Otras novedades sobre vacunas en fases clínicas avanzadas: SPUTNIK V
Habiendo descrito en detalle las principales evidencias disponibles para las tres vacunas ya autorizadas en la Unión Europea y otras consideradas en la Estrategia de Vacunación del Ministerio de Sanidad1 (véase sección Medicamentos en España y Vacunas del presente número), se resumen a continuación los hallazgos más relevantes divulgados para una vacuna autorizada en Rusia, llamada SPUTNIK V o Gam-COVID-Vac: una vacuna heteróloga compuesta por dos vectores recombinantes de adenovirus humanos –uno de tipo 26 (rAd26-S) y otro de tipo 5 (rAd5-S)– que portan un gen codificante para la proteína S del SARS-CoV-2 de cadena completa. Inicialmente, reinó cierto escepticismo en la comunidad científica en torno a esta vacuna por la opacidad de los resultados (en el momento de su aprobación solo se conocía públicamente el perfil de seguridad y de inmunogenicidad en ensayos de fase 1/2). Pero, ahora que diversos países empiezan a valorar su uso, se han publicado los resultados preliminares de un ensayo pivotal en fase 3 que parecen esclarecer sus posibilidades terapéuticas.
Se trata de un estudio aleatorizado, doble ciego, multicéntrico (realizado en 25 hospitales y centros sanitarios de Moscú) y controlado por placebo, en el que un total de 21.977 voluntarios adultos sin antecedentes de infección por SARS-CoV-2 (negativos en una PCR y en una prueba de anticuerpos IgG e IgM) fueron asignados al azar (3:1) a recibir la vacuna (N= 16.501) o placebo (N= 5.476), con estratificación por grupos de edad, entre el 7 de septiembre y el 24 de noviembre de 2020. Se excluyeron participantes que habían sufrido una enfermedad infecciosa en los 14 días anteriores a la aleatorización, y que hubieran recibido otras vacunas en los 30 días previos. La vacuna se administró por vía intramuscular (0,5 ml/dosis) en una pauta de dos dosis (cebado con rAd26 y refuerzo con rAd5) separadas por un intervalo de 21 días.
Para el análisis de eficacia se usaron los datos de 19.866 sujetos que recibieron 2 dosis de vacuna o placebo. A los 21 días después de la primera dosis, el día pre-establecido de administración de la segunda dosis, se confirmaron un total de 16 casos de COVID-19 en el grupo experimental (de 14.964 participantes: un 0,1%) y 62 casos en el grupo placebo (de 4.902: un 1,3%). Esto se traducía en una eficacia global de la vacuna del 91,6% (IC95% 85,6-95,2) en la prevención de la COVID-19 sintomática, que se mantenía en valores similares con independencia de la mayor edad (> 60 años). Los autores refieren también una eficacia del 100% en la prevención de COVID-19 moderada o grave, ya que no se registró ningún caso entre los vacunados frente a 20 en el grupo placebo. En relación a la seguridad, la vacuna fue bien tolerada, ya que la mayoría (94%) de los eventos adversos notificados fueron leves (grado 1), siendo los más frecuentes: fiebre y síntomas gripales, astenia, reacciones en el sitio de inyección y cefalea. Además, la tasa de eventos adversos graves fue muy similar entre el grupo vacunal y en el brazo placebo (0,3% vs. 0,4%), y ninguno de ellos fue considerado por el comité independiente de seguimiento como asociado con la vacunación. Durante el estudio se notificaron 4 muertes (3 en el grupo de la vacuna y 1 en el grupo de placebo), ninguna de las cuales se consideró relacionada con la vacuna.
Estamos ante un nuevo ejemplo de que la mejor forma de despejar incertidumbres sanitarias es la realización de ensayos controlados, aleatorizados y amplios que aporten una evidencia científica de calidad suficiente como para extraer conclusiones sólidas. Estos resultados pueden abrir la vía a su aprobación y utilización en la UE. De manera interesante, se acaba de conocer que científicos de la Universidad de Oxford/AstraZeneca y los responsables del desarrollo de SPUTNIK V van a impulsar ensayos clínicos conjuntos que combinarán sus dos vacunas –basadas en la misma tecnología: vectores virales no replicativos que portan un gen codificante para la proteína S– en voluntarios adultos, sobre la hipótesis de que tal asociación podría aumentar considerablemente la eficacia de las vacunas. Si se demuestra que la administración de una sola dosis de cada vacuna aporta la misma o superior inmunoprotección que la administración de dos dosis de cada una de ellas, puede permitir flexibilizar los programas de inmunización poblacionales.
