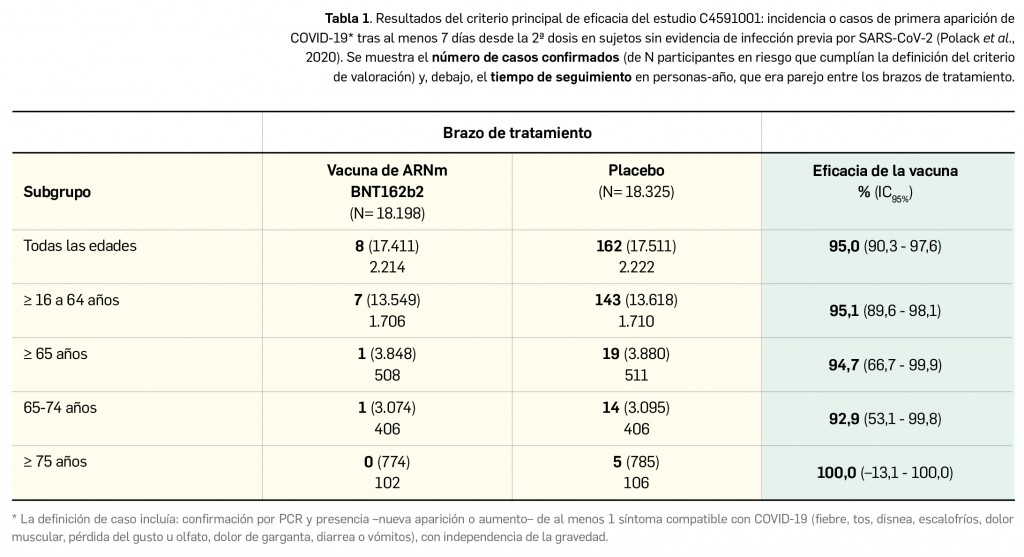
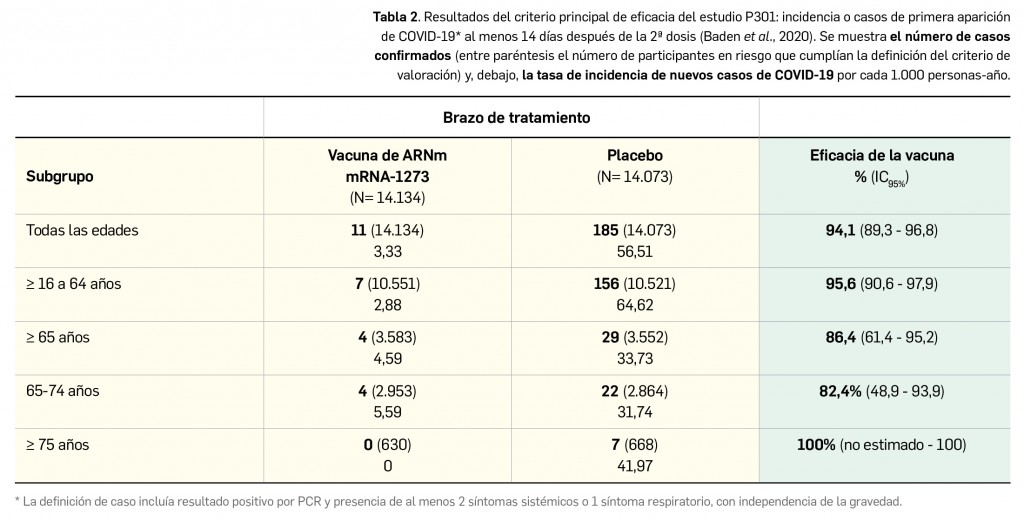
Como continuación de la referencia en el número 436 de Panorama Actual del Medicamento a los primeros hallazgos del Estudio Nacional de Seroprevalencia ENE-COVID, desarrollado por el Ministerio de Sanidad para conocer de forma más detallada la verdadera difusión del virus SARS-CoV-2 en España, se deben citar brevemente los últimos resultados del citado estudio1 divulgados hacia mediados del mes de diciembre de 2020 y pertenecientes a la cuarta ronda de la investigación (que coincidió con el “fin” de la popularmente llamada segunda ola de contagios), también basada en la realización de test rápidos de anticuerpos. En líneas generales, los nuevos datos –relativos a 51.409 participantes– revelan que la prevalencia global de anticuerpos IgG frente al SARS-CoV-2 desde el inicio de la epidemia se sitúa en el 9,9% (IC95% 9,4-10,4), siendo bastante similar en mujeres (10,1%; IC95% 9,5-10,7) y en hombres (9,6%; IC95% 9,0-10,2). Es decir, que prácticamente una de cada diez personas en España habría sido infectada por el virus a lo largo de todo el periodo de estudio (más de 4,5 millones de personas si se extrapola en base al número de habitantes), lo que supone el doble de lo calculado en las primeras fases del estudio. A pesar de haberse observado en esta ronda una mayor dispersión de la epidemia en España, todavía se aprecia una marcada variabilidad geográfica: mientras Canarias y algunas provincias gallegas (Coruña y Lugo) presentan prevalencias inferiores o cercanas al 4%, la zona centro del país (Comunidad de Madrid y provincias limítrofes como Cuenca, Guadalajara o Toledo) muestra cifras cercanas o superiores al 15%. La proporción de personas con anticuerpos IgG frente a SARS-CoV-2 es mayor en residentes de grandes ciudades, situándose en el 11,6% en el total del periodo de estudio.
