85% de los cálculos, la parótida el 14%, y la sublingual el 11%.
En cuanto a la génesis del cálculo, se cree que los tapones mucosos o detritus celulares forman el nido para el depósito de calcio inorgánico y sales fosfatadas que formarán luego el cálculo. La aparición del cálculo se ve favorecida por la existencia de procesos disquílicos transitorios (alteraciones cuantitativas o cualitativas de la secreción salival).
Existen dos grandes formas de presentación de esta enfermedad: aguda y crónica. En la forma aguda el paciente sufre una súbita hinchazón muy dolorosa de la glándula que, típicamente, aparece en el momento de la ingestión alimentaria; al continuar comiendo, la distensión sigue aumentando. En la forma crónica, el paciente presenta tumefacción recidivante de la glándula afectada durante las comidas (debido a la obstrucción del conducto excretor) y después de finalizadas éstas. El diagnóstico en ambos casos se basa en la observación del cuadro clínico y puede corroborarse al palpar el cálculo (palpación bimanual del conducto afecto); cuando el contenido en calcio del cálculo es alto puede observarse en la radiografía simple.
El tratamiento inicial se basa en el uso de analgésicos, relajantes y medidas locales. Cuando es posible debe realizarse la extirpación de la litiasis (lo que comporta una rápida desaparición del dolor), mediante intentos de dilatación del conducto excretor con pequeñas sondas preparadas con un balón inflable para que el cálculo se expulse espontáneamente. Otra posibilidad es la incisión endo-oral del conducto en la región sublingual, si existen cálculos situados en la porción anterior del conducto excretor. En casos de litiasis profundas o intraglandulares con inflamación de la glándula, esta deberá ser extirpada por vía externa, procurando respetar la rama marginal de la mandíbula del nervio facial (o nervio submentoniano) que transcurre horizontalmente a lo largo de la mandíbula. En los casos recidivantes, también debe procederse a la extirpación de la glándula salival correspondiente.
Sialosis
Constituyen un grupo de enfermedades de naturaleza distrófica, nutricionales, a menudo denominadas sialoadenosis, en las que frecuentemente existe una afectación sistémica, caracterizada por la existencia de un infiltrado linfocitario. Clínicamente se trata de enfermedades que dan lugar a tumefacciones recidivantes no inflamatorias e indoloras de las glándulas salivales, con disminución posterior de la salivación (hiposialia) y sequedad oral (xerostomía). La sialoadenosis más frecuente afecta a la parótida y entre las causas más frecuente se encuentra el alcoholismo (una hipertrofia en el 80% de los sujetos alcohólicos), el exceso de alimentos ricos en almidón, la hiperproteinemias tipo IV y los estados desnutricionales importantes, como la anorexia nerviosa en el mundo desarrollado.
Entre las causas sistémicas de la sialoadenosis podemos destacar las siguientes:
- Síndrome de Gougerot-Sjögren o síndrome de Sjögren: de causa desconocida, es una enfermedad de carácter autoinmune caracterizada por la inflamación y destrucción de las glándulas lagrimales y salivales. Se produce una infiltración linfocitaria que conlleva un aumento del tamaño de las glándulas salivales, en especial la parótida. Presenta un claro predominio en mujeres, con un inicio entre los 40 y 60 años.
Existen dos formas del síndrome de Sjögren: primario y secundario. La forma primaria se denomina síndrome seco, presentando afectación exclusiva de glándulas salivales y/o lacrimales, mientras que en la forma secundaria este síndrome se acompaña de afectación del tejido conectivo (especialmente artritis reumatoide, xerostomía, queratoconjuntivitis seca, rinofaringitis seca), junto a la mencionada tumefacción de las glándulas salivales; más adelante aparecerá atrofia glandular. Hay hipertrofia parotídea en el 80% de las formas primarias y en el 35% de las secundarias.
El diagnóstico se confirma por la presencia de anticuerpos específicos, así como por la biopsia de las glándulas salivales menores. El tratamiento es problemático, debido a que se desconoce la etiología, y está encaminado a combatir la sequedad, la afectación glandular salival y las alteraciones sistémicas. Para la sequedad de la boca y laringe se administra lágrima artificial. - Sarcoidosis: esta enfermedad autoinmune afecta en el 50% de los casos a las glándulas salivales menores y tan solo en un 4% a la parótida, de ahí la importancia de la biopsia labial para el diagnóstico. El síndrome de Heerfordt o fiebre uveoparotidea es una forma clínica rara de sarcoidosis, consistente en la asociación de uveítis, parálisis facial, tumefacción1 parotídea y fiebre.
- Síndrome de Inmunodeficiencia Adquirida: la infección por el virus de la inmunodeficiencia humana (VIH) puede producir una hipertrofia linfoproliferativa con quistes en el espesor de las glándulas salivales. Se desconoce si la lesión se produce directamente por la propia acción del virus o bien por un incremento linfoproliferativo, pero es común la aparición de una tumoración difusa de una o ambas parótidas que, a menudo, se acompaña de adenopatías cervicales; en muchos pacientes aparece también xerostomía.
Respecto a su diagnóstico, si en la exploración por imágenes se detecta la presencia de quistes múltiples de parótida, a menudo bilaterales, obliga a descartar una infección por VIH. El diagnóstico definitivo se obtiene mediante la correlación de la historia clínica con los hallazgos de laboratorios típicos de esta infección. Se debe considerar un diagnóstico diferencial con el Síndrome de Mikulicz (no tiene etiología unitaria ni se considera una enfermedad independiente) y con la hipertrofia maseterina. Esta última se produce al morder o cerrar la boca, se manifiesta con una tumefacción de tipo muscular, que se endurece y acentúa, cuando se compara con palpación bimanual endoral y exterior.
El tratamiento es el propio de los individuos infectados por el VIH, esto es, con fármacos antirretrovirales. En ocasiones, frente a los quistes parótideos se utiliza la cirugía o la radioterapia a dosis bajas.
4. TRAUMATISMOS
El compromiso más importante en un traumatismo que afecta a las glándulas salivales es la afectación nerviosa, tanto del nervio facial o del hipogloso como del lingual, debiendo procederse a la reparación quirúrgica inmediata. Las lesiones del conducto excretor solo requieren de cirugía si afectan a los grandes conductos excretores. Las fístulas salivales postraumáticas suelen ceder de forma espontánea.
El síndrome auriculotemporal o síndrome de Frey se caracteriza por sudoración y enrojecimiento de la región preauricular durante la masticación y la deglución; durante la ingesta de la comida aparece rubor (por vasodilatación) y sudoración de la piel de la mejilla delante de la oreja (sudoración gustativa). Debuta a los meses de un accidente traumático o postoperatorio de la parótida, cuando en la regeneración aberrante de los nervios secretores parotídeos se produce una anastomosis entre fibras parasimpáticas postganglionares (destinadas a la glándula) y fibras simpáticas vegetativas (destinadas a la piel); de este modo se llega a inervar las glándulas sudoríparas de la piel de la mejilla. Es por ello que aparece una hipersensibilidad de las glándulas sudoríparas cutáneas frente a impulsos colinérgicos. El tratamiento se realiza con pomadas de escopolamina o solución de cloruro de aluminio, recomendándose solo en ocasiones la neurectomía del nervio timpánico en la caja timpánica. Actualmente también se utiliza la toxina botulínica.
5. SIALORREA (PTIALISMO)
Sialorrea y ptialismo son sinónimos, y designan la secreción salival excesiva y no el derrame de saliva fuera de la boca (esto puede producirse por sialorrea, pero también en pacientes con disfagia). Entre las distantes causas que pueden producirla, destacan las lesiones digestivas, las intoxicaciones, causas neurológicas, causas gineco-obstétricas (como el ptialismo gravídico), endocrinopatías o efectos secundarios de medicamentos.
6. FÍSTULA SALIVAL
Se puede considerar de forma independiente debido a su etiología tan variada. Su origen puede ser secundario a un traumatismo de la glándula o del conducto excretor, intervenciones quirúrgicas, inflamaciones específicas o inespecíficas, malformación congénita, etc. Aparecen síntomas cuando se produce el drenaje salival hacia el exterior, sobre todo durante la ingesta de los alimentos. Las fístulas glandulares se cierran espontáneamente, mientras que las ductales deben ser intervenidas (conversión quirúrgica de la fístula externa en interna). Existen otras posibilidades de tratamiento como es la reducción de la secreción salival mediante radioterapia, o bien, como último recurso, la exéresis de la glándula.
7. ESTENOSIS
Pueden ser extrínsecas, por alteraciones odontológicas principalmente, o intrínsecas, consecutivas a infecciones, traumatismos, cálculos o incluso neoplasias del conducto. Se caracterizan por la aparición de inflamación dolorosa y periódica asociada a la ingesta de alimentos. En el diagnóstico se utiliza la sialografía y el tratamiento consiste en la supresión de los factores causales.
Patología tumoral
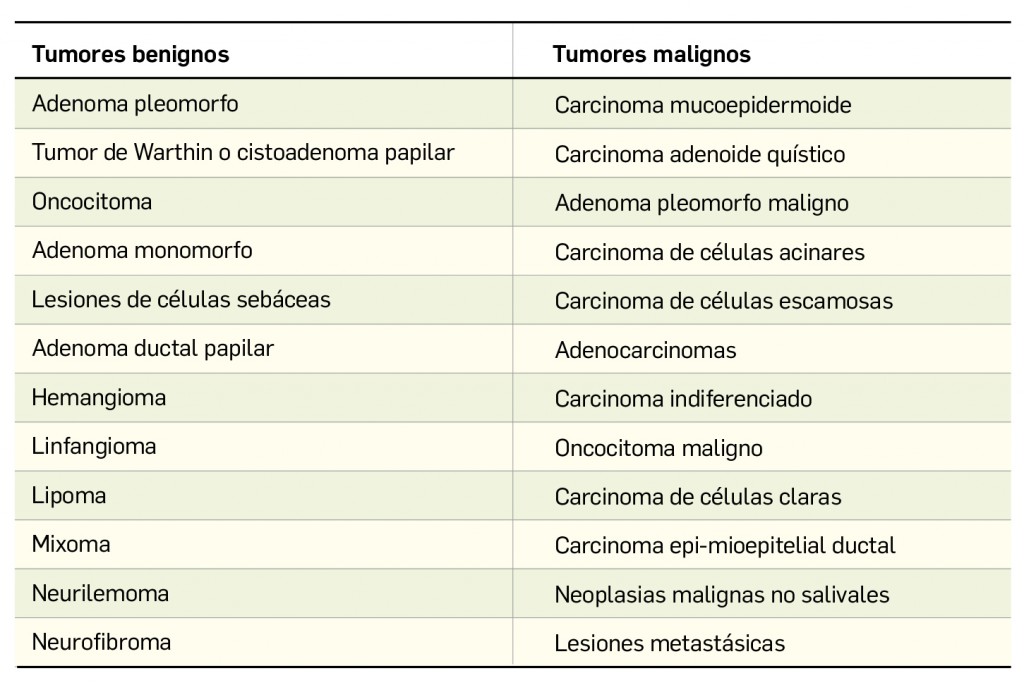
GENERALIDADES
El 80% de los tumores de las glándulas salivales se localizan en la parótida (la mayoría de ellos en la cola de la glándula o en el lóbulo superficial), el 10% tiene localización submaxilar y el otro 10% restante corresponden al resto de las glándulas. Afortunadamente, son benignos un 80% de los tumores parotídeos, 70% de los submaxilares y 50% de los de glándulas salivales menores (localizados éstos más frecuentemente en el paladar), si bien la mayoría de los tumores de la glándula sublingual son malignos. Se cumple la regla de que, según disminuye el tamaño de la glándula, aumentan las probabilidades de malignidad del tumor.
Se suelen presentar como un bulto o tumoración, duro a la palpación, y posiblemente asociados a parálisis facial (es raro, pero si se observa, es casi patognomónico de malignidad, ya que hay invasión nerviosa). Otros datos que podemos observar en la exploración son la ulceración de la piel o mucosas, la fijación a planos profundos, o un rápido crecimiento. Para llegar al diagnóstico es necesario la palpación cuidadosa de la lesión, observando sus características de dureza, tamaño, adherencia a planos profundos, etc. Está indicada la realización de una punción aspiración con aguja fina (PAAF) si la localización del tumor lo permite. Además, pueden ser útiles pruebas de imagen como TAC o RNM, pero no es posible realizar biopsia parotídea ya que se produce como consecuencia una inflamación de la glándula y la consiguiente alteración de estructuras.
El tratamiento de estas lesiones suele ser la parotidectomía superficial o total, intentando siempre que sea posible la conservación del nervio facial. A veces, es necesario realizar tratamiento adyuvante con radioterapia, dependiendo de la estirpe tumoral.
Se definirán a continuación las características principales de los tumores clínicamente más relevantes de las glándulas salivales.
Adenoma pleomorfo o tumor mixto
Es el más frecuente de todos los tumores de glándulas salivales: el “rey” de los tumores de la glándula parótida. Más del 80% (≈84%) de estos tumores se suelen localizar en el lóbulo superficial parotideo, apareciendo en la anatomía patológica habitualmente “encapsulados”, conformados por una mezcla de células ductales, mioepiteliales y mesenquimales; el estroma está compuesto por tejidos condroides, mucoides, hialinos y mixoides.
El tratamiento es exclusivamente quirúrgico, siendo la parotidectomía superficial la técnica de elección. Durante la cirugía se suele respetar parte de la glándula, ya que la recurrencia es rara (tasa de recurrencia del 2%) y se trata de un tumor benigno de buen pronóstico, si bien se debe realizar una exéresis del mismo (ya que, aunque es raro, se puede malignizar).
Tumor de Warthin o cistoadenoma papilar linfomatoso
Es el segundo tumor benigno más frecuente en glándulas salivales, casi exclusivo de parótida (2-6% de los tumores parotídeos), y más frecuente en varones. En la anatomía patológica se visualiza circunscrito, blando, quístico y con contenido mucoide, observándose elementos papilares con epitelio en dos capas y estroma linfoide. El tratamiento es quirúrgico mediante enucleación: se extirpa exclusivamente la tumoración, sin tocar ni parótida ni el nervio facial. Otros autores abogan por realizar una parotidectomía total, aunque es una opción muy discutible. Puede presentar hasta un 10% de recidivas por multicentralidad, aunque la malignización es excepcional.
Carcinoma mucoepidermoide
Es el carcinoma maligno de glándulas salivales más frecuente en la infancia, siendo de los tumores malignos más frecuentes de la parótida (60-70%), aunque también puede presentarse en la cavidad oral (15-20%) o en las submaxilares (6-10%). Pero donde se dan con más frecuencia es en las glándulas pequeñas: labiales, palatinas, etc.
Existen varios grados de malignidad en este tipo de tumores: a) bajo grado de malignidad, con un comportamiento casi benigno; b) alto grado de malignidad; y c) grado intermedio (el más frecuente).
En la anatomía patológica suelen aparecer células secretoras de mucina, epidermoides e intermedias. Es frecuente la invasión perineural y linfática en los de alto grado. El tratamiento va a depender del grado: en los de bajo grado, el tratamiento de elección suele ser la cirugía mediante una parotidectomía total conservadora; en los de grado intermedio se realiza la parotidectomía total preservando el facial si es posible, y radioterapia postoperatoria; y en los de alto grado se opta por cirugía con amplios márgenes mediante parotidectomía total radical, acompañado de radioterapia postoperatoria. Para estos tumores se ha descrito un alto índice de recurrencias.