- Logunov DY, Dolzhikova IV, Zubkova OV, Tukhvatulin AI, Shcheblyakov DV, Dzharullaeva AS et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia. Lancet. 2021; DOI: https://doi.org/10.1016/S0140-6736(21)00234-8.
Una vacuna ideada y producida en España también progresa
Uno de los candidatos vacunales frente a la COVID-19 desarrollados en el Centro Superior de Investigaciones Científicas (CSIC) de España, en concreto, el diseñado por el grupo de investigación que dirige el Dr. Mariano Estaban, acaba de obtener resultados prometedores de inmunogenicidad y eficacia en la fase pre-clínica. Se trata de la vacuna denominada MVA-CoV-2-S, basada en el derivado de poxvirus vaccinia de Ankara (MVA), que ha sido modificado para expresar la proteína S completa del SARS-CoV-2; esa proteína se produce tan pronto como el virus penetra en la célula y es la que inducirá la respuesta del sistema inmunitario.
Los autores desarrollaron dos candidatos similares –una de ellas basada en ácido nucleico de la proteína S (ADN-S)– que evaluaron en ratones a través de dos estrategias de inmunización de cebado y refuerzo (2 dosis): ADN/MVA y MVA/MVA. Sus resultados revelan que ambas vacunas y ambas estrategias indujeron respuestas robustas, amplias y polifuncionales, específicas frente a la proteína S, tanto de células T CD4+ (principalmente Th1) como de CD8+, con un fenotipo de memoria efectora T; la inmunización ADN/MVA indujo una respuesta de células T más potente. Todos los regímenes estudiados desencadenaron la generación de títulos elevados de anticuerpos IgG específicos para la proteína S y para el dominio de unión al receptor; el predominio del isotipo IgG2c era indicativo de una respuesta inmunitaria Th1. De forma interesante, el suero de los ratones vacunados demostró una capacidad significativa de neutralización frente al SARS-CoV-2 en cultivos celulares, siendo la inmunización MVA/MVA la que evidenció la mayor actividad neutralizante. Además, tanto una como dos dosis de la vacuna MVA-CoV2-S protegieron a los ratones humanizados (modelo K18-hACE2, susceptible de infección por el coronavirus) de la infección con una dosis letal de SARS-CoV-2, esto es, un 100% de eficacia en la prevención de la letalidad asociada a COVID-19; el régimen de dos dosis confierió incluso una inhibición total de la replicación del virus en los pulmones.
Para definir el espectro de acción de la vacuna y cumplir con los requisitos de las agencias reguladoras para los ensayos clínicos, se están llevando a cabo actualmente experimentos de inmunogenicidad y eficacia en un modelo de hámster. A expensas de que pueda avanzar a la evaluación de seguridad y eficacia en primates no humanos y hasta las fases clínicas de investigación, estos resultados son esperanzadores: la posibilidad de obtener una vacuna de producción nacional puede contribuir significativamente a cubrir las carencias de dosis de otras vacunas que ya se están produciendo en la cadena de distribución a nivel internacional.
- García-Arriaza J, Garaigorta U, Pérez P, Lázaro-Frías A, Zamora C, Gastaminza P et al. COVID-19 vaccine candidates based on modified vaccinia virus Ankara expressing the SARS-CoV-2 spike induce robust T- and B-cell immune responses and full efficacy in mice. J Virol. 2021: JVI.02260-20. DOI: 10.1128/JVI.02260-20.
Nuevos datos clínicos de bamlanivimab en enfermedad leve-moderada o como profiláctico
Según se avanzaba en esta sección del anterior número de Panorama Actual del Medicamento, el anticuerpo monoclonal neutralizante bamlanivimab recibió a finales del año pasado la autorización de uso de emergencia por la FDA estadounidense para el tratamiento –en dosis única por infusión intravenosa– de pacientes de ≥ 12 años no hospitalizados con un diagnóstico reciente de COVID-19 de leve a moderado, y que tengan alto riesgo de progresar a enfermedad grave y/o ser hospitalizados, incluyendo a pacientes con ≥ 65 años de edad o que tienen ciertas comorbilidades crónicas. Dicha autorización se basó en los resultados de eficacia clínica del ensayo de fase 2/3 BLAZE-1.
