Archive
Revista PAM: 450
Número 450, Enero-febrero 2022
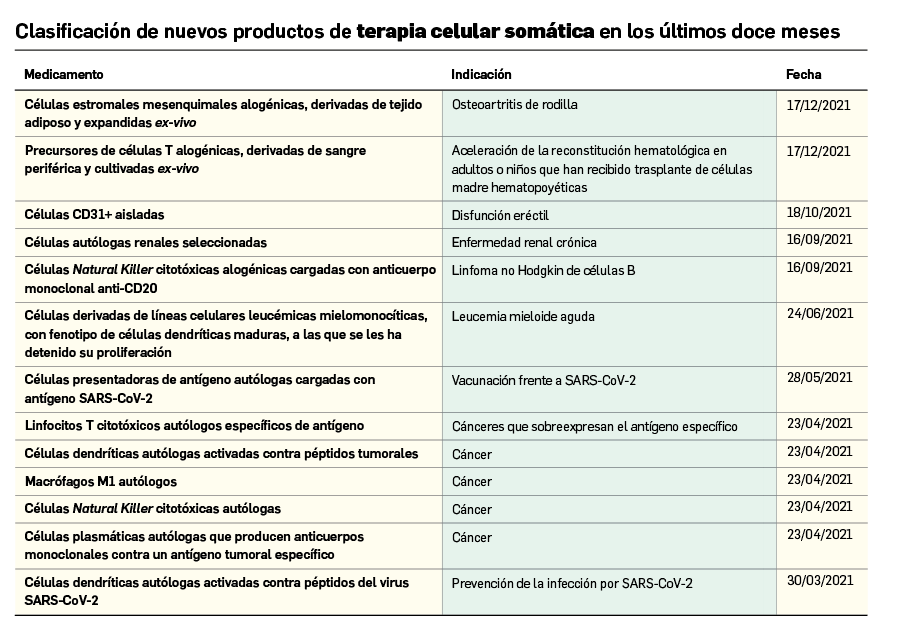
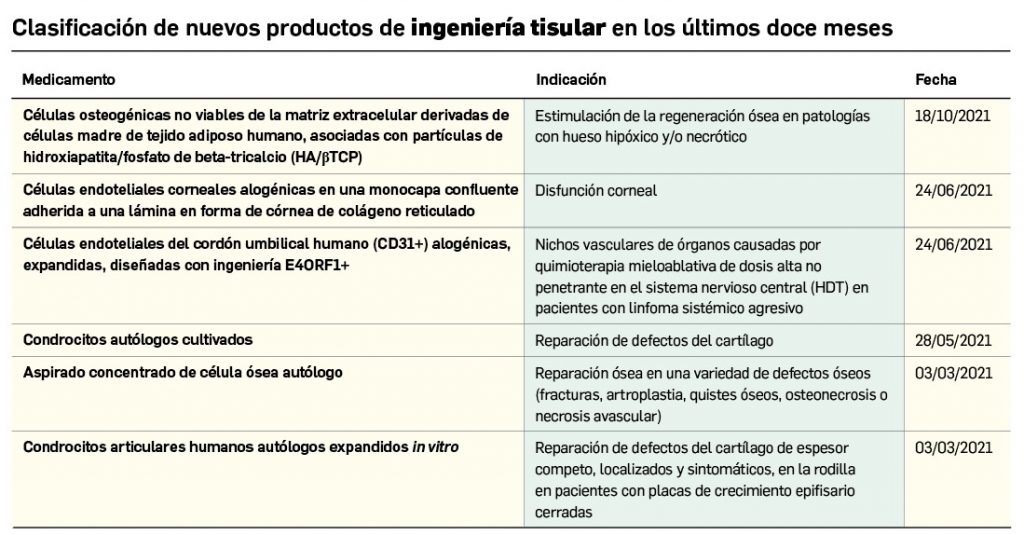
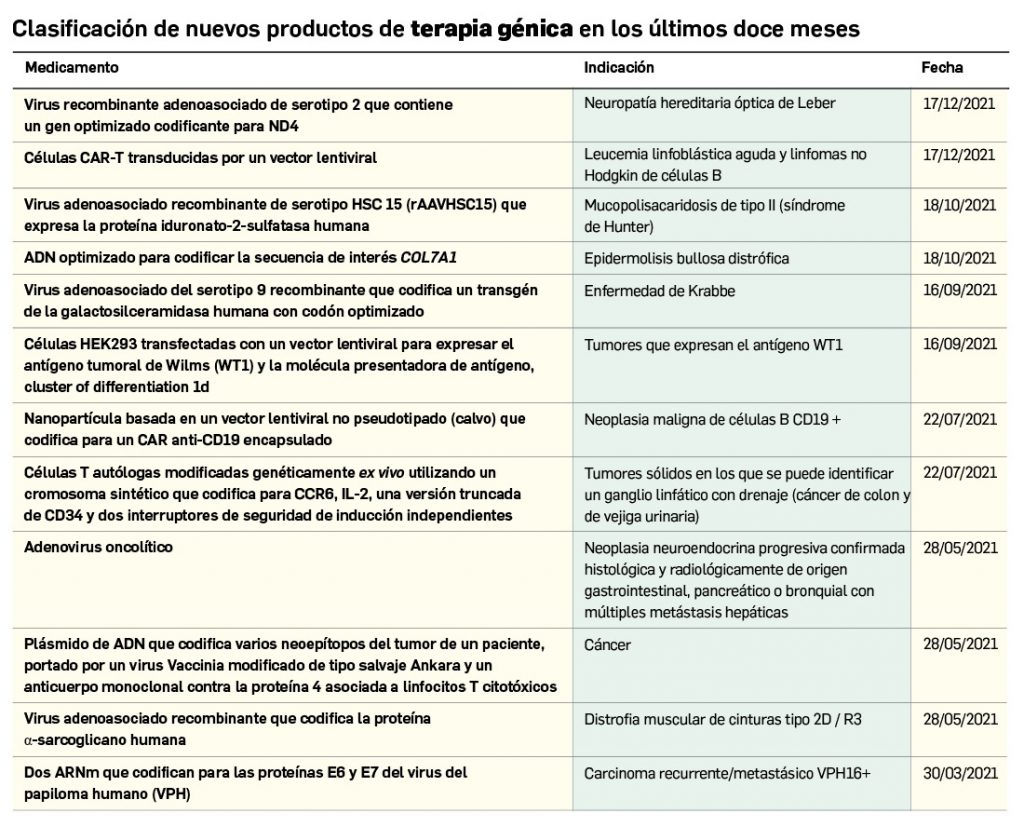
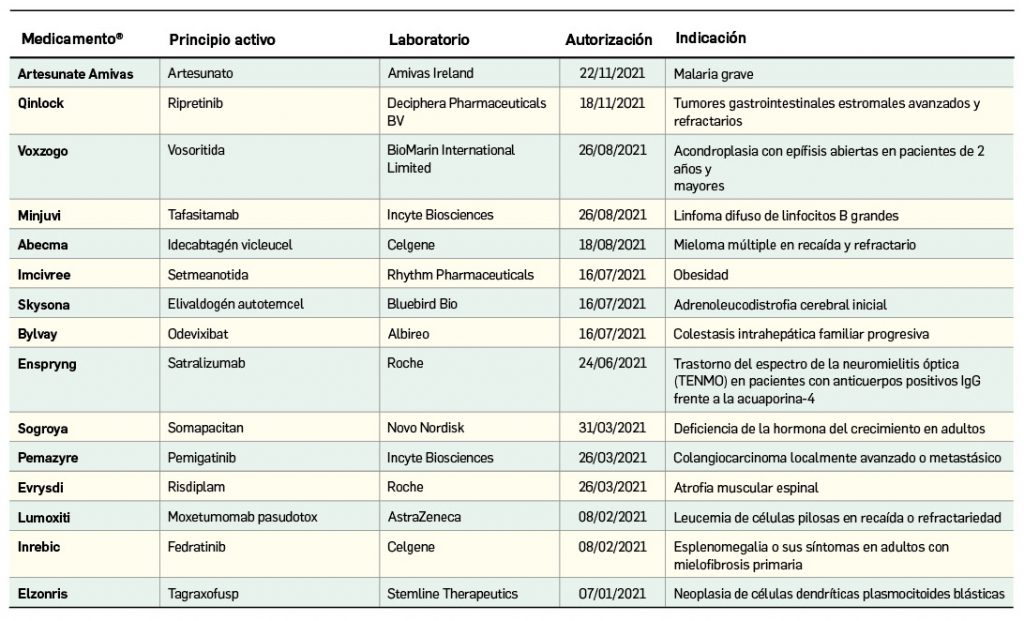
Nuevos medicamentos de terapias avanzadas
Los medicamentos de terapia avanzada (MTA o Advanced Therapy Medicinal Products, ATMP) ofrecen nuevos e innovadores tratamientos para las enfermedades. Están basados en la terapia génica, la terapia celular somática o la ingeniería tisular. El marco legal para las ATMP en la Unión Europea está establecido en la Regulation (EC) No 1394/2007 on advanced therapy medicinal products que asegura el libre movimiento de estas medicinas dentro de la Unión Europea y el acceso a los mercados. La regulación (EC) nº 