Carcinoma Adenoide Quístico O Cilindroma
Se trata del tumor maligno más frecuente de las glándulas salivales menores. A diferencia de la mayoría de los tumores de cabeza y cuello, que suelen dar metástasis linfoganglionares, pero rara vez metastatizan a distancia, el cilindroma es de los pocos tumores que dan metástasis a distancia2. Su crecimiento e invasión de los tejidos adyacentes es lenta. Histológicamente se presenta como células ductales y no ductales o mioepiteliales alrededor de espacios ovales conformando un “patrón cribiforme” con invasión perineural. El tratamiento consiste en la maxilectomía en los de paladar, o bien parotidectomía radical y radioterapia postoperatoria. Dado el riesgo de metástasis a distancia, se precisa un largo periodo de seguimiento.
Variaciones en medicamentos previamente comercializados
Cambio de nombre de medicamentos
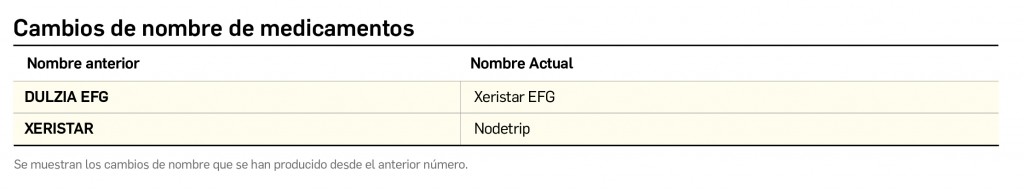
Cambios de laboratorio titular de autorización
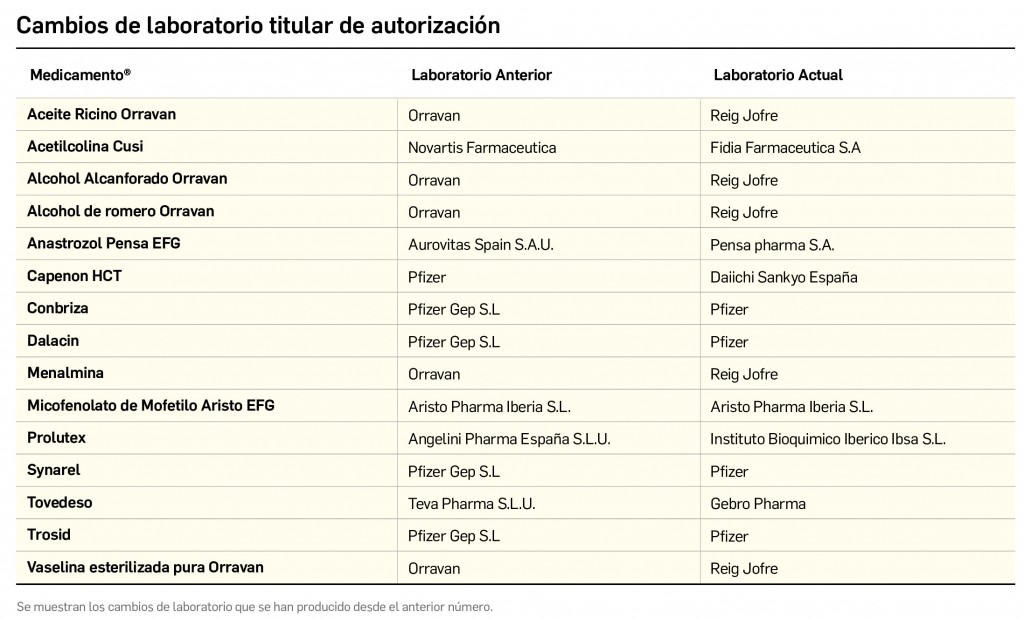
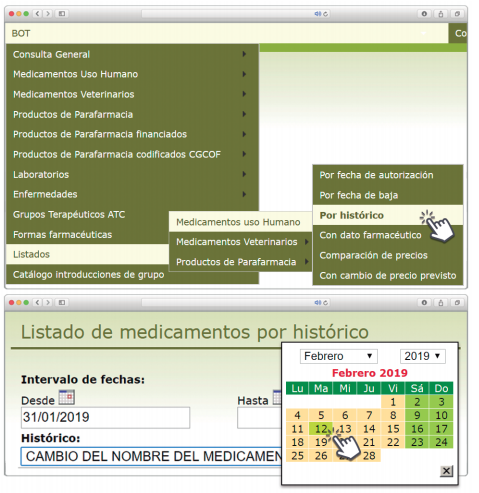
Cómo localizar cambios de nombre y de laboratorio con BOT PLUS
Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
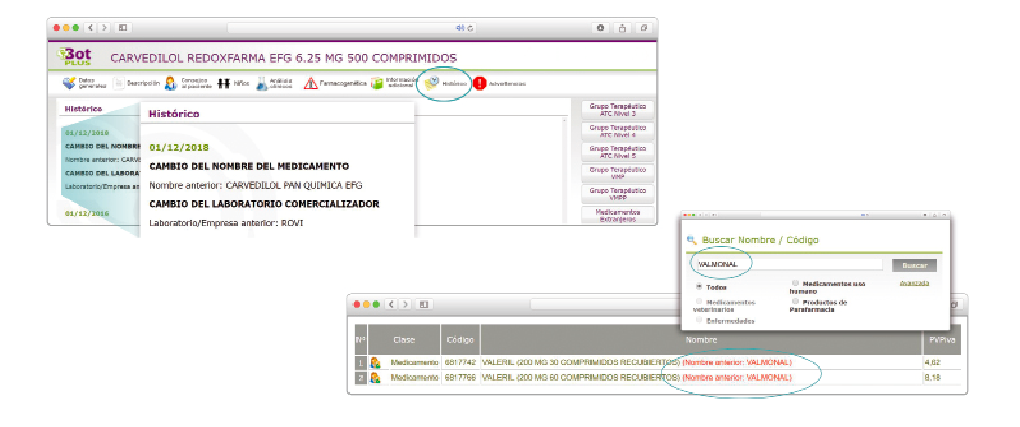
Posibilidad de generar listados por histórico
Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.
Servicio profesional farmacéutico de cesación tabáquica
Servicio de Indicación farmacéutica
> Segundo Accésit, 8ª Edición Premios FORO de Atención Farmacéutica en Farmacia Comunitaria.
Presentación del servicio
El Servicio Profesional Farmacéutico de Cesación Tabáquica es un servicio de seguimiento y apoyo en la Farmacia Comunitaria (FC) realizado por un farmacéutico especializado en una Zona de Atención Personalizada (ZAP) que se le ofrece al paciente que manifiesta su deseo y solicita ayuda para dejar de fumar.
Este Servicio permite motivar al paciente para conseguir sus objetivos mediante la utilización de distintas guías, equipos, prospectos informativos que ayudan a reforzar los mensajes y consejos del farmacéutico comunitario, junto con visitas rutinarias y/o tratamientos farmacológicos.
Motivo del servicio
El motivo del Servicio era poder transmitir y concienciar al paciente que se estaba planteando dejar de fumar, de todos los inconvenientes que conlleva fumar y los beneficios que se consiguen mediante el abandono del consumo de tabaco, tanto a corto como a largo plazo, con el fin de apoyar y conseguir que el propio paciente fuera capaz de dejar de fumar con la ayuda y los consejos del farmacéutico comunitario.
Perfil de paciente – Evaluación
Durante la dispensación, el paciente nos comunicó que su dependencia al tabaco había ido aumentando en estos últimos años debido a la ansiedad que tenía por varios problemas personales. Anteriormente había intentado dejar de fumar, con periodos de abstinencia de 1 a 3 meses con cigarrillo electrónico.
Nos dejó entrever su predisposición para dejar de fumar, motivada por su entorno familiar y su estado de salud. Seguidamente, se procedió a informarle con varios prospectos informativos y se concretó una primera visita para hacerle una valoración general.
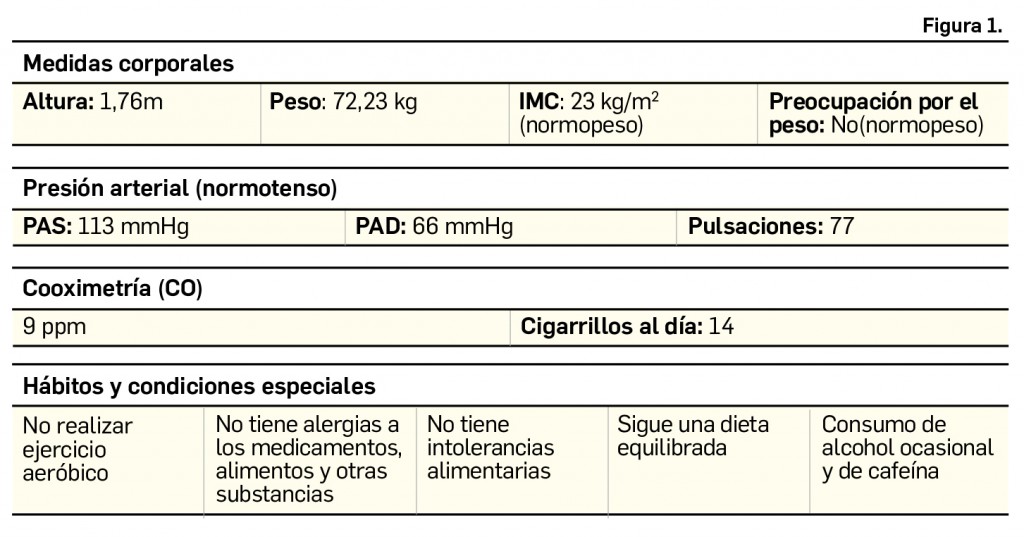
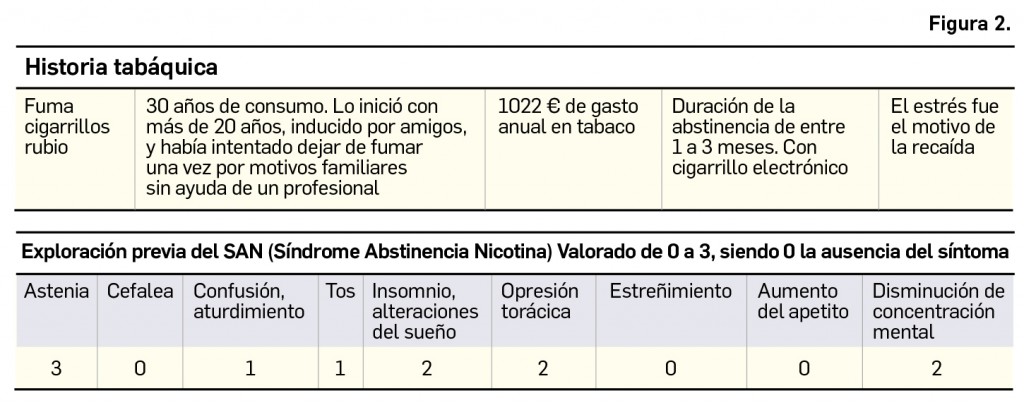
En esa primera visita se valora el paciente siguiendo el programa CESAR de SEFAC. Las características personales en el momento basal se recogen en la Figura 1, y aquellas relativas a su hábito tabáquico, en la Figura 2.
Actuación/Intervención realizada
Inicialmente, se somete al paciente a dos cuestionarios diferentes.
TEST DE RICHMOND (grado de motivación):
- ¿Le gustaría dejar de fumar si pudiera hacerlo fácilmente? Sí.
- ¿Intentará dejar de fumar en las próximas 2 semanas? Definitivamente, sí.
- ¿Cuánto interés tiene usted en dejarlo? Muy seriamente.
- ¿Cuál es la posibilidad de que usted dentro de los próximos 6 meses sea un no fumador? Definitivamente, sí.
La puntuación Richmond fue de 10 (alta motivación), pudiendo concluir que el paciente estaba preparado para comenzar la cesación en un corto periodo de tiempo.
TEST DE FAGERSTRÖM (grado de dependencia):
- ¿Cuánto tiempo pasa entre que se levanta y se fuma su primer cigarrillo? De 31 a 60 minutos.
- ¿Encuentra difícil no fumar en lugares donde está prohibido (hospital, cine, biblioteca)? No.
- ¿Qué cigarrillo le desagrada más dejar de fumar? El primero de la mañana.
- ¿Cuántos cigarrillos fuma al día? Entre 11 a 20 cigarrillos al día.
- ¿Fuma con más frecuencia durante las primeras horas después de levantarse que durante el resto del día? Sí.
- ¿Fuma, aunque esté tan enfermo que tenga que guardar cama la mayor parte del día? Sí.
La puntuación Fagerström fue de 5 (dependencia media): el fumador tiene una dependencia media a la nicotina.
A continuación, se citó al paciente el día D y se le derivó a su médico con una carta en la que se aconsejaba el tratamiento farmacológico con vareniclina. En el día D, el paciente acudió a la farmacia con la prescripción de vareniclina en la receta de su médico. Desde la farmacia, se le dieron las recomendaciones para un adecuado proceso de uso de vareniclina.
Las siguientes visitas se concertaron 8 días después del día D, un mes después, dos meses después, tres meses después y seis meses después del día D. Durante la cuarta visita, el farmacéutico comunitario observó que el paciente manifestaba síntomas de ansiedad y se le derivó al médico. Como consecuencia de esta nueva derivación, le recetaron citalopram 10 mg comprimidos (0-0-0-1-0).
Resultados
Con el Servicio Profesional Farmacéutico de Cesación Tabáquica y el seguimiento del farmacéutico se consiguieron los objetivos planteados y se constató, en las sucesivas visitas, que el paciente dejó de fumar. El tratamiento con vareniclina fue el adecuado y las derivaciones al médico fueron cruciales para mantener el abandono del consumo de tabaco por parte del paciente, justificando aún más la labor del farmacéutico comunitario. En todo momento, el médico aceptó las recomendaciones y el tratamiento aconsejado por el farmacéutico comunitario.
En definitiva, la labor del farmacéutico fue crucial para derivar al paciente en los momentos oportunos y poderlo motivar durante todo el proceso de cesación, ayudándole a superar los síntomas de abstinencia a la nicotina.
Comentarios
El tabaco, desde un punto de vista clínico, y debido a la gran variedad de componentes tóxicos que contiene, es un factor de riesgo que puede ocasionar distintas patologías, como por ejemplo: enfermedad pulmonar obstructiva crónica (EPOC), cáncer, asma y cataratas. Adicionalmente, el tabaquismo puede repercutir directa y negativamente en el nivel económico del paciente, e indirectamente mediante el incremento del gasto sanitario.
El Servicio Profesional Farmacéutico de Cesación Tabáquica es una gran oportunidad para eliminar este mal hábito en los pacientes que se están planteando dejar de fumar, con la ayuda de un farmacéutico comunitario, que les podrá aconsejar, derivar y apoyar durante todo el seguimiento.
Casos ganadores y finalistas 8ª edición premios FORO AF-FC
El objetivo de la edición anual de Premios FORO de Atención Farmacéutica en Farmacia Comunitaria (Foro AF-FC) es reconocer el compromiso asistencial de los farmacéuticos que ofrecen a la población Servicios Profesionales Asistenciales de acuerdo a los procedimientos consensuados1, contando con la colaboración de laboratorios Cinfa. La convocatoria de la 8ª edición se realizó entre los meses de marzo a julio de 2019, periodo tras el cual el jurado, compuesto por seis representantes de las instituciones que forman Foro AF-FC, evaluó los 30 casos recibidos de acuerdo a los modelos editados en las webs de todas las instituciones para la descarga, cumplimentación y remisión on line, por parte de farmacéuticos voluntariamente interesados.