Se divulgan ahora en una revista científica esos y otros resultados complementarios –correspondientes al análisis final– del citado estudio, que tuvo un diseño aleatorizado y se realizó en 49 centros sanitarios de EE.UU. Incluyó un total de 613 pacientes ambulatorios con confirmación de COVID-19 y uno o más síntomas moderados o leves, quienes fueron asignados a recibir una administración única de bamlanivimab de 700 mg (N= 101), 2.800 mg (N= 107) o 7.000 mg (N= 101), de una combinación de 2.800 mg de bamlanivimab y 2.800 mg del nuevo anticuerpo neutralizante etesevimab (N= 112), o bien de placebo (N= 156). Los resultados demuestran que para los 533 pacientes que recibieron una infusión y fueron evaluables hasta el día 29 (media de edad de 44,7 años, 55% mujeres), el cambio en el logaritmo de la carga viral al día 11 en comparación con el estado basal –variable primaria– fue de -3,72 para la dosis de 700 mg de bamlanivimab, de -4,08 para la de 2.800 mg, -3,49 para la de 7.000 mg, de -4,37 para el tratamiento combinado y de -3,8 para el grupo control. Así pues, en comparación con placebo, las diferencias en esa variable fueron de +0,09 (p= 0,69) para 700 mg, -0,27 (p= 0,21) para 2.800 mg, y 0,31 (p= 0,16) para 7.000 mg de bamlanivimab, y de -0,57 (p= 0,01) para la combinación de los dos anticuerpos; solo esta última comparación alcanzó significación estadística. Entre las variables secundarias, destaca que la proporción de pacientes con hospitalización o visita a urgencias relacionadas con la COVID-19 fue de: 5,8% para placebo, 1,0% para 700 mg, 1,9% para 2.800 mg y 2,0% para 7.000 mg de bamlanivimab, y de 0,9% para el tratamiento en combinación. En relación a la seguridad, es preciso señalar que se notificaron reacciones de hipersensibilidad inmediata en 9 pacientes: 6 tratados con bamlanivimab, 2 con la combinación de anticuerpos, y 1 con placebo. No se reportó ninguna muerte durante el periodo de estudio.
La principal novedad de estos datos es, pues, que solo en combinación con etesevimab es cuando banlanivimab induce una reducción estadísticamente significativa de la carga viral del SARS-CoV-2, y no así cuando se administra en monoterapia. Futuros resultados que determinen el impacto de dicho aclaramiento viral sobre variables primarias de beneficio clínico (reducción de hospitalizaciones y muerte, fundamentalmente) permitirán confirmar si la combinación de anticuerpos puede ser una opción terapéutica interesante como tratamiento de casos leves en el ámbito comunitario. A la vista de los resultados de las variables secundarias, es también previsible que la monoterapia con bamlanivimab sea capaz de reducir el riesgo de complicaciones de una COVID-19 leve-moderada.
Adicionalmente, la compañía comercializadora de bamlanivimab ha divulgado2 datos de otro estudio diferente, el BLAZE 2, en el que se ha administrado de forma profiláctica el fármaco (4.200 mg) o placebo a adultos mayores que viven en residencias (N= 299) y al personal sanitario de estos centros (N= 666), incluyendo 132 participantes en los que se confirmó la infección por SARS-CoV-2 en el inicio. En el análisis primario de los datos, a las 8 semanas de seguimiento, se observó una frecuencia significativamente menor de casos sintomáticos de COVID-19 en el grupo de tratamiento con bamlanivimab en comparación con placebo (OR= 0,43; p= 0,00021); en el subgrupo de residentes mayores, esa reducción fue más notable (OR= 0,20; p= 0,00026), lo que sugería una reducción del 80% del riesgo de desarrollar la enfermedad. Se notificaron 4 fallecimientos atribuidos a COVID-19 en el grupo placebo, ninguno entre los tratados con el fármaco.
Todo ello, junto al proceso de rolling review que ha iniciado la EMA3 para evaluar la posibilidad de una autorización condicional de la combinación de otros dos anticuerpos monoclonales (casirivimab / imdevimab) muestra las posibilidades de este tipo de terapias en la COVID-19.
- Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA. 2021: e210202. DOI: 10.1001/jama.2021.0202.
El pontencial terapéutico de colchicina
En una nota de prensa4 –la tendencia de dudoso rigor en la comunicación científica que se ha generalizado con la pandemia– publicada el pasado 23 de enero, el Instituto de Salud de Montreal (Canadá) anunciaba los resultados del ensayo clínico que ha promovido, denominado COLCORONA, para evaluar el potencial del antiinflamatorio colchicina en el tratamiento de la COVID-19. Es éste un fármaco ampliamente conocido y autorizado en España para el tratamiento de la gota y de la fiebre mediterránea familiar. El empleo en investigación clínica de agentes antiinflamatorios se justifica por su posible capacidad para atenuar la tormenta de citocinas proinflamatorias que media la patogénesis de las complicaciones de la enfermedad.