1394/2007 también establece el nuevo Comité en Terapias avanzadas (CAT) cuya responsabilidad fundamental consiste en preparar un proyecto de opinión sobre cada nueva solicitud de medicamento de terapia avanzada planteada a la Agencia Europea de Medicamentos, antes de que el Comité de Medicamentos de Uso Humano (CHMP, Committee for Medicinal Products for Human Use) de la misma adopte una opinión definitiva sobre la concesión, modificación, suspensión o revocación de una autorización de comercialización para el medicamento en cuestión.
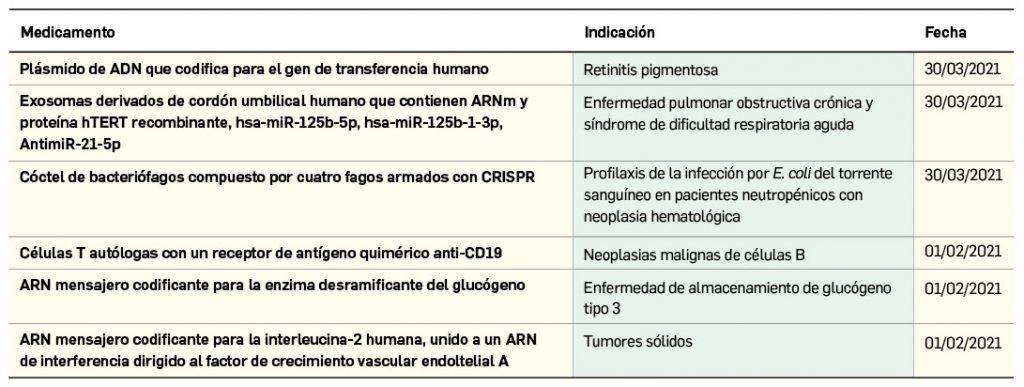
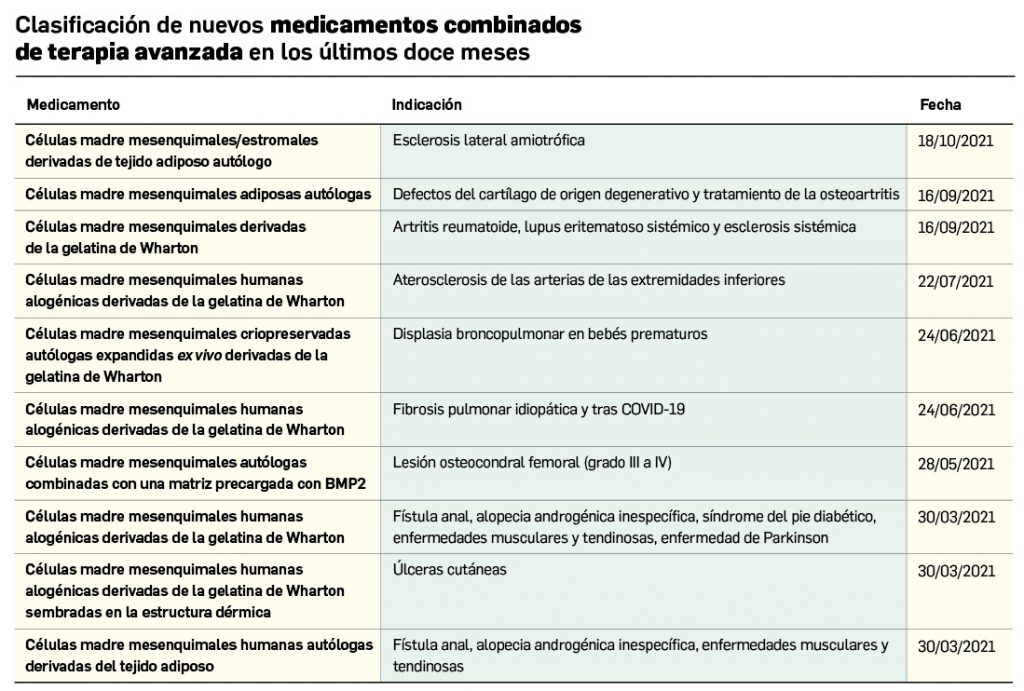
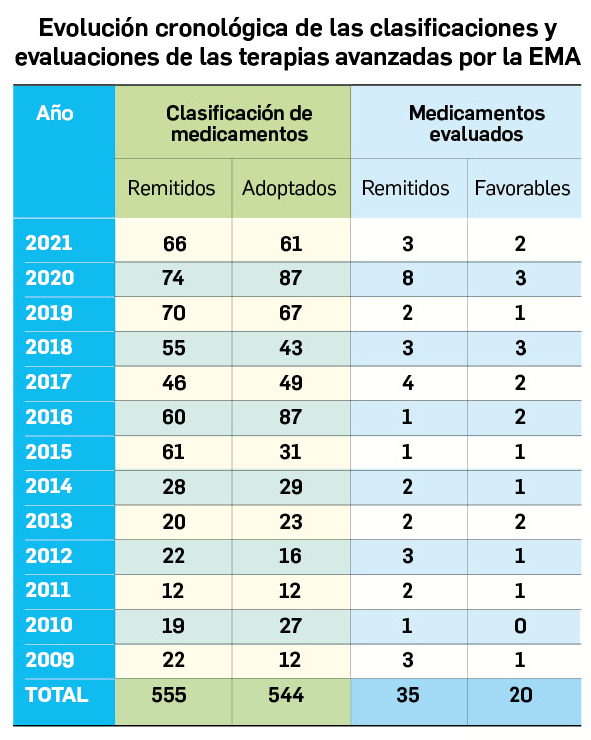
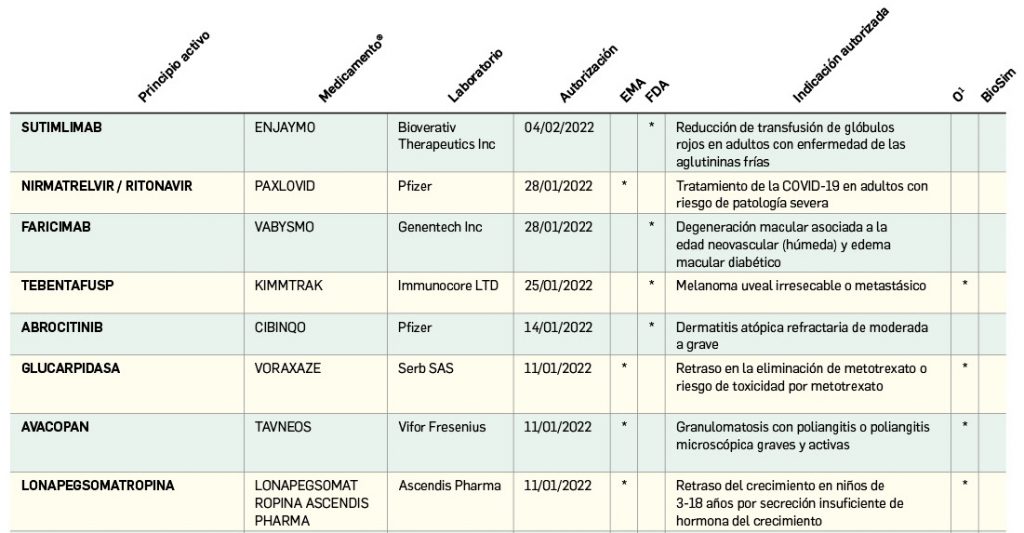
Nuevos medicamentos huérfanos
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