Se mantuvo un modelo abierto para aquellos casos que no se correspondieran con los Servicios descritos. Como novedad, en esta edición se posibilitó una categoría específica para casos de alumnos y tutores en prácticas tuteladas o máster. Los casos recibidos se clasificaron en: 8 casos del Servicio de Dispensación, 1 del Servicio de Indicación Farmacéutica, 3 del Servicio de SFT, 15 de modelo abierto y 4 de alumnos. El jurado valoró en cada caso once apartados relacionados con la adecuación de la terminología y metodología de Foro AF-FC, la existencia de práctica colaborativa, la utilización de herramientas informáticas, el interés clínico y el profesional del caso en la Farmacia Comunitaria, entre otros.
Los casos ganadores y finalistas de la citada edición se irán presentando periódicamente en esta sección de PAM. No obstante, puede acceder a la publicación íntegra de todos los casos a través del siguiente enlace al espacio de los Premios de FORO AF-FC:
Foro de Atención Farmacéutica en Farmacia Comunitaria (Foro AF-FC) es un grupo de trabajo y debate constructivo creado en 2009. Actualmente está constituido por farmacéuticos representantes de las siguientes instituciones:
- Consejo General de Colegios Oficiales de Farmacéuticos (CGCOF).
- Conferencia Nacional de Decanos.
- Fundación Pharmaceutical Care España.
- Grupo de Investigación en Atención Farmacéutica de la Universidad de Granada.
- Sociedad Española de Farmacia Familiar y Comunitaria (SEFAC).
La principal función de Foro AF-FC es contribuir a la implantación de los Servicios Profesionales Farmacéuticos
Asistenciales (SPFA) en la farmacia comunitaria, para lo cual:
- Mantiene el consenso adoptado y la homogeneidad de los procedimientos de Foro AF 2008 en los proyectos, conjunta o individualmente; de esta manera, se consigue un objetivo común transmitiendo el mismo mensaje con una terminología consensuada.
- Apoya la máxima difusión de los SPFA para alcanzar su implantación en la Farmacia Comunitaria.
- Incrementa la colaboración entre las organizaciones del Grupo.
- Constituye un agente de referencia en SPFA coordinado por el Consejo General.
Principales actividades:
- Elaboración de documentos consensuados; definición y procedimientación de Servicios Profesionales Farmacéuticos
- Asistenciales.
- Definición y clasificación de SPFA abordando tanto los relacionados con el medicamento (AF) como con los relacionados con la Salud Comunitaria.
- Edición de la actualización de la Guía practica de SPFA
- Edición anual de Premios a los mejores casos de farmacéuticos implicados en SPFA y participación en eventos/congresos para su divulgacion.
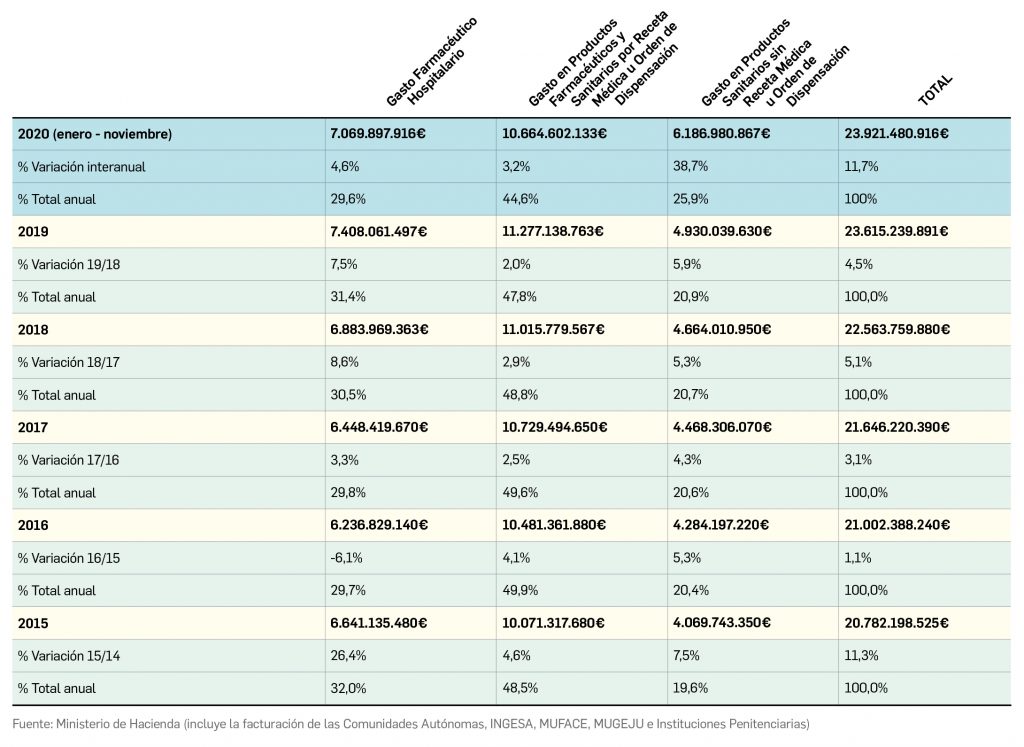
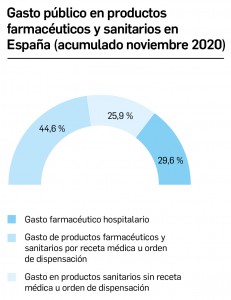
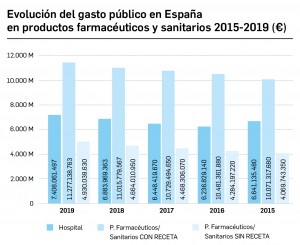
Gasto público en productos farmacéuticos y sanitarios
Estadísticas de altas y bajas, composición, precio y características de medicamentos


La necesidad de perseverar en el uso racional de antimicrobianos
Un interesante trabajo transversal desarrollado por un grupo de 47 farmacéuticos de Atención Primaria en España, distribuidos en 30 áreas de salud de 12 Comunidades Autónomas (que abarcaban casi 6 millones de habitantes), ha puesto de manifiesto una mayor frecuencia en la exposición a tratamientos antibióticos de larga duración en los pacientes adultos mayores con pluripatología, como demuestra el hecho de que el 51% de ellos recibiese antibióticos a lo largo de todo el año.
Se consideraron todos los pacientes a quienes se les dispensó durante el año 2017 al menos 30 envases de medicamentos antibacterianos para uso sistémico y se evaluó la prevalencia del uso de antibióticos, las patologías para las que se prescribieron, las características clínicas de los pacientes (incluyendo comorbilidades y tratamientos concomitantes) y las cepas de los microorganismos aislados. Para ellos, se obtuvo una edad promedio de 72 años, el 52% eran hombres, el 60% fumadores o ex-fumadores, y la práctica totalidad presentaban al menos una patología crónica, con 4 comorbilidades de media, destacando las cardiovasculares (67%, con o sin hipertensión), las respiratorias –asma, EPOC o bronquiectasias– (62%), obesidad (54%), afecciones neurológicas/mentales (32%), diabetes (23%) y enfermedades urológicas (21%); el 29% estaban inmunosuprimidos y el 10% había fallecido en el momento de la toma de datos. Respecto al uso de antibióticos, esos pacientes recibieron una media de 3 tratamientos durante el año, principalmente fluoroquinolonas (28%), macrólidos (21%), penicilinas (19%) o cefalosporinas (12%), los cuales se usaban mayoritariamente para patologías del tracto respiratorio inferior (48%), infecciones urinarias (27%) e infecciones de piel o tejidos blandos (11%). En hasta el 21% de los casos fueron aplicados con fines profilácticos. Además, destaca el hecho de que solo el 35% de los pacientes obtuvo un diagnóstico microbiológico definitivo, siendo Pseudomonas aeruginosa (30%) y Escherichia coli (16%) los patógenos aislados con mayor frecuencia.
Se concluye, en definitiva, que los “grandes consumidores” de antibióticos en el ámbito comunitario son pacientes mayores, pluripatológicos y polimedicados, quienes son en muchos casos tratados con antibióticos de amplio espectro durante periodos prolongados y presentan infecciones frecuentes por bacterias multirresistentes; asimismo, el riesgo de procesos complicados que requieran hospitalización se puede ver incrementado, pero también el de interacciones farmacológicas, contraindicaciones o reacciones adversas al tratamiento. Por tanto, parece razonable aceptar que el abordaje de las infecciones en este perfil de pacientes debería personalizarse y diferenciarse de los pacientes “sanos” que reciben antibióticos ocasionalmente, todo ello a fin de minimizar el grave problema de salud pública que representan las resistencias a antimicrobianos y la aparición de bacterias multirresistentes. Los autores sugieren que la mejora del uso de antibióticos en este grupo de población pasa necesariamente por una adecuada coordinación entre los equipos PROA (programas de optimización de uso de antimicrobianos) de Atención Primaria y Hospitalaria que consiga una mejora en la selección del tipo de antibiótico más adecuado y un ajuste de la duración del tratamiento al menor tiempo posible, siendo especialmente conveniente evitar terapias profilácticas prolongadas.
Medicamentos con nuevos principios activos o biosimilares
COMERCIALIZADOS EN ESPAÑA EN LOS ÚLTIMOS DOCE MESES
Valoración de la innovación terapéutica en PAM
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
- SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
- INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
- INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
- Evidencia clínica: mediante estudios controlados, específicamente diseñados y desarrollados para demostrar la eficacia y la seguridad del nuevo medicamento, con demostración fehaciente de lo que puede ser un avance o mejora sobre la terapia estándar hasta ese momento, en el que caso de que exista.
- Plausibilidad científica (potencialidad): existencia de aspectos en el medicamento que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostrados mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
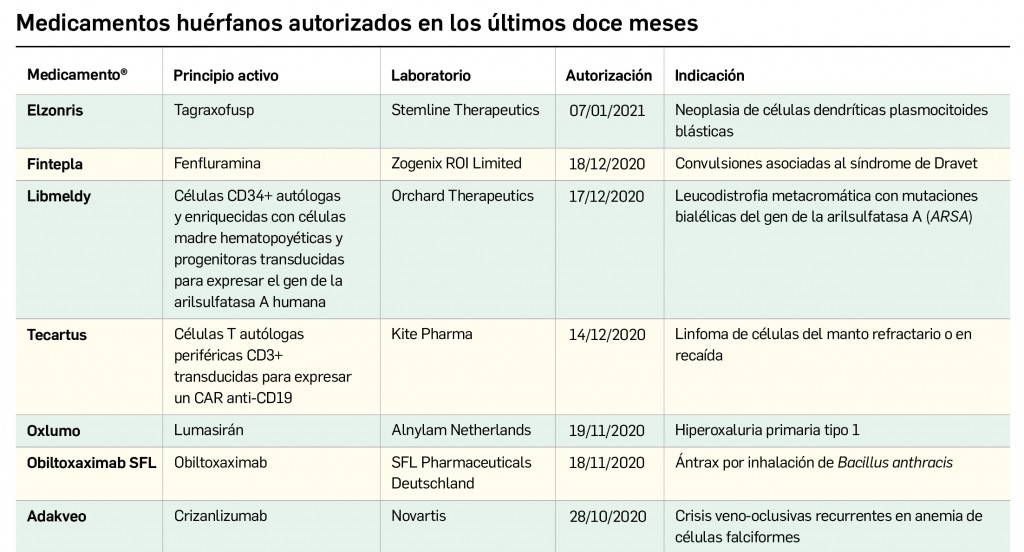
Nuevos medicamentos huérfanos
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
Instituciones y redes españolas
- Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
– Instituto de Enfermedades Raras. Más información en este enlace.
– CIBERER(Centro de Investigación Biomédica en Red de Enfermedades Raras). Más información en este enlace.
- Instituto de Mayores y Servicios Sociales (IMSERSO, MINISTERIO DE SANIDAD, CONSUMO Y BIENESTAR SOCIAL). Más información en este enlace.
- Federación Española de Enfermedades Raras (FEDER). Más información en este enlace.
- Asociaciones de pacientes en España. Más información en este enlace.
Instituciones y redes europeas
- Agencia Europea de Medicamentos (EMA; EuropeaN Medicines Agency). Apartado de Medicamentos Huérfanos (Inglés). Más información en este enlace, y en este.
- Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español). Más información en este enlace.
- Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español). Más información en este enlace.
- Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). Más información en este enlace.
Otras instituciones y redes internacionales
- Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés). Más información en este enlace.
- Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés). Más información en este enlace.
Nuevos medicamentos en 2020
Resumen
Como viene siendo habitual, inauguramos el primer número anual de PAM con un resumen de todos los medicamentos con nuevos principios activos comercializados por primera vez en España en el año previo. En comparación con los anteriores, el difícil 2020 ha sido un año menos prolífico en cuanto a la incorporación al arsenal terapéutico de innovaciones farmacológicas, habiéndose comercializado un total de 11 nuevos principios activos, 20 menos que en el año anterior; casi la mitad de ellos (5) se enmarcan dentro del grupo terapéutico ATC L (terapia antineoplásica y agentes inmunomoduladores), continuando con la tendencia del último lustro en que la mayor parte de los nuevos principios activos se dirigen al tratamiento de patologías oncológicas o de naturaleza autoinmune. En 2020, solo se han comercializado 2 nuevos principios activos en medicamentos designados como huérfanos.
A nivel de volumen de medicamentos, hay que destacar que por cuarto año consecutivo se reduce el número de presentaciones de medicamentos comercializadas, pues en 2020 se han comercializado 827 nuevas presentaciones –tanto de nuevos principios activos como de los ya existentes– frente a las 949 que se han dado de baja. A finales del año, el mercado de medicamentos en España contaba con casi 18.000 formatos o presentaciones comerciales de medicamentos. En la última década se han incorporado 9.221 presentaciones, lo que supone un 51,5% del total, y han desaparecido 9. 586, con un balance negativo de 365 formatos.
En cuanto al grado de innovación terapéutica, destaca sobremanera la comercialización –y financiación– en España de dos principios activos nuevos: dupilumab, un anticuerpo monoclonal frente a la subunidad α del receptor de la IL-4, que representa el primer tratamiento específicamente dirigido a contrarrestar la inflamación tipo 2 mediada por células Th2, inaugurando una prometedora vía terapéutica en dermatitis atópica y asma; y patisirán, el primer ARN pequeño de interferencia autorizado –en un medicamento huérfano– para el tratamiento de una enfermedad en humanos (la polineuropatía en adultos con amiloidosis familiar por transtiretina), y que se erige como cabeza de serie de una vasta clase de fármacos con gran potencial en diversas enfermedades; el primero de ellos fue merecedor del Premio Panorama 2020, y patisirán recibió una Mención de Honor en los citados galardones. Pero éstos no han sido los únicos, habiéndose comercializado también interesantes medicamentos frente al cáncer de pulmón no microcítico o el acné, entre otras patologías. Las principales características farmacoclínicas de todos los nuevos principios activos de 2020 se resumen en el presente artículo.