En el ensayo clínico controlado de fase 3, doble ciego, multicéntrico y multinacional (incluyendo España), se aleatorizaron un total de 4.488 pacientes no hospitalizados con COVID-19 –diagnosticada por PCR o criterios clínicos– a recibir colchicina (0,5 mg dos veces al día durante 3 días, y posteriormente una vez al día) o placebo durante 30 días. La variable primaria de eficacia –una medida compuesta de muerte u hospitalización por COVID-19– se verificó en el 4,7% de los pacientes tratados con colchicina y en el 5,8% de los pacientes en el grupo placebo (OR= 0,79; IC95% 0,61-1,03; p= 0,08). Si se consideran solo los pacientes con confirmación de la infección por PCR, el diagnóstico más preciso, la variable primaria ocurrió en el 4,6% de los tratados con colchicina frente al 6,0% de los pacientes en el grupo control (OR= 0,75; IC95% 0,57-0,99; p= 0,04). Concretamente, para ese grupo los autores refieren una reducción del 25% en las probabilidades de hospitalización (OR= 0,75; IC95% 0,57-0,99), del 50% en el riesgo de ventilación mecánica (OR= 0,50; IC95% 0,23-1,07) y del 44% en el riesgo de muerte (OR= 0,56; IC95% 0,19-1,66). Respecto al perfil toxicológico, se reportó una frecuencia de eventos adversos significativamente menor en el grupo de colchicina que en el grupo placebo (4,9% vs. 6,3%; p= 0,05), notificándose con menor frecuencia, por ejemplo, casos de neumonía (2,9% vs. 4,1% en el grupo control); no obstante, la diarrea fue más común en el grupo de la colchicina (13,7% vs. 7,3%; p< 0,0001).