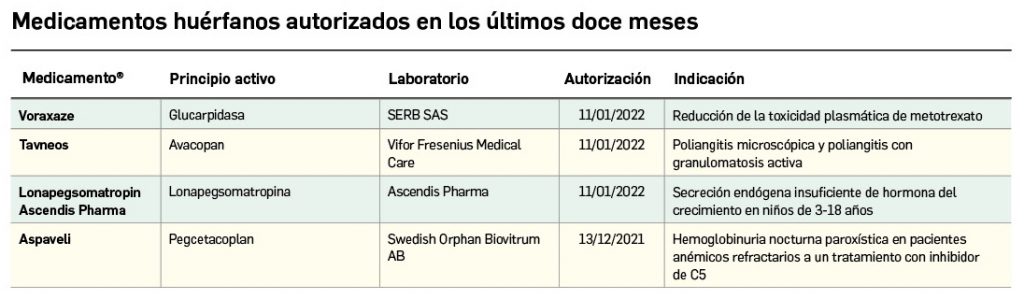
Instituciones y redes españolas
► Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
Instituto de Investigación en Enfermedades Raras:
https://www.isciii.es/QuienesSomos/CentrosPropios/IIER/Paginas/default.aspx
CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras): https://www.ciberer.es/
► Instituto de Mayores y Servicios Sociales (IMSERSO, Ministerio de Derechos Sociales y Agenda 2030):
http://www.imserso.es/imserso_01/index.htm
► Federación Española de Enfermedades Raras (FEDER): www.enfermedades-raras.org
► Asociaciones de pacientes en España: https://enfermedades-raras.org/index.php/asociaciones/nuestros-socios
Instituciones y redes europeas
► Agencia Europea de Medicamentos (EMA; Europea Medicines Agency). Apartado de Medicamentos Huérfanos