- Fernández Moriano C. Nuevos Medicamentos en 2020. Panorama Actual Med. 2021; 45(440): 7-19
Nuevos medicamentos
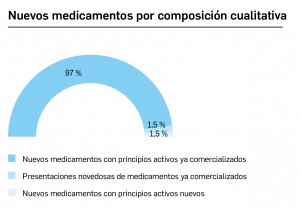
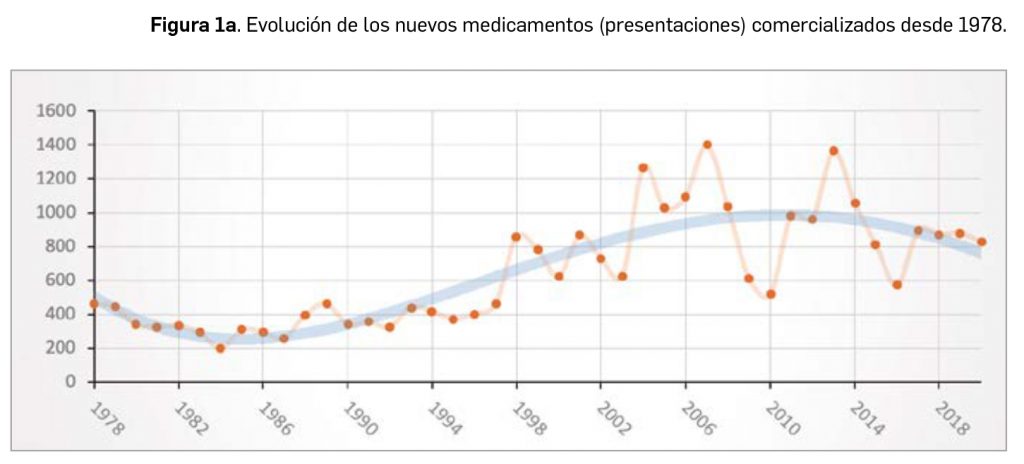
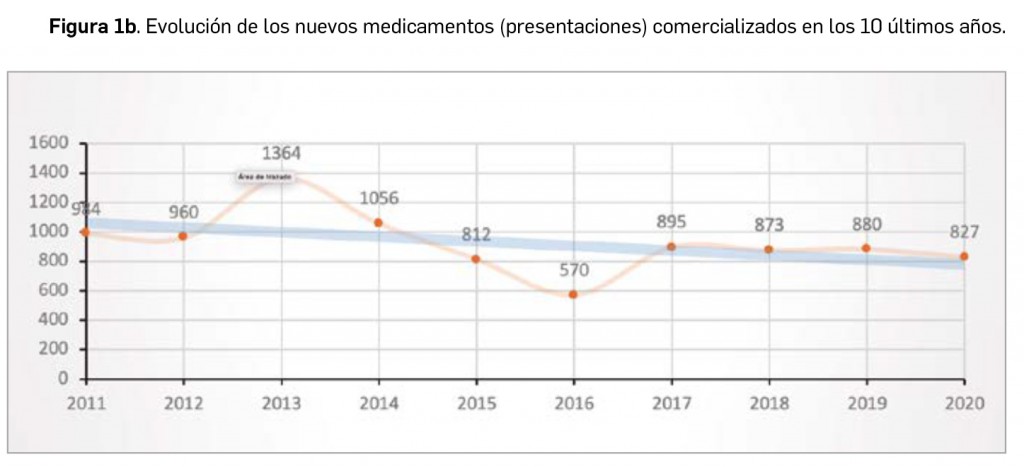
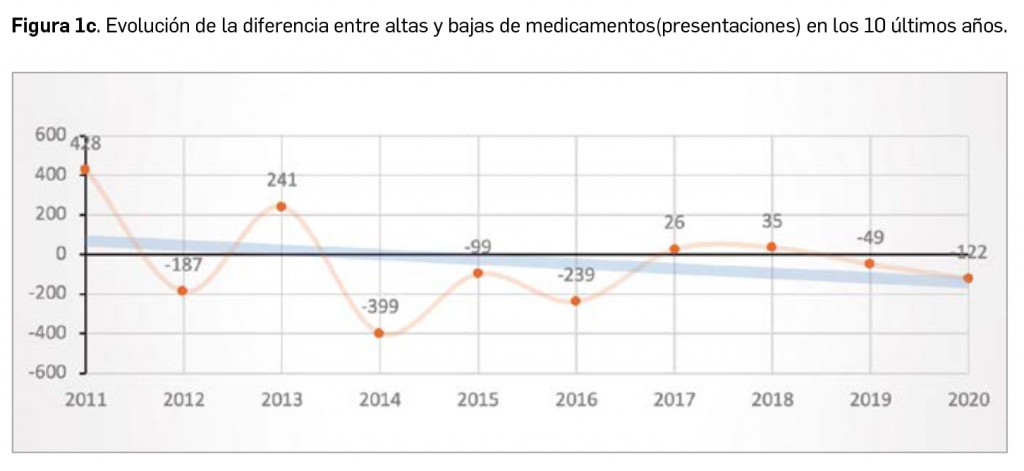
Durante el año 2020, se registraron en España 827 nuevas presentaciones comerciales o formatos de medicamentos, tal y como se recoge en la Tabla 1, incluyendo el 96% de ellas principios activos previamente comercializados. El dato se alinea con la tendencia mostrada en los periodos analizados, en los últimos 42 años (Figura 1a) y en los últimos 10 años (Figura 1b). Cabe destacar, por ejemplo, que un total de 500 presentaciones corresponden a medicamentos genéricos, lo cual representa el 60,5% del total de nuevas presentaciones. Sin embargo, en el último año también se dieron de baja o anularon un total de 949 presentaciones, lo que representa un balance negativo en el número de medicamentos comercializados.
 Por lo que se refiere a los nuevos principios activos comercializados en 2020 en nuestro país, han sido un total de 11 –de los cuales, solo 2 han sido incluidos en medicamentos designados como como huérfanos (un 18%)–
Por lo que se refiere a los nuevos principios activos comercializados en 2020 en nuestro país, han sido un total de 11 –de los cuales, solo 2 han sido incluidos en medicamentos designados como como huérfanos (un 18%)–
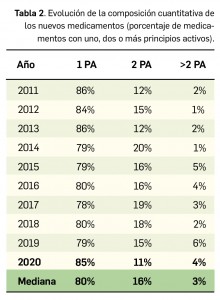
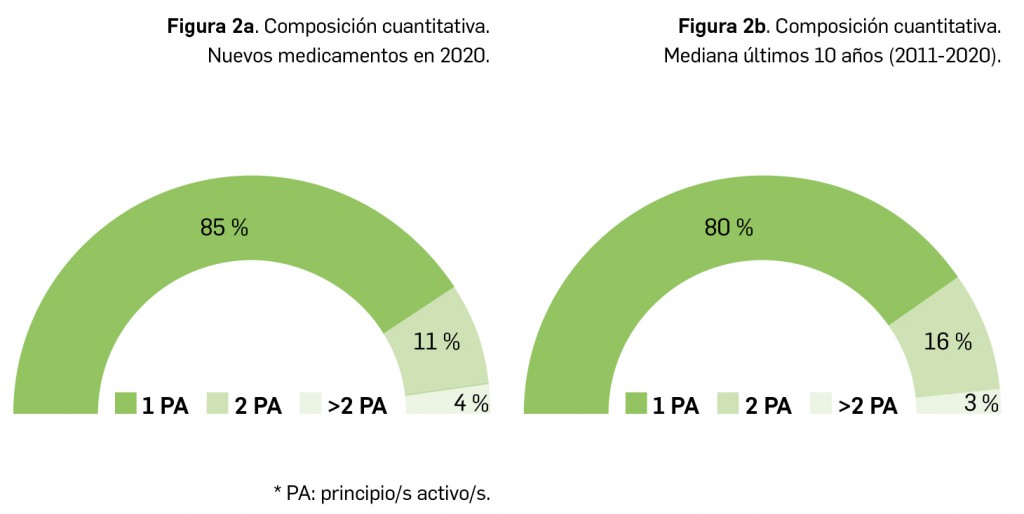
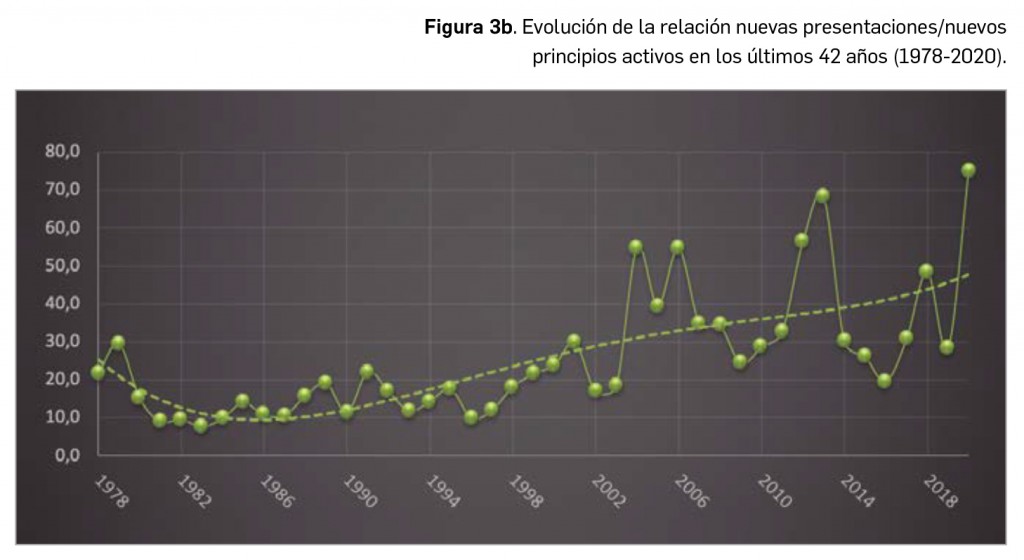
(Tabla 3), 20 menos que el año pasado, lo que representa un número significativamente inferior a la mediana de 29 nuevos principios activos/año correspondiente a la década 2011-2020 (el promedio de nuevos principios activos/año se sitúa en 25,1). Teniendo en cuenta que se han comercializado1 un total de 827 formatos de medicamentos en 2020, se obtiene un promedio de 75,2 presentaciones nuevas de medicamentos por cada nuevo principio activo comercializado1, ampliamente superior a la mediana de la década (30,6). En este sentido, cabe destacar que el 98% de los nuevos medicamentos comercializados durante 2020 incluyeron principios activos autorizados y comercializados en años previos; de ellos, un 2% son presentaciones novedosas de medicamentos ya comercializados previamente, entendiendo como tal aquellas que suponen una innovación en forma farmacéutica y/o vía de administración.
A grandes rasgos, las tendencias de la incorporación de nuevos principios activos en los últimos 42 años (1978-2020) (Figura 3a) y de la relación nuevas presentaciones/nuevos principios activos (Figura 3b) son moderadamente fluctuantes2. Si bien en los últimos 10 años se aprecia una ligera estabilización en la incorporación de nuevos principios activos en el mercado español (Figura 3c), ésta ha sido interrumpida por el citado año: 2020 ha sido el año en el que se ha producido la menor comercialización de principios activos nuevos desde que se tienen registros en PAM (y, por ello mismo, el año en que la relación global nuevas presentaciones/nuevos principios activos ha adquirido el valor más alto de la serie histórica).
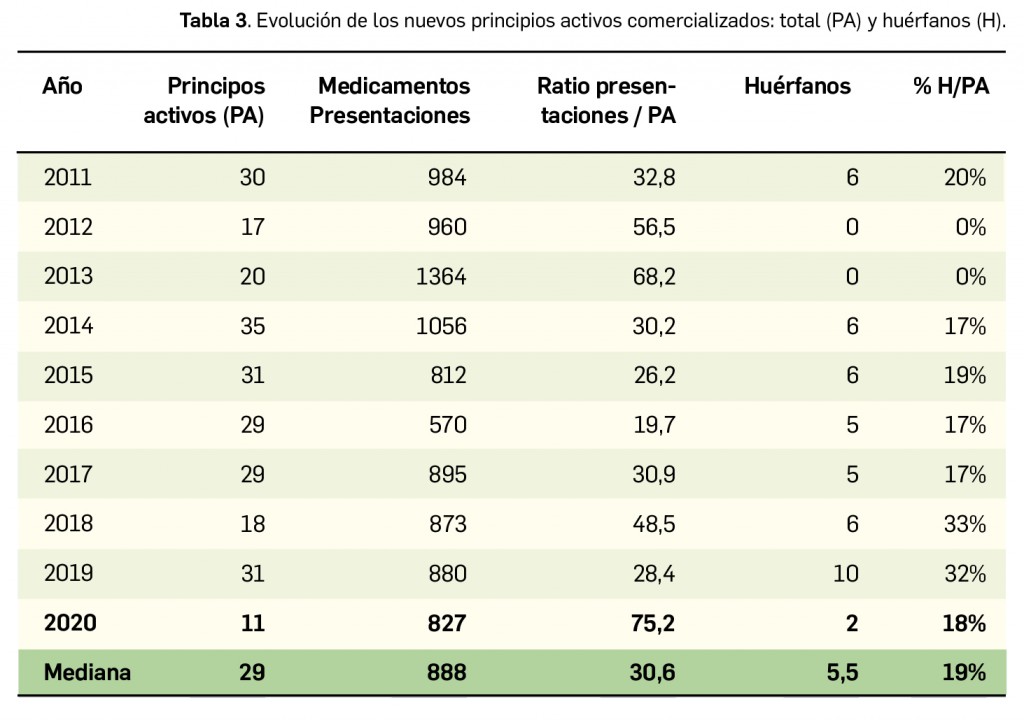
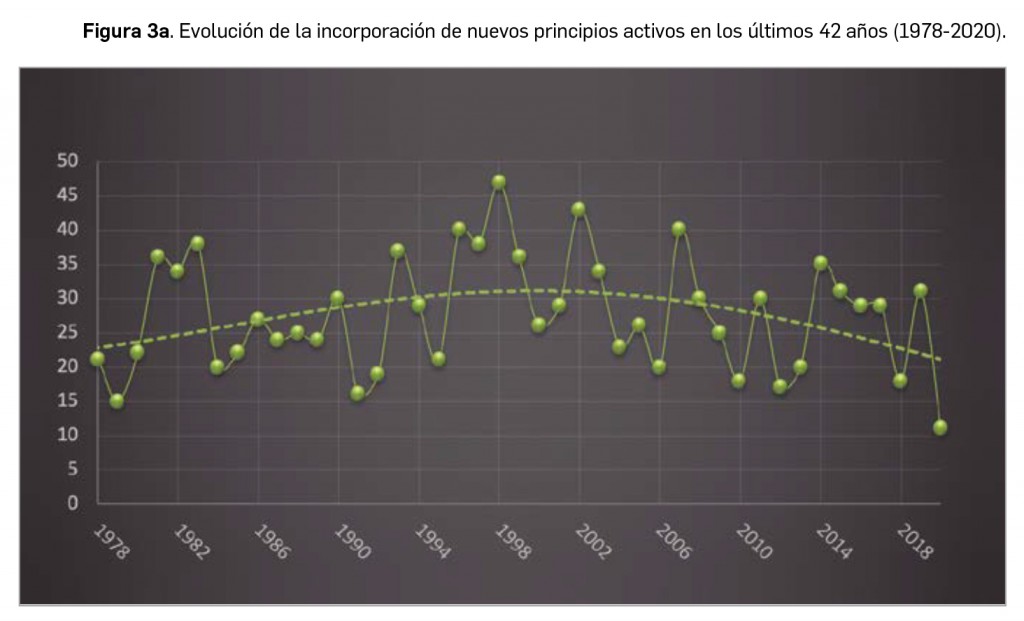
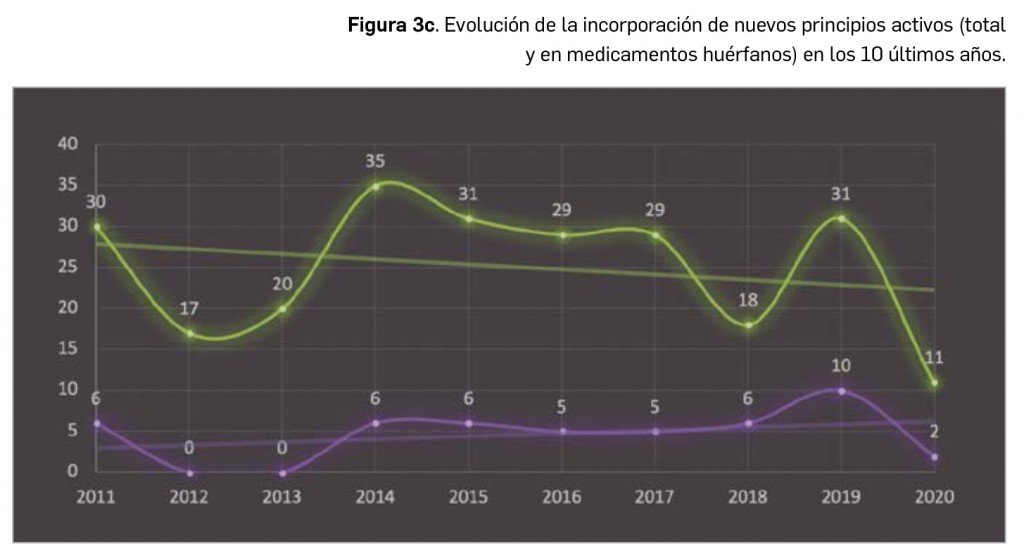
Desde el año 2002, se han comercializado en España 93 nuevos principios activos como medicamentos huérfanos3, lo que supone un 19% de los nuevos principios activos incorporados en ese periodo (un total de 510). La tendencia ha sido ligeramente ascendente hasta 2019 gracias a los 32 nuevos medicamentos huérfanos incorporados en los cinco años previos (2015-2019), pero también interrumpida en 2020 por los solo 2 nuevos principios activos incluidos en medicamentos que han sido designados como huérfanos. En cualquier caso, parece mantenerse cierta proporcionalidad entre el número total de nuevos principios activos y el de los incluidos específicamente en medicamentos huérfanos en cada año, lo que queda reflejado en el paralelismo entre ambas líneas de tendencia (Figura 3c).