Estos resultados se han recogido en un artículo aún no publicado en una revista científica con revisión por pares, por lo que deben ser tratados con cautela. Pero se trata del primer fármaco de administración oral, y fácilmente accesible en términos económicos, para el que se demuestra que su uso en pacientes con enfermedad leve previene significativamente las complicaciones de la COVID-19 en al menos 1 de cada 5 casos. Se debe recordar, no obstante, que la colchicina tiene un margen terapéutico estrecho y es extremadamente tóxica en caso de sobredosis, lo que requiere un ajuste muy riguroso de su pauta posológica. Solo en España hay a día de hoy 9 ensayos clínicos en marcha con este fármaco, que aportarán más luz sobre sus posibilidades de autorización con indicación en COVID-19.
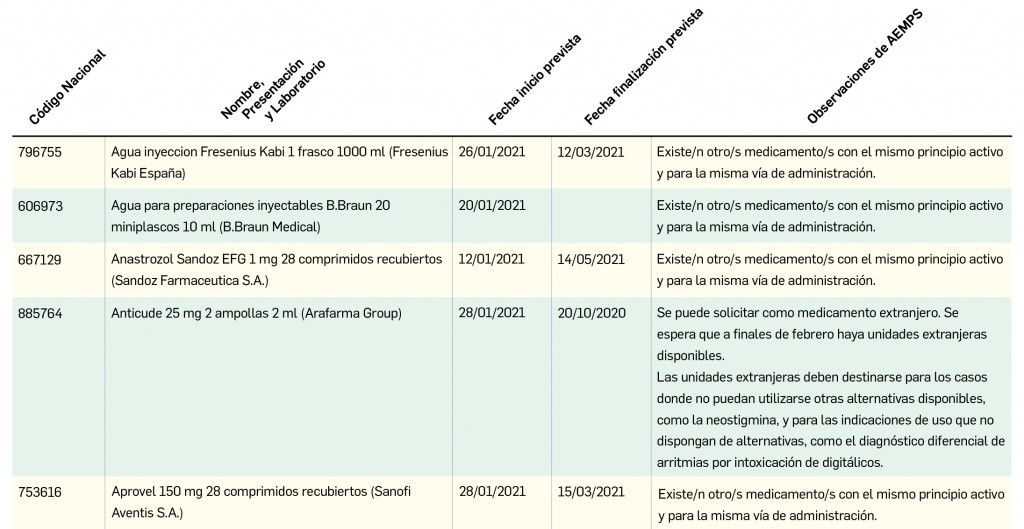
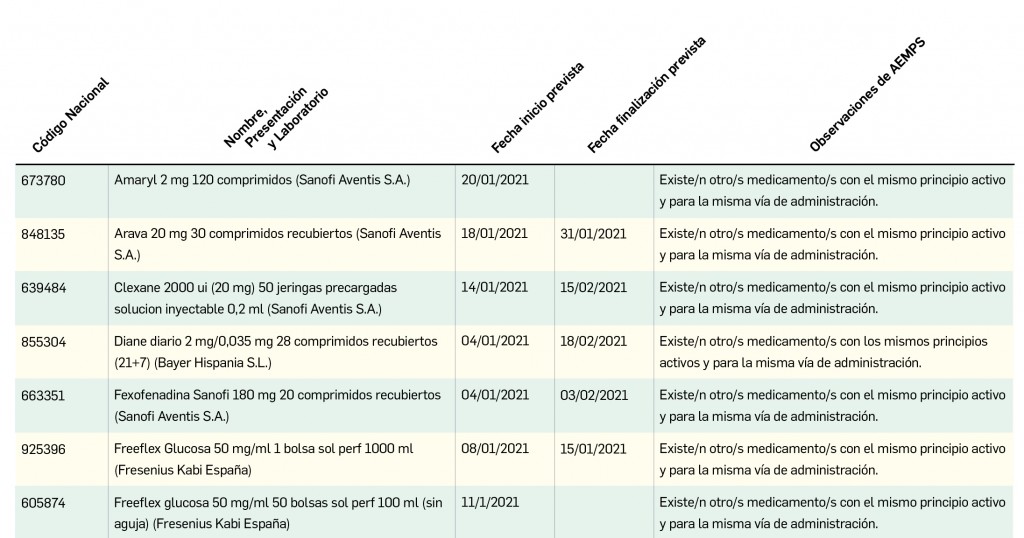


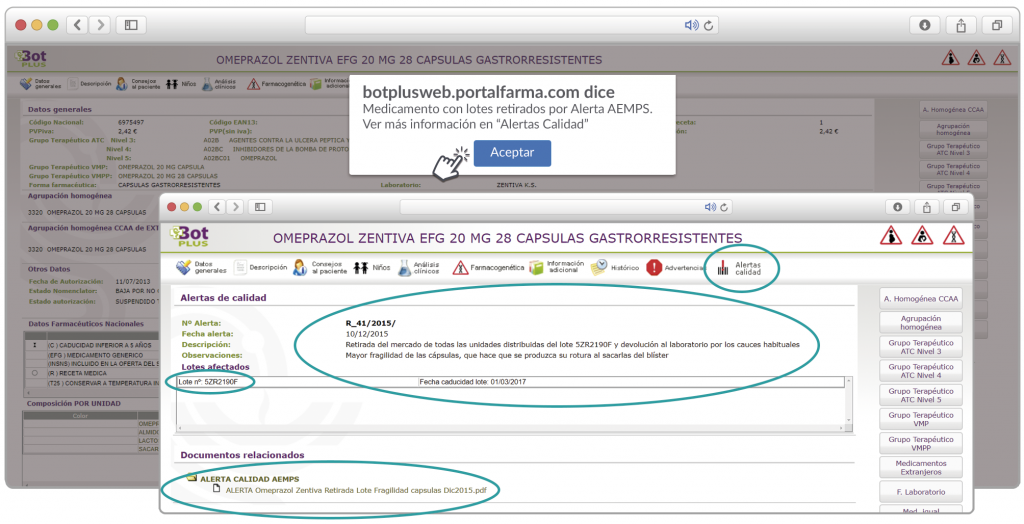
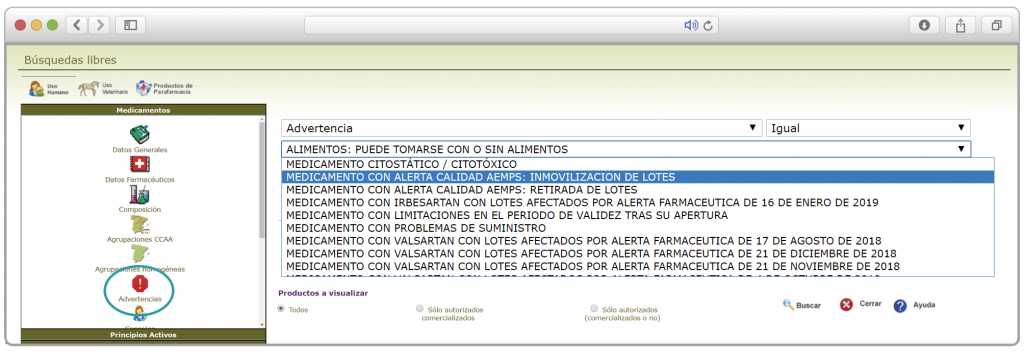
 Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
Además, se incluye una pestaña específica en la que se pueden consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.