Cabe destacar que, de nuevo, se revela que el personal sanitario y las mujeres que cuidan a personas dependientes en el domicilio son los grupos poblacionales que presentan una prevalencia más alta, del 16,8 y el 16,3%, respectivamente. Esas cifras se ven también elevadas en mayor medida entre las personas que en algún momento han sido convivientes de un caso sintomático de COVID-19 (tasa de prevalencia del 31% en la 4ª ronda). En relación al diagnóstico de COVID-19, entre las personas con síntomas compatibles la prevalencia aumenta con el número de síntomas, siendo particularmente alta entre quienes refieren anosmia (43%), similar a lo encontrado en la primera fase. Además, se detectan anticuerpos IgG en un 3,3% de los participantes que no han referido síntomas en ninguna de las rondas y, entre las personas negativas en la primera fase del estudio, la tasa de seroconversión se sitúa en el 3,8% (IC95% 3,5-4,1), mayor que el 0,7-0,9% detectado previamente. El porcentaje de asintomáticos en relación con el total de personas positivas para IgG se estima en torno al 30%.
Aunque estos resultados son preliminares, representan una aproximación a la verdadera expansión del virus y el número de infecciones reales, que distan notablemente de las cifras de casos confirmados manejadas por las autoridades sanitarias. Afortunadamente, los autores del estudio estiman que en la segunda ola de la epidemia ha mejorado la capacidad diagnóstica del sistema sanitario, habiéndose confirmado el diagnóstico microbiológico de 6 de cada 10 pacientes infectados (frente a 10% de diagnósticos en las primeras rondas).




 vez pinnatisectas, con 3 a 7 segmentos de 1 a 5 cm, profundamente dentados y en ocasiones pinnatífidos, de contorno deltado o rómbico. El peciolo es endeble y envainador en la mayor parte de su longitud. Las hojas superiores son en su mayoría profundamente trilobuladas o trisectas, con peciolo envainador en casi toda su longitud. Las flores se disponen en umbelas compuestas, generalmente opuestas a las hojas, con 4 a 12 radios de 1 a 3 cm, desiguales, con pedúnculos más cortos que los radios o inexistentes. Las flores presentan un cáliz sin dientes y pétalos de color blanco. En la fructificación, los estilos, recurvados, son tan largos como el estilopodio. Los frutos, demasiadas veces confundidos con semillas y denominados así en casi todas las publicaciones científicas, son esquizocarpos anchamente ovoides, constituidos por dos mericarpos con costillas muy visibles pero delgadas (Figura 3). Las plantas de A. graveolens desprenden un olor intenso característico.
vez pinnatisectas, con 3 a 7 segmentos de 1 a 5 cm, profundamente dentados y en ocasiones pinnatífidos, de contorno deltado o rómbico. El peciolo es endeble y envainador en la mayor parte de su longitud. Las hojas superiores son en su mayoría profundamente trilobuladas o trisectas, con peciolo envainador en casi toda su longitud. Las flores se disponen en umbelas compuestas, generalmente opuestas a las hojas, con 4 a 12 radios de 1 a 3 cm, desiguales, con pedúnculos más cortos que los radios o inexistentes. Las flores presentan un cáliz sin dientes y pétalos de color blanco. En la fructificación, los estilos, recurvados, son tan largos como el estilopodio. Los frutos, demasiadas veces confundidos con semillas y denominados así en casi todas las publicaciones científicas, son esquizocarpos anchamente ovoides, constituidos por dos mericarpos con costillas muy visibles pero delgadas (Figura 3). Las plantas de A. graveolens desprenden un olor intenso característico.

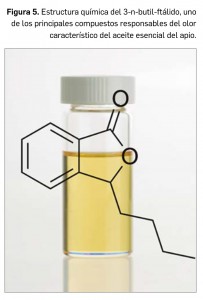 Los ftálidos, componentes lactónicos derivados del ácido benzoico, aislados de diferentes partes de esta especie vegetal, y algunos de sus análogos de síntesis, han mostrado efectos favorables en el tratamiento de la isquemia cerebral pues podrían incrementar el riego cerebral y proteger frente al daño neuronal provocado por la isquemia. Los dos enantiómeros del 3-n-butilftálido (Figura 5), principalmente (S)-NBP aunque también la forma racémica, que es más abundante (RS)-NBP, han sido ampliamente investigados en esta patología, razón por la cual fueron aprobados para su uso terapéutico en China en el año 2002 y la FDA americana ha autorizado los ensayos clínicos de fase II. Sus efectos son consecuencia principalmente de sus actividades antiagregante plaquetaria, antioxidante y neuroprotectora.
Los ftálidos, componentes lactónicos derivados del ácido benzoico, aislados de diferentes partes de esta especie vegetal, y algunos de sus análogos de síntesis, han mostrado efectos favorables en el tratamiento de la isquemia cerebral pues podrían incrementar el riego cerebral y proteger frente al daño neuronal provocado por la isquemia. Los dos enantiómeros del 3-n-butilftálido (Figura 5), principalmente (S)-NBP aunque también la forma racémica, que es más abundante (RS)-NBP, han sido ampliamente investigados en esta patología, razón por la cual fueron aprobados para su uso terapéutico en China en el año 2002 y la FDA americana ha autorizado los ensayos clínicos de fase II. Sus efectos son consecuencia principalmente de sus actividades antiagregante plaquetaria, antioxidante y neuroprotectora.