Por otro lado, resulta reseñable que en el año 2020 se han comercializado en España 7 nuevos medicamentos biosimilares: tres del principio activo bevacizumab, dos de la teriparatida, uno de rituximab y un biosimilar de la insulina glargina.
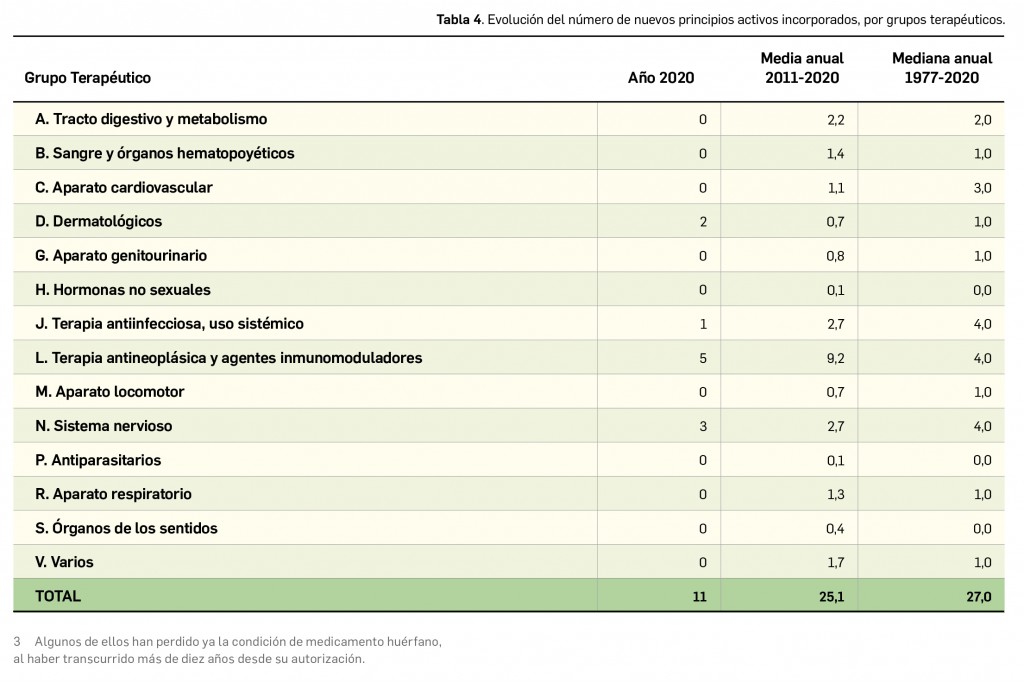
Si se considera la clasificación terapéutica ATC de los nuevos principios activos comercializados en 2020, se han incorporado principios activos a 4 de los 14 grupos terapéuticos existentes. El grupo con mayor número de nuevos principios activos durante el año ha sido, como ya venía ocurriendo en los últimos años, el grupo L (Terapia antineoplásica y agentes inmunomoduladores), con un total de 5.
A modo de resumen, desde el año 1977, en que apareció por vez primera Panorama Actual del Medicamento, se han incorporado un total de 1.207 nuevos principios activos al mercado farmacéutico español, con independencia de su clasificación terapéutica ATC.
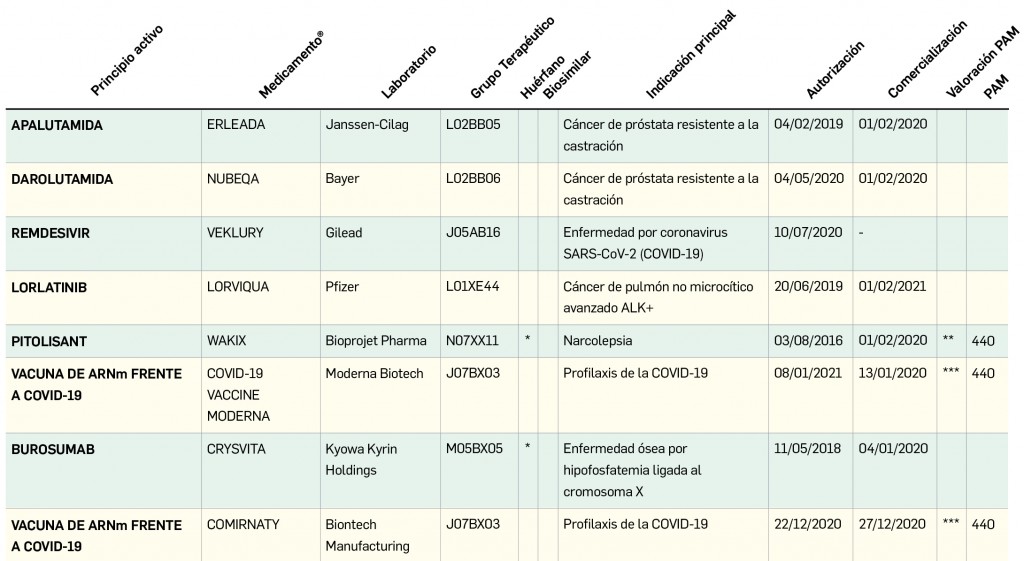
En la siguiente tabla (Tabla 5) se muestran los nombres de los nuevos principios activos comercializados durante el año 2020, junto a los nombres de los medicamentos en que se incluyen, su grupo terapéutico ATC e indicación principal. Seguidamente, se resumen las principales características farmacoclínicas de cada uno de ellos, clasificados por grupos terapéuticos, en base a la información disponible en el momento de su primera comercialización en España.
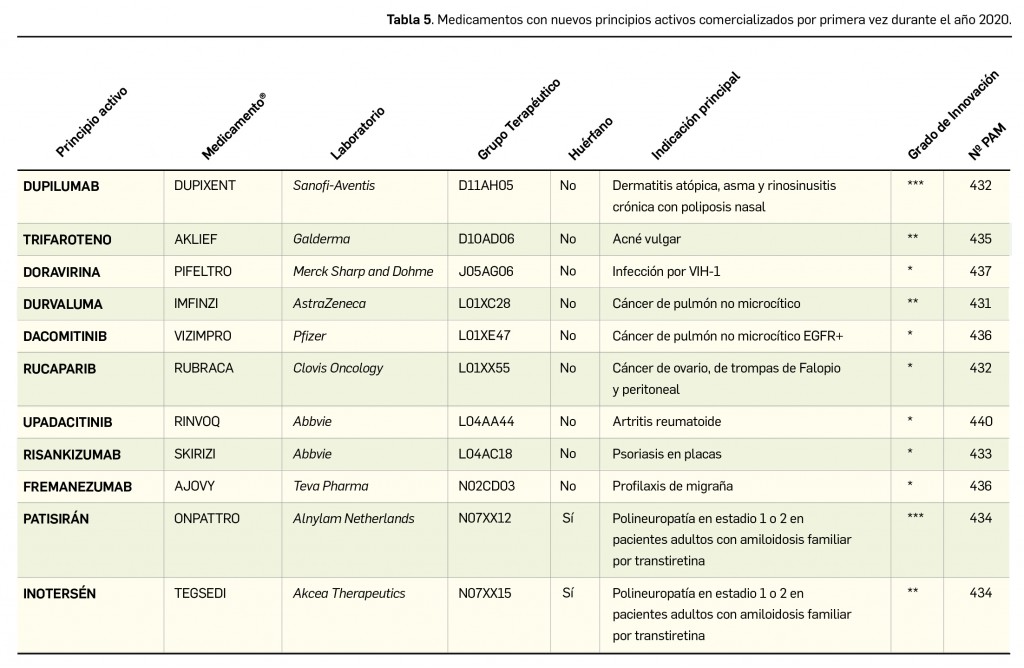
D. DERMATOLÓGICOS
DERMATITIS ATÓPICA
Dupilumab
▼DUPIXENT® (Sanofi-Aventis) PAM 432
Dupilumab es un nuevo anticuerpo monoclonal de administración subcutánea dirigido frente a la subunidad α del receptor de la IL-4 (IL-4Rα), que impide la señalización mediada por la unión de esa citocina a su receptor tipo I (IL-4Rα/γc) y también por IL-4 e IL-13 a través del receptor tipo II (IL4Rα/IL-13Rα). Puesto que IL-4 e IL-13 son los principales mediadores de la inflamación tipo 2, dupilumab ejerce interesantes efectos antiinflamatorios. En base a ello, el medicamento ha sido autorizado para el tratamiento de la dermatitis atópica (DA) moderada-grave en pacientes adultos y adolescentes de ≥12 años candidatos a tratamiento sistémico, y también como tratamiento de mantenimiento adicional para el asma grave con inflamación de tipo 2 (caracterizada por eosinófilos elevados en sangre y/o FeNO elevado), en adultos y adolescentes de ≥12 años que no están adecuadamente controlados con corticosteroides inhalados en dosis altas en combinación con otro medicamento.
El fármaco ha demostrado superioridad frente a placebo en el tratamiento de la DA tanto en pacientes que inician tratamiento sistémico tras fracaso al tratamiento tópico como en pacientes pre-tratados con ciclosporina: en monoterapia, el fármaco induce aumentos significativos en la proporción de pacientes respondedores con aclaramiento de la piel (+27-28% según escala IGA y +37-44% que alcanzan EASI-50), que también se verifican en combinación con un corticoide o inhibidor de calcineurina tópicos (+24-28% en IGA y +40-48% con EASI-50); un estudio en pacientes adolescentes confirmó resultados similares. En el tratamiento adicional del asma no controlada, evidenció una eficacia notable en pacientes con biomarcadores inflamatorios de tipo 2 elevados. Así, en presencia de altos niveles de eosinófilos en sangre y de FeNO en aire exhalado, reduce el riesgo de exacerbaciones en un 66-67% y mejora la funcionalidad pulmonar (duplicando el valor de VEF1 pre-broncodilatador) en comparación con placebo. Además, los resultados de un estudio específico demuestran que dupilumab permite reducir/suprimir la dosis de corticoides orales –y, con ello, su toxicidad– en un 70% de pacientes (vs. 42% con placebo). En ambas patologías, el efecto es de inicio rápido (2 semanas) y duradero en periodos de hasta 1 año, aportando beneficio en síntomas y calidad de vida reportados por los pacientes.
A grandes rasgos, dupilumab muestra una buena tolerabilidad a corto-medio plazo, con un perfil toxicológico aceptable y manejable. En línea con la seguridad de otras proteínas terapéuticas, entre las reacciones adversas más frecuentes destacan reacciones en el lugar de la inyección (eritema, edema y prurito) e infecciones (conjuntivitis, blefaritis, nasofaringitis, infecciones respiratorias del tracto superior, sinusitis y herpes oral), en su mayoría leves-moderadas. El riesgo de desarrollo de reacciones alérgicas e inmunogenicidad parece bajo, si bien se requieren aún datos de seguridad a largo plazo que permitan esclarecer los potenciales riesgos de desarrollo de neoplasias malignas.
Dupilumab inaugura una nueva vía farmacológica en sus dos indicaciones, patologías de curso crónico-recurrente que afectan a población pediátrica y adulta y que en ciertos casos pueden resultar incapacitantes y tener un elevado impacto socio-laboral. Aporta, además, un beneficio clínico superior a placebo en casos graves-moderados, siendo quizás más relevante su indicación en dermatitis atópica, pues supone el primer avance terapéutico desde hace mucho tiempo (primer tratamiento biológico autorizado) y será una alternativa adecuada en pacientes sin respuesta o no candidatos a tratamiento con ciclosporina.
ACNÉ
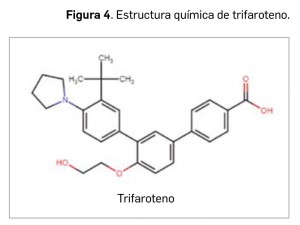
Trifaroteno
▼AKLIEF® (Galderma) PAM 435
El trifaroteno es un nuevo derivado de retinoide de cuarta generación (Figura 4) que actúa como un potente agonista del receptor γ del ácido retinoico (RARγ), con una especificidad muy elevada y una afinidad 50 veces mayor que sobre RARα y 8 veces mayor que sobre RARβ; no presenta, en cambio, actividad sobre receptores X de retinoides (RXR). Así, puesto que los RARγ son los receptores de retinoides más abundantes en la piel, trifaroteno será capaz de modular la expresión de genes diana implicados en el crecimiento y la diferenciación celular, la respuesta al estrés, cascadas antiinflamatorias y la apoptosis, entre otros procesos. Si bien el mecanismo exacto por el cual mejora el acné no se conoce completamente, el medicamento ha sido autorizado para el tratamiento (por vía tópica en forma de crema) de acné vulgar en la cara, en el pecho y/o en la espalda en pacientes mayores de 12 años, en presencia de numerosos comedones, pápulas y pústulas.
 Su eficacia y seguridad clínica han sido bien definidas en 2 ensayos pivotales de fase 3 de igual diseño, doble ciegos, de grupos paralelos y controlados por vehículo, que aleatorizaron un total de 2.420 pacientes de > 9 años de edad con acné facial y troncal moderado (grado 3). Con una aplicación tópica diaria durante 12 semanas, el fármaco demostró una superioridad significativa frente a la crema vehículo en pacientes de ≥ 12 años, evidenciada por tasas de éxito terapéutico del 29-42% en acné facial (vs. 19-26% con vehículo) y del 36-43% en acné troncal (vs. 25-30%). Además, la reducción de las lesiones inflamatorias (en un 54-66%) y no inflamatorias (en un 49-58%) en ambas localizaciones también fue significativamente superior al vehículo; el beneficio clínico era evidente incluso desde la primera-segunda semana de tratamiento. Un estudio adicional de un único brazo con datos de tratamiento durante 1 año en 342 pacientes confirmó los resultados de los estudios pivotales y el beneficio continuo del tratamiento con trifaroteno: las tasas de éxito aumentaban hasta el 65% para el acné en cara (desde el 27% a la semana 12) y hasta el 67% para el acné en tronco (desde el 39%), con una amplia proporción de pacientes (54%) que reportaban que el acné no afectaba a su calidad de vida.
Su eficacia y seguridad clínica han sido bien definidas en 2 ensayos pivotales de fase 3 de igual diseño, doble ciegos, de grupos paralelos y controlados por vehículo, que aleatorizaron un total de 2.420 pacientes de > 9 años de edad con acné facial y troncal moderado (grado 3). Con una aplicación tópica diaria durante 12 semanas, el fármaco demostró una superioridad significativa frente a la crema vehículo en pacientes de ≥ 12 años, evidenciada por tasas de éxito terapéutico del 29-42% en acné facial (vs. 19-26% con vehículo) y del 36-43% en acné troncal (vs. 25-30%). Además, la reducción de las lesiones inflamatorias (en un 54-66%) y no inflamatorias (en un 49-58%) en ambas localizaciones también fue significativamente superior al vehículo; el beneficio clínico era evidente incluso desde la primera-segunda semana de tratamiento. Un estudio adicional de un único brazo con datos de tratamiento durante 1 año en 342 pacientes confirmó los resultados de los estudios pivotales y el beneficio continuo del tratamiento con trifaroteno: las tasas de éxito aumentaban hasta el 65% para el acné en cara (desde el 27% a la semana 12) y hasta el 67% para el acné en tronco (desde el 39%), con una amplia proporción de pacientes (54%) que reportaban que el acné no afectaba a su calidad de vida.
Con respecto a la seguridad, su perfil toxicológico está en línea con el ya conocido para otros retinoides tópicos. Ha sido bien caracterizado y parece aceptable (tasas de discontinuación bajas, de < 2% a las 12 semanas) y clínicamente manejable con ajustes posológicos o el uso de hidratantes/limpiadores. El tratamiento con la crema de trifaroteno se asocia con la aparición signos y síntomas locales leves-moderados (eritema, descamación, sequedad y ardor/quemazón), de incidencia variable (10-30%) y transitorios; la tolerabilidad fue mejor en tronco que en cara. Las reacciones adversas más frecuentes son: irritación (7,5% vs. 0,3% con vehículo) y prurito en el lugar de aplicación (2,4% vs. 0,8%) y quemaduras solares (2,6% vs. 0,5%). Su escasa absorción sistémica y rápido metabolismo hepático hacen que carezca de eventos adversos sistémicos, permitiendo su uso en amplias áreas cutáneas del tronco. Pero se mantiene la contraindicación de su uso en embarazo por el riesgo de teratogenicidad.
En ausencia de comparaciones directas con otras alternativas de tratamiento tópico del acné en adolescentes y adultos y, en particular, frente a los otros retinoides disponibles desde hace décadas (tretinoína y adapaleno), resulta difícil su posicionamiento. Parece que trifaroteno podría situarse como una alternativa más en primera línea del tratamiento tópico del acné moderado, aportando un beneficio clínico similar o ligeramente superior a otros fármacos disponibles. Aunque no representa una innovación importante en términos mecanísticos, aporta como novedad respecto al resto de retinoides su selectividad por el receptor RARγ cuya significación clínica no está del todo clara; su reducida afinidad sobre los RARβ dérmicos podría relacionarse con una menor incidencia de irritación cutánea, lo cual aún debe ser comprobado. Conviene subrayar que es la primera incorporación en las últimas dos décadas al grupo bien conocido de los retinoides y el primer fármaco que ha demostrado eficacia significativamente superior a placebo –en estudios bien diseñados– en acné del tronco, que hasta ahora representaba una necesidad médica no cubierta. No representa una cura del acné pero permite un buen control de los síntomas con una tolerabilidad aceptable.
J. ANTIINFECCIOSOS PARA USO SISTÉMICO
INFECCIÓN POR VIH
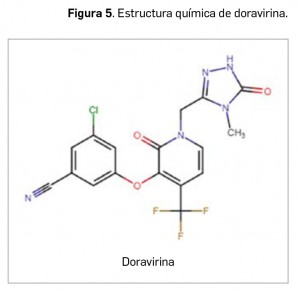
Doravirina
▼PIFELTRO® (MSD) PAM 437
Doravirina es un nuevo inhibidor de la enzima transcriptasa inversa del VIH-1 de tipo no nucleosídico (ITINN) (Figura 5) que inhibe de forma potente la replicación del virus en la célula hospedadora. Se une específicamente a la enzima viral de forma no competitiva (en una zona cercana pero diferente del centro catalítico de la enzima, usado por los ITIAN) y sin incorporarse a la cadena de ADN viral en formación, pero no inhibe las ADN polimerasas celulares α, ß ni la ADN polimerasa γ mitocondrial del ser humano. En base a ello, el medicamento ha sido autorizado, en combinación con otros medicamentos antirretrovirales, para el tratamiento por vía oral de adultos infectados por el VIH-1 sin evidencia pasada o presente de resistencia a otros ITINN.
 Su eficacia ha sido adecuadamente contrastada en pacientes sin tratamiento antirretroviral previo (naïve) mediante dos amplios estudios pivotales de fase 3, controlados y doblemente ciegos. En el primero de ellos, demostró una no inferioridad frente al IP darunavir (potenciado con ritonavir), ambos en combinación con 2 ITIAN, con tasas de pacientes respondedores (< 40 copias de ARN viral/ml) similares a la semana 48 (83% vs. 79%). El segundo estudio reveló que el tratamiento con doravirina/lamivudina/tenofovir ejerce una eficacia pareja a la de otro régimen a base de ITINN (efavirenz/emtricitabina/tenofovir): las tasas de respuesta a la semana 48 fueron, respectivamente, del 84% y 80%. En ambos estudios, el patrón de eficacia se mantuvo constante hasta la semana 96, y se mostró independiente de los parámetros basales típicamente considerados (viremia, recuento de células CD4+ o subtipo viral). Adicionalmente, un tercer ensayo pivotal demostró que la combinación a dosis fija de doravirina, lamivudina y tenofovir es también significativamente eficaz en pacientes virológicamente suprimidos y sin antecedentes de fracaso virológico ni de mutaciones de resistencia a dichos fármacos, con independencia del TAR previo; de manera interesante, ningún paciente desarrolló resistencias al régimen con doravirina.
Su eficacia ha sido adecuadamente contrastada en pacientes sin tratamiento antirretroviral previo (naïve) mediante dos amplios estudios pivotales de fase 3, controlados y doblemente ciegos. En el primero de ellos, demostró una no inferioridad frente al IP darunavir (potenciado con ritonavir), ambos en combinación con 2 ITIAN, con tasas de pacientes respondedores (< 40 copias de ARN viral/ml) similares a la semana 48 (83% vs. 79%). El segundo estudio reveló que el tratamiento con doravirina/lamivudina/tenofovir ejerce una eficacia pareja a la de otro régimen a base de ITINN (efavirenz/emtricitabina/tenofovir): las tasas de respuesta a la semana 48 fueron, respectivamente, del 84% y 80%. En ambos estudios, el patrón de eficacia se mantuvo constante hasta la semana 96, y se mostró independiente de los parámetros basales típicamente considerados (viremia, recuento de células CD4+ o subtipo viral). Adicionalmente, un tercer ensayo pivotal demostró que la combinación a dosis fija de doravirina, lamivudina y tenofovir es también significativamente eficaz en pacientes virológicamente suprimidos y sin antecedentes de fracaso virológico ni de mutaciones de resistencia a dichos fármacos, con independencia del TAR previo; de manera interesante, ningún paciente desarrolló resistencias al régimen con doravirina.
Por otro lado, el perfil toxicológico de doravirina es aceptable, con buena tolerabilidad, y similar o mejor que el de darunavir potenciado y el de efavirenz, con una baja tasa de interrupción del tratamiento por eventos adversos (~3%, vs. 6% con regímenes con efavirenz). Entre las reacciones adversas relacionadas con doravirina, por lo general escasas y leves, sobresalen las náuseas (4%) y la cefalea (3%). Tiene un perfil lipídico neutro e insignificante efecto sobre los niveles de bilirrubina, asociándose con un menor riesgo de eventos neuropsiquiátricos y rash cutáneo que efavirenz, y con una menor tasa de eventos gastrointestinales que darunavir.
En definitiva, doravirina ha demostrado que, en combinación con 2 ITIAN, aporta un beneficio clínico relevante y similar al del IP darunavir y al del ITINN efavirenz en pacientes naïve sin antecedentes de resistencia viral a fármacos de su clase, presentando, además, una buena tolerabilidad que mejora el perfil de los antirretrovirales usados como comparadores activos. Por superar algunas limitaciones de los ITINN hasta ahora disponibles, puede ser el fármaco de elección dentro de su grupo, en caso de que se decida un TAR de inicio a base de 2 ITIANN más 1 ITINN. Sin embargo, parece que el beneficio clínico que pueda aportar la doravirina no supera, con la evidencia hasta ahora disponible (y a pesar de que no se han realizado comparaciones directas), al de los antirretrovirales preferidos en el TAR de inicio (los INI), ni supone ninguna innovación a nivel mecanístico. A expensas de las consideraciones del IPT de la AEMPS, se puede concluir que doravirina se posicionará en regímenes de TAR alternativos a los de primera línea, sin implicar una innovación sustancial frente a las opciones ya disponibles para pacientes con infección por VIH-1 sin tratamiento previo. No se empleará, por ahora, en regímenes de cambio de TAR tras un fracaso virológico ni en pacientes con virus resistentes a ITINN.
L. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
CÁNCER DE PULMÓN NO MICROCÍTICO
Durvalumab
▼INFINZI® (AstraZeneca) PAM 431
El durvalumab es un anticuerpo monoclonal humanizado que se une selectivamente a la proteína PD-L1 (expresada en células tumorales e inflamatorias) y bloquea la interacción con sus receptores de membrana PD1 (en la superficie de linfocitos T efectores) y CD80 (en diversos tipos de células inmunitarias), potenciando las respuestas inmunitarias antitumorales, fundamentalmente la citotoxicidad mediada por células T activadas. Se diferencia de otros anticuerpos monoclonales que actúan sobre la vía PD-1/PD-L1, como nivolumab y pembrolizumab (anti-PD-1), en que al bloquear solo PD-L1 se conserva la interacción de PD-L2 con PD-1, lo cual es una ventaja potencial que puede minimizar la autoinmunidad. Durvalumab continúa la vía mecanística inaugurada por atezolizumab (otro anti-PD-L1 previamente comercializado) en el tratamiento de CPNM. El medicamento ha sido autorizado para el tratamiento en monoterapia de pacientes adultos con cáncer de pulmón no microcítico (CPNM) localmente avanzado, no resecable, cuyos tumores expresan PD-L1≥ 1% en las células tumorales y cuya enfermedad no haya presentado progresión después de quimiorradioterapia basada en platino. A diferencia con atezolizumab y nivolumab, no ha recibido aprobación en CPNM metastásico.
En el ensayo pivotal PACIFIC, durvalumab ha demostrado prolongar la supervivencia libre de progresión tumoral en 11,6 meses frente a placebo (mediana de 17,2 vs. 5,6 meses), reduciendo el riesgo de progresión en un 49%. Tras más de 2 años de seguimiento, los resultados de supervivencia global (mediana no alcanzada vs. 28,7 meses con placebo) también alcanzaron significación estadística, con una reducción de riesgo de muerte del 32%; la tasa de SG a los 24 meses fue del 66% en el grupo de durvalumab en comparación con el 56% en el grupo de placebo. La eficacia del nuevo fármaco se confirmó con independencia de factores como raza, edad, género, historia de tabaquismo, estado mutacional del EGFR e histología del tumor, pero parece que es considerablemente menor en pacientes con niveles de PD-L1< 1% que en aquellos con PD-L1≥ 1% (mediana de SLP de 10,7 y de 17,8 meses, respectivamente). En cuanto a la seguridad clínica, el perfil toxicológico de durvalumab es importante (toxicidad superior a placebo) pero clínicamente manejable y está en línea con lo esperado a la vista de la seguridad clínica mostrada por otros inhibidores de PD-L1. Entre las reacciones adversas más frecuentes destaca la tos (35%), neumonitis o neumonitis por radiación (34%), fatiga (24%) y disnea (22%); también se describió alta incidencia de pirexia, neumonía, prurito y alteraciones tiroideas. La tasa de eventos de grado 3-4 relacionados con el fármaco fue del 8%, destacando por su gravedad los casos de neumonía. Las muertes relacionadas con el tratamiento fueron raras.
En resumen, los resultados de supervivencia sin progresión permiten considerar a durvalumab como una opción de tratamiento preferente –durante 12 meses o hasta progresión o toxicidad inaceptable– frente al seguimiento clínico activo (sin farmacoterapia) como terapia de consolidación en pacientes que no progresan y no muestran toxicidades acumulativas tras quimiorradiación con intención curativa. No se dispone de evidencia de beneficio en el mantenimiento del tratamiento más allá de los 12 meses. Por tratarse de una necesidad médica no cubierta (la mayoría de pacientes con CPNM no operable recaen relativamente rápido y tienen mal pronóstico, con tasas de supervivencia a los 5 años del ~15%), los resultados aquí comentados se consideran clínicamente relevantes.
Dacomitinib
▼VIZIMPRO® (Pfizer) PAM 436
Dacomitinib es un nuevo antineoplásico (Figura 6) que actúa como inhibidor universal, selectivo, irreversible y competitivo con el ATP, del dominio tirosina cinasa del receptor del factor de crecimiento epidérmico humano (EGFR/HER1, pero también de HER2 y HER4), con actividad contra el EGFR mutado con deleciones en el exón 19 o la sustitución L858R en el exón 21. En base a ello, atenúa los procesos de crecimiento celular neoplásico (incluyendo, entre otros, la proliferación celular incontrolada, la migración celular, la invasión del estroma, la angiogénesis y la resistencia a la apoptosis), por lo que el medicamento ha sido autorizado, en monoterapia, para el tratamiento por vía oral de primera línea de pacientes adultos con cáncer de pulmón no microcítico (CPNM) localmente avanzado o metastásico con mutaciones activadoras del EGFR.
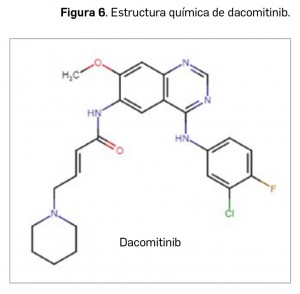 En el ensayo pivotal de fase 3 y multicéntrico (ARCHER 1050; N= 452), el tratamiento con dacomitinib (45 mg/día) ha demostrado su capacidad de prolongar en 5,5 meses la mediana de la supervivencia libre de progresión frente al comparador activo gefitinib (14,7 vs. 9,2 meses; p < 0,0001). Sin diferencias destacables en la tasa de respuesta, el nuevo fármaco también aporta mejoras en la mediana de duración de la respuesta (14,8 vs. 8,3 meses) y en el tiempo hasta el fallo del tratamiento (11,1 vs. 9,2 meses), lo cual se traduce en un aumento de la supervivencia global con una prolongación de la mediana de 7,3 meses (34,1 vs. 26,8 meses con gefditinib; p < 0,05), si bien estos resultados se consideran exploratorios. Su eficacia se mostró consistente en todos los subgrupos de pacientes con independencia de factores como la edad (salvo > 75 años), el género, el hábito tabáquico, el tipo de mutación de EGFR o el estado funcional; la excepción de los pacientes de raza no asiática, en que no se verificó mejora de SLP, es considerado por la EMA como un hallazgo debido al azar. Además, un estudio de soporte de un solo brazo también apunta a interesantes resultados de eficacia con dacomitinib (mediana de SLP de 18,2 meses y de SG de 42,3 meses) en pacientes con mutación activadora de EGFR.
En el ensayo pivotal de fase 3 y multicéntrico (ARCHER 1050; N= 452), el tratamiento con dacomitinib (45 mg/día) ha demostrado su capacidad de prolongar en 5,5 meses la mediana de la supervivencia libre de progresión frente al comparador activo gefitinib (14,7 vs. 9,2 meses; p < 0,0001). Sin diferencias destacables en la tasa de respuesta, el nuevo fármaco también aporta mejoras en la mediana de duración de la respuesta (14,8 vs. 8,3 meses) y en el tiempo hasta el fallo del tratamiento (11,1 vs. 9,2 meses), lo cual se traduce en un aumento de la supervivencia global con una prolongación de la mediana de 7,3 meses (34,1 vs. 26,8 meses con gefditinib; p < 0,05), si bien estos resultados se consideran exploratorios. Su eficacia se mostró consistente en todos los subgrupos de pacientes con independencia de factores como la edad (salvo > 75 años), el género, el hábito tabáquico, el tipo de mutación de EGFR o el estado funcional; la excepción de los pacientes de raza no asiática, en que no se verificó mejora de SLP, es considerado por la EMA como un hallazgo debido al azar. Además, un estudio de soporte de un solo brazo también apunta a interesantes resultados de eficacia con dacomitinib (mediana de SLP de 18,2 meses y de SG de 42,3 meses) en pacientes con mutación activadora de EGFR.
Por otra parte, el perfil toxicológico de dacomitinib es importante y desfavorable en comparación con gefitinib (aumento de la diarrea y de toxicidades cutáneas), pero clínicamente manejable y en línea con lo esperado por los resultados de seguridad de afatinib, el otro EGFR-TKI de 2ª generación disponible. Casi todos los pacientes reportan algún evento adverso durante el tratamiento (de grado 3-4 en más de la mitad), destacando por su frecuencia las reacciones adversas del tracto gastrointestinal (diarrea y estomatitis), de la piel (dermatitis, sequedad, rash o prurito), de las uñas (paroniquia) y una reducción del apetito. La tasa de eventos adversos graves relacionados con dacomitinib alcanzó el 7% (sobre todo, diarrea, enfermedad pulmonar intersticial, dermatitis cutánea y reducción del apetito), lo cual se asocia con una tasa de discontinuación permanente por toxicidad del fármaco a tener en cuenta (9,7%); también notable es la proporción de pacientes que requiere reducciones de dosis (52%).
En resumen, el retraso en la progresión del tumor que aporta dacomitinib se considera clínicamente relevante, especialmente por demostrarlo frente a gefitinib, un fármaco comúnmente empleado en la práctica clínica en primera línea de CPNM avanzado o metastásico con mutaciones de EGFR. A pesar de un peor perfil de seguridad, éste se considera manejable, y los aspectos de eficacia pueden convertirlo en una alternativa interesante para el tratamiento de pacientes no pre-tratados. Sin embargo, no implica novedad mecanística alguna ni es una opción curativa, y los más favorables resultados clínicos divulgados para otros inhibidores de tirosina cinasa de EGFR, especialmente osimertinib, sugieren que dacomitinib no sería la opción preferente en la mayoría de pacientes, por lo que no parece aportar una innovación clínica sustancial.
CÁNCER DE OVARIO
Rucaparib
▼RUBRACA® (Clovis Oncology) PAM 432
El rucaparib (Figura 7) es un nuevo inhibidor selectivo de poli-ADP-ribosa polimerasas (PARP-1, -2 y -3), enzimas implicadas en los mecanismos de reparación del ADN, tanto en células normales como tumorales: mediante su inhibición, la citotoxicidad de rucaparib –que ha demostrado ser independiente de la presencia de mutaciones en los genes BRCA– se debe a un aumento de la formación de complejos PARP-ADN, conducente a un daño del ADN y a la activación de la apoptosis y la muerte celular. Por ello, el medicamento ha sido autorizado en el tratamiento de mantenimiento, en monoterapia, de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, en recidiva, sensible al platino, que responde completa o parcialmente a la quimioterapia con platino. También está indicado en monoterapia para el tratamiento de pacientes adultas con cáncer de ovario epitelial, de trompa de Falopio o peritoneal primario, de alto grado, con mutación BRCA (germinal y/o somática), sensible al platino, en recaída o progresión, que hayan sido tratadas con dos o más líneas previas de quimioterapia con platino y que no son capaces de tolerar más quimioterapia a base de platino.
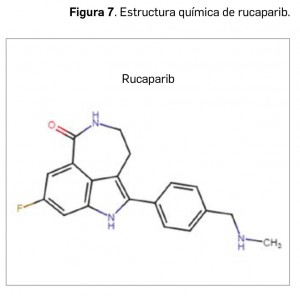 En un amplio ensayo pivotal de fase 3, administrado como monoterapia de mantenimiento, rucaparib ha conseguido prolongar la supervivencia libre de progresión en 5,4 meses frente a placebo (hasta más de 8 meses en revisión por comité independiente), reduciendo en un 64% el riesgo de progresión de la patología; ese beneficio, que parece inferior en pacientes sin mutación BRCA, aumentaba hasta los 11,2 meses en la cohorte de pacientes con BRCA mutado. No obstante, la ausencia de datos maduros impide confirmar un beneficio en supervivencia global con el fármaco. Por otra parte, los datos clínicos relativos a su indicación en tratamiento de pacientes pre-tratadas en recaída derivan de 2 ensayos abiertos no controlados de fase 2 y no son confirmatorios. El análisis conjunto de los datos reveló una tasa de respuesta objetiva con el tratamiento de rucaparib del 54,7% (del 44% en revisión por comité independiente), la mayoría (46%) respuestas parciales, y la mediana de la duración de la respuesta fue de 9,7 meses; la eficacia fue mayor en el subgrupo de pacientes sensibles a platino (TRO de 64,6%), el único incluido en la indicación autorizada.
En un amplio ensayo pivotal de fase 3, administrado como monoterapia de mantenimiento, rucaparib ha conseguido prolongar la supervivencia libre de progresión en 5,4 meses frente a placebo (hasta más de 8 meses en revisión por comité independiente), reduciendo en un 64% el riesgo de progresión de la patología; ese beneficio, que parece inferior en pacientes sin mutación BRCA, aumentaba hasta los 11,2 meses en la cohorte de pacientes con BRCA mutado. No obstante, la ausencia de datos maduros impide confirmar un beneficio en supervivencia global con el fármaco. Por otra parte, los datos clínicos relativos a su indicación en tratamiento de pacientes pre-tratadas en recaída derivan de 2 ensayos abiertos no controlados de fase 2 y no son confirmatorios. El análisis conjunto de los datos reveló una tasa de respuesta objetiva con el tratamiento de rucaparib del 54,7% (del 44% en revisión por comité independiente), la mayoría (46%) respuestas parciales, y la mediana de la duración de la respuesta fue de 9,7 meses; la eficacia fue mayor en el subgrupo de pacientes sensibles a platino (TRO de 64,6%), el único incluido en la indicación autorizada.
A grandes rasgos, rucaparib muestra una tolerabilidad similar a los otros dos inhibidores de PARP comercializados (olaparib y niraparib), con un perfil toxicológico importante pero clínicamente manejable. La mayoría de reacciones adversas son leves-moderadas y destacan, por su frecuencia, los eventos adversos de naturaleza gastrointestinal y hematológica (náuseas, vómitos, anemia, neutropenia); entre los eventos graves, sobresale la anemia, la neutropenia febril y la fatiga. Con los tres fármacos se han descrito casos de síndrome mielodisplásico y/o leucemia mieloide aguda, y se hace necesario considerar este riesgo y conocer, en futuros estudios, más datos sobre los posibles efectos a medio y largo plazo del tratamiento con rucaparib.
En resumen, rucaparib ha demostrado un beneficio relevante frente a placebo en el mantenimiento pos-quimioterapia y parece que viene a representar una alternativa terapéutica más a olaparib y niraparib entre dos intervalos de quimioterapia en pacientes con buen estado funcional (permitirá que las pacientes se beneficien en mayor medida de un nuevo régimen posterior basado en platino); los datos clínicos disponibles no avalan el uso secuencial de los inhibidores de PARP ni permiten establecer entre ellos una superioridad en términos supervivencia (sobre todo en pacientes con BRCA no mutado) o diferencias importantes en el balance beneficio/riesgo. En tratamiento de pacientes pre-tratadas en recaída, los datos de rucaparib son limitados y requieren confirmación, pero sugieren que puede dar lugar a tasas de respuesta similares a otras alternativas de quimioterapia disponibles (doxorubicina liposomal pegilada con o sin trabectedina, o topotecán) con un mejor perfil de seguridad y de adherencia terapéutica. En definitiva, sin incorporar ninguna novedad mecanística respecto a otros inhibidores de PARP usados frente al cáncer de ovario, tampoco parece que vaya a aportar una innovación clínica notable en su tratamiento.
ARTRITIS REUMATOIDE
Upadacitinib
▼RINVOQ® (Abbvie) PAM 440
Upadacitinib es un nuevo inmunosupresor activo por vía oral que actúa mediante la inhibición selectiva y reversible de las cinasas Janus o JAK, preferentemente sobre las JAK-1 o JAK-1/3, de manera que modula la respuesta inflamatoria e inmunitaria mediada por citocinas o factores de crecimiento. Estrechamente relacionado (Figura 8) con otros dos inhibidores orales de JAK previamente comercializados en España (tofacitinib y baricitinib), en base a dicho efecto farmacológico el medicamento ha sido autorizado para el tratamiento de la artritis reumatoide (AR) activa de moderada a grave en pacientes adultos con respuesta inadecuada o intolerancia a uno o más fármacos antirreumáticos modificadores de la enfermedad (FAME), pudiendo ser empleado en monoterapia o en combinación con metotrexato.
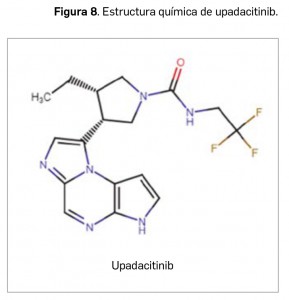 Su eficacia ha sido adecuadamente contrastada en 5 ensayos clínicos de fase 3, aleatorizados y doble-ciego, que han aleatorizado casi 4.400 pacientes, y en los que ha demostrado una superioridad estadísticamente significativa frente a placebo en pacientes con fracaso a FAME convencionales o biológicos, y también mayor que metotrexato (en pacientes naïve o con respuesta inadecuada a dicho FAME) y adalimumab (en pacientes con fracaso previo a metotrexato u otros FAME biológicos). Tanto en segunda como en tercera línea de tratamiento, y tanto en monoterapia como en combinación con FAME convencionales, la dosis de 15 mg/día por vía oral de upadacitinib permitió alcanzar tasas de remisión clínica (DAS28-PCR < 2,8 puntos) a las 12-14 semanas entre 28% y 36% (ascendiendo a 48% a los 4 meses), hasta 20 puntos porcentuales superiores a los controles (6-10% con placebo, 8-14% con metotrexato, 18% con adalimumab). La respuesta clínica fue consistente en términos de baja actividad de la enfermedad (DAS28-PCR < 3,2 puntos), alcanzada por una proporción de pacientes significativamente mayor con udapatinib (43-53% vs. 14-17% con placebo, 19-28% con metotrexato y 29% con adalimumab). Esa eficacia, de inicio rápido y verificada en todos los subgrupos de pacientes evaluados, se completó con los resultados positivos en otras variables como la progresión radiológica de la AR, la función física de los pacientes o la calidad de vida, que mejoraron significativamente frente a placebo y metotrexato. Las fases de extensión de los estudios, ahora en marcha, permitirán confirmar o descartar un beneficio clínico a largo plazo.
Su eficacia ha sido adecuadamente contrastada en 5 ensayos clínicos de fase 3, aleatorizados y doble-ciego, que han aleatorizado casi 4.400 pacientes, y en los que ha demostrado una superioridad estadísticamente significativa frente a placebo en pacientes con fracaso a FAME convencionales o biológicos, y también mayor que metotrexato (en pacientes naïve o con respuesta inadecuada a dicho FAME) y adalimumab (en pacientes con fracaso previo a metotrexato u otros FAME biológicos). Tanto en segunda como en tercera línea de tratamiento, y tanto en monoterapia como en combinación con FAME convencionales, la dosis de 15 mg/día por vía oral de upadacitinib permitió alcanzar tasas de remisión clínica (DAS28-PCR < 2,8 puntos) a las 12-14 semanas entre 28% y 36% (ascendiendo a 48% a los 4 meses), hasta 20 puntos porcentuales superiores a los controles (6-10% con placebo, 8-14% con metotrexato, 18% con adalimumab). La respuesta clínica fue consistente en términos de baja actividad de la enfermedad (DAS28-PCR < 3,2 puntos), alcanzada por una proporción de pacientes significativamente mayor con udapatinib (43-53% vs. 14-17% con placebo, 19-28% con metotrexato y 29% con adalimumab). Esa eficacia, de inicio rápido y verificada en todos los subgrupos de pacientes evaluados, se completó con los resultados positivos en otras variables como la progresión radiológica de la AR, la función física de los pacientes o la calidad de vida, que mejoraron significativamente frente a placebo y metotrexato. Las fases de extensión de los estudios, ahora en marcha, permitirán confirmar o descartar un beneficio clínico a largo plazo.
Por otro lado, el perfil toxicológico del fármaco, consistente en su uso en monoterapia y en combinación con FAME convencionales, es complejo, pero concuerda con los riesgos ya conocidos de los fármacos inhibidores de las JAKs. Entre los eventos adversos asociados al tratamiento destacan por su frecuencia las infecciones (más frecuentes en pacientes de > 75 años) –especialmente del tracto respiratorio superior y herpes zóster–, neutropenia, alteraciones lipídicas, hepatotoxicidad, síntomas gastrointestinales e incremento de enzimas musculares.
La tasa de mortalidad no difiere sustancialmente de la de los comparadores activos empleados, ni tampoco si se compara indirectamente con la de baricitinib o tofacitinib. Así pues, el seguimiento que requieren los pacientes tratados con upadacitinib es similar a las medidas de monitorización comunes en el abordaje de la AR.
Aun considerando que es necesario conocer su perfil de seguridad a largo plazo, que no se dispone de comparaciones directas frente a otros tratamientos de los que podría ser una alternativa (con excepción de adalimumab) y que la evidencia derivada de comparaciones indirectas es escasa y no concluyente, upadacitinib se posiciona como una alternativa válida más dentro de los FAME sintéticos dirigidos, similar a otros inhibidores de JAK autorizados (tofacitinib o baricitinib), entre los cuales todavía no se puede establecer diferencias sustanciales. Así pues, sin innovación en el plano mecanístico, los usos autorizados constituyen una 2ª o 3ª línea de tratamiento, en pacientes con respuesta inadecuada o intolerancia a FAME convencionales o a anti-TNF, y no van a suponer ninguna modificación sustancial de la terapéutica estándar de la artritis reumatoide.
PSORIASIS EN PLACAS
Risankizumab
▼SKYRIZI® (Abbvie) PAM 433
Risankizumab es un anticuerpo monoclonal que se une con alta afinidad y especificidad a la subunidad p19 de la proteína interleucina 23 (IL-23) e inhibe su interacción con su receptor específico en la superficie celular (IL-23R), bloqueando las acciones biológicas mediadas por esta citocina proinflamatoria: inhibe la diferenciación de linfocitos Th17 y la secreción de la IL-17 y otras citocinas efectoras (como IL-22), las cuales desencadenarían la respuesta inmunitaria implicada en la patogénesis de la psoriasis. Comparte, así, mecanismo con guselkumab y tildrakizumab, diferenciándose de ustekinumab por su ausencia de selectividad y de unión a la IL-12. En base a ello, el medicamento ha sido oficialmente autorizado para el tratamiento por vía subcutánea de la psoriasis en placas de moderada a grave en adultos candidatos a tratamiento sistémico.
Risankizumab ha demostrado una robusta y elevada eficacia en el tratamiento de la psoriasis en placas crónica moderada-grave en 4 amplios ensayos pivotales de fase 3 con diseño aleatorizado, doble ciego y controlados, bien por placebo o bien por comparadores activos (adalimumab y ustekinumab). En global, tras 4 meses de tratamiento, la proporción de pacientes tratados con risankizumab que lograron aclaramiento de la piel con respuesta de PASI 90 fue del 72-75%, significativamente superior (p< 0,001) a los comparadores: aumentos de 25 puntos porcentuales frente a adalimumab, de 27-33 puntos frente a ustekinumab y de 70-73 frente a placebo. Resultados comparables se obtuvieron para la proporción de pacientes con puntuación sPGA 0 o 1: 84-88% con risankizumab, 60% con adalimumab, 62-63% con ustekinumab y 5-8% con placebo. Se hallaron también mejoras notables en la calidad de vida y en las manifestaciones en áreas difíciles de tratar (uñas o cuero cabelludo). Estos beneficios se mantienen con magnitud similar al año de tratamiento, con independencia de factores como edad, sexo, gravedad basal de la psoriasis o tratamiento previos (risankizumab también mostró eficacia destacable en pacientes con respuesta inadecuada a adalimumab).
El perfil de seguridad, bien definido y relativamente benigno, es similar al de otros biológicos usados para tratar la psoriasis, con una la tasa de eventos adversos relacionados con el fármaco baja (12%), comparable al placebo y, en general, más favorable que ustekinumab (15%) y adalimumab (20%); solo un 1,6% de pacientes tratados con risankizumab abandonó el tratamiento por motivos de seguridad. Las reacciones adversas más frecuentes fueron las infecciones del tracto respiratorio superior (incluidas infecciones víricas), artralgia y cefalea, en su mayoría leves y autolimitadas; entre las graves, destacan las infecciones (sepsis, celulitis o neumonía), los eventos adversos cardiovasculares o la hepatotoxicidad. Con un bajo potencial de inmunogenicidad, se debe aún caracterizar el riesgo de desarrollo de neoplasias malignas asociado al uso a largo plazo de risankizumab.
No se dispone de comparaciones directas de risankizumab con los otros inhibidores de IL-23, y no se puede establecer su superioridad. Así, a pesar de que ha demostrado una eficacia robusta, duradera –al menos 1 año– y clínicamente relevante, con blanqueamiento notable de la piel (destacan los altos porcentajes de pacientes que alcanzan PASI 100 y sPGA 0) y mejora de otras manifestaciones psoriásicas, incluyendo la afectación de la calidad de vida, no supone ninguna innovación mecanística ni implica ventajas adicionales en términos de adherencia, por lo que se incorpora como una opción más al grupo de los anti-IL-23 (donde ya están guselkumab y tildrakizumab) y no parece que vaya a aportar ninguna mejora en el tratamiento de la psoriasis frente a las opciones ya existentes. Se posicionará, pues, como una alternativa terapéutica a otros biológicos de alta eficacia, en segunda línea de tratamiento de pacientes con psoriasis en placas moderada-grave tras respuesta inadecuada, contraindicación o intolerancia a tratamientos sistémicos convencionales, o PUVA y que sean candidatos a tratamiento sistémico.
N. SISTEMA NERVIOSO
MIGRAÑA
Fremanezumab
▼AJOVY® (Teva) PAM 436
Fremanezumab es un nuevo anticuerpo monoclonal con actividad antimigrañosa por su capacidad de inhibir las acciones del neuropéptido CGRP (péptido relacionado con el gen de la calcitonina). Comparte mecanismo de acción con el previamente comercializado galcanezumab: se une con alta especificidad y afinidad al péptido CGRP (en sus dos isoformas, α y β) y evita los efectos biológicos –fundamentalmente, vasodilatación y modulación de señales nociceptivas– mediados por su unión al receptor en áreas cerebrales relevantes en la patogénesis de la migraña, como el ganglio del trigémino. Pese a que se desconoce el mecanismo exacto por el que previene las crisis migrañosas, el medicamento ha sido oficialmente autorizado para la profilaxis de la migraña en adultos con al menos 4 días de migraña al mes.
Dicha autorización se basó en los datos derivados de dos amplios ensayos pivotales de fase 3, doblemente ciegos y controlados por placebo de 12 semanas de duración; uno de ellos en pacientes con migraña crónica (N= 1.130) y el otro en pacientes con migraña episódica (N= 875) con medias de 21 y 9 días con migraña al mes, respectivamente. En los primeros, las dos pautas evaluadas de fremanezumab –mensual y trimestral– demostraron la capacidad de reducir significativamente el número de días mensuales con cefalea al menos moderada (unos 2 días adicionales respecto a placebo), con tasas de respuesta del 50% del doble que placebo (38-41% vs. 18%). En pacientes con migraña episódica, la reducción del número de días mensuales con migraña fue de 1,5 días adicionales, siendo las tasas de respuesta también mayores que con placebo (44-48% vs. 28%). La superioridad del fármaco se verificó desde el primer mes, y se mantuvo constante durante el periodo de tratamiento y consistente en todos los subgrupos evaluados, aunque el beneficio parece limitado: es necesario tratar al menos a 4-5 pacientes con migraña crónica o 5-6 con migraña episódica para conseguir una profilaxis eficaz en uno de ellos. Un ensayo de fase 3 a largo plazo no controlado permitió confirmar que la eficacia de fremanezumab perdura al menos por periodos superiores a 1 año, con tasa de respuesta del 50% en > 60% de los pacientes.
En relación a la seguridad, fremanezumab parece un fármaco bien tolerado, con una incidencia de eventos adversos sin grandes diferencias frente a placebo y un perfil toxicológico similar al descrito para galcanezumab. A grandes rasgos, las reacciones adversas relacionadas con el tratamiento (47-51% vs. 37-42% con placebo) fueron leves-moderadas y transitorias, asociándose a bajas tasas de abandono (< 3%). Destacan por su frecuencia las reacciones en el lugar de inyección (43-45% vs. 38% con placebo) y, entre ellas, principalmente dolor (26-30% vs. 26-28%), induración (20-25% vs. 15-18%) y eritema (18-20% vs. 14-16%). La limitación de la evidencia a largo plazo plantea aún incertidumbres sobre los riesgos potenciales derivados de la inhibición crónica de la acción vasodilatadora del CRPG, en especial los efectos cardiovasculares.
No se dispone de comparaciones directas de fremanezumab frente al resto de alternativas terapéuticas, pero las comparaciones indirectas podrían sugerir una similar eficacia a la del resto de opciones tanto en migraña episódica como crónica (incluyendo erenumab y galcanezumab), y no se puede aún concluir sobre su posible superioridad. En resumen, sin aportar novedad en términos mecanísticos, fremanezumab ha demostrado una eficacia significativa y temprana frente a placebo en la disminución de la frecuencia de crisis de migraña y sobre la calidad de vida de los pacientes, tanto naïve como pre-tratados sin éxito con una opción terapéutica. Si bien la magnitud del beneficio clínico es modesta, fremanezumab se incorpora al grupo de antagonistas del CGRP como una alternativa a los ya disponibles erenumab y galcanezumab en pacientes refractarios o intolerantes a otras opciones de profilaxis antimigrañosa. Su uso en primera línea se ve aún limitado por la falta de datos de eficacia y seguridad a largo plazo.
AMILOIDOSIS FAMILIAR POR TRANSTIRETINA
Inotersén
▼TEGSEDI® (Akcea Therapeutics) PAM 434
Patisirán
▼ONPATTRO® (Alnylam Netherlands) PAM 434
Inotersén es un oligonucleótido antisentido diseñado para su unión selectiva a la región no traducida en 3’ del ARNm de la transtiretina (TTR) humana, tanto de tipo mutante como normal o salvaje, y provoca su degradación a través de la escisión mediada por la RNAsa H1. Por su parte, patisirán es el primer ARN de interferencia autorizado en Europa (Figura 9): un ARN bicatenario pequeño formulado en nanopartículas lipídicas que se une específicamente a una secuencia conservada en la región 3’-UTR del ARNm de la TTR, también tanto en su forma mutante como salvaje, y a través de la interferencia de ARN y con mediación de la endonucleasa argonauta-2, produce la degradación catalítica de dicho ARNm. En consecuencia, ambos fármacos inhiben la síntesis hepática y la secreción a sangre de la proteína TTR, reduciendo de forma sustancial sus niveles séricos circulantes, lo que se traduce en una mayor estabilización o aclaramiento de los depósitos de TTR amiloidótica y de las manifestaciones de la polineuropatía. Designados por la EMA como medicamentos huérfanos, ambos han sido autorizados para el tratamiento de la polineuropatía en estadios 1 o 2 en pacientes adultos con amiloidosis familiar (o hereditaria) por transtiretina (ATTRh). Inotersén se administra por vía subcutánea una vez por semana y patisirán por vía intravenosa una vez cada 3 semanas.
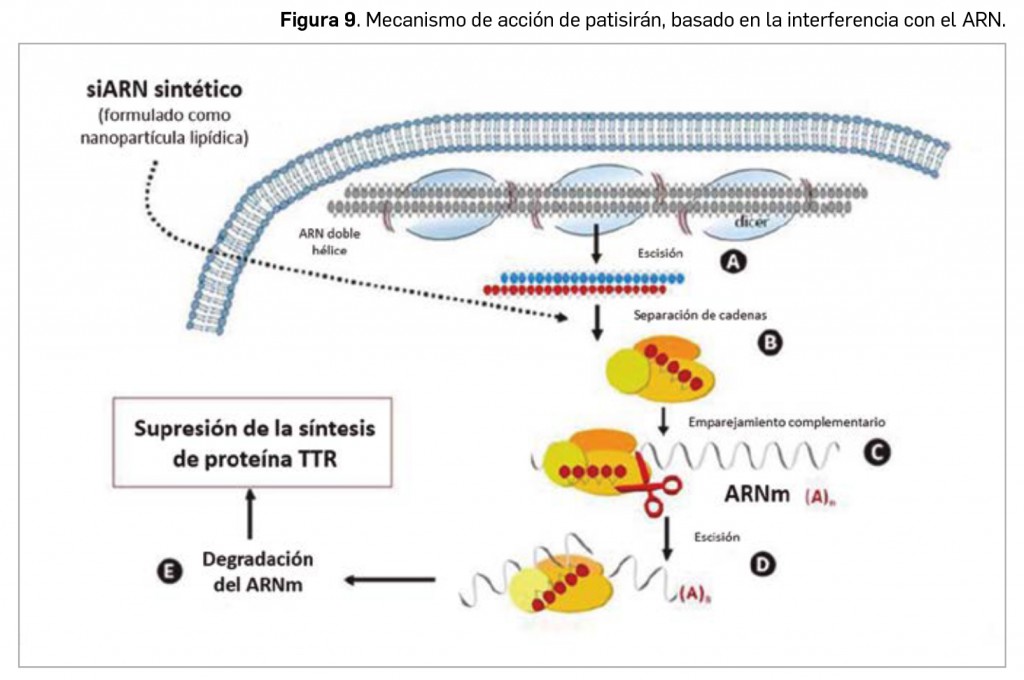
La eficacia y seguridad clínicas de inotersén han sido contrastadas en un ensayo aleatorizado de fase 2/3 (N= 173), doble ciego y controlado por placebo. El tratamiento durante 15 meses demostró una eficacia en la estabilización de los síntomas neurológicos periféricos significativamente superior a placebo (diferencia de 15 puntos en la escala mNIS+7) y en la calidad de vida de los pacientes (diferencia de 9 puntos en el cuestionario Norfolk QoL-DN). De forma similar, la superioridad de patisirán frente a placebo para frenar la lesión nerviosa progresiva fue demostrada en un ensayo pivotal de fase 3 (N= 225), también doble ciego y aleatorizado. Tras 18 meses, el fármaco redujo una media de 6 puntos la puntuación de mNIS+7 (diferencia de 34 puntos respecto a placebo), indicando una ligera mejoría de los pacientes, significativa desde el 9º mes de tratamiento; también indujo una modesta mejora de su calidad de vida (diferencia de 21 puntos en el cuestionario Norfolk QoL-DN a favor de patisirán). Adicionalmente, las variables secundarias respaldaron el beneficio aportado por los dos fármacos sobre la fuerza motora, el estado nutricional o la velocidad de la marcha; una eficacia que se mostraba consistente entre subgrupos de pacientes e independiente de factores como el tipo de mutación, gravedad del paciente, tratamiento previo o presencia de cardiopatía.
El perfil toxicológico a corto-medio plazo de ambos fármacos, más benigno para patisirán, es clínicamente manejable, siendo la mayoría de eventos adversos leves-moderados y autolimitados. Las reacciones adversas más frecuentes con inotersén son reacciones en el lugar de inyección (51%) –sobre todo, eritema, dolor y prurito–, náuseas (31%), anemia (28%) o cefalea (23%), si bien sobresale, por su gravedad, el riesgo de trombocitopenia (13%, grave en el 7%) y de glomerulonefritis (3%), así como una inmunogenicidad no desdeñable. Los eventos adversos más comunes con el mejor tolerado patisirán son: edema periférico (30%) y reacciones relacionadas con la perfusión (19%), como dolor de espalda, rubefacción, dolor abdominal o náuseas. Los estudios de extensión ahora en marcha aportarán más datos a largo plazo.
En definitiva, son dos nuevos tratamientos etiológicos que inauguran una vía terapéutica en el tratamiento de la ATTRh y aportan un beneficio clínicamente relevante en el manejo de la polineuropatía: aunque se exceptúan los pacientes más graves (estadio 3), amplían las posibilidades terapéuticas de tafamidis (pacientes en estadio 1 exclusivamente) y serán opciones de tratamiento útiles, especialmente si se considera que hasta dos tercios de los pacientes no son susceptibles de trasplante hepático. No se dispone aún de comparaciones directas o indirectas entre las distintas opciones de tratamiento ni se puede concluir acerca de la superioridad de una sobre otra, pero los resultados divulgados podrían sugerir que inotersén y patisirán son más eficaces que tafamidis. Patisirán parece aportar un mayor beneficio clínico y una mayor innovación terapéutica.