(inglés): https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp
► Comisión Europea: web oficial de la Comisión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://ec.europa.eu/health/rare_diseases/policy/index_es.htm
► Orphanet: Portal de información oficial de la Unión Europea sobre enfermedades raras y medicamentos huérfanos (español).
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
► Eurordis: Federación Europea de Asociaciones de Pacientes con Enfermedades Raras (español). http://www.eurordis.org/es
Otras instituciones y redes internacionales
► Food & Drug Administration (FDA, Estados Unidos). Apartado de Medicamentos Huérfanos (inglés):
http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/default.htm
► Pharmaceuticals & Medical Devices Agency. Agencia de Medicamentos y Dispositivos Médicos, de Japón (inglés):
http://www.pmda.go.jp/english/index.html
Casos de parestesia, mielitis, miocarditis y vasculitis: actualización europea de la información sobre seguridad de las diversas vacunas frente a la COVID-19
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) describe en sus últimos Informes de Farmacovigilancia de Vacunas contra la COVID-19 los avances en el seguimiento de la seguridad de dichas vacunas y los cambios en sus fichas técnicas tras la revisión de los datos de seguridad disponibles, que ha evaluado el PRAC (Comité Europeo de Evaluación de Riesgos en Farmacovigilancia).
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha publicado en sus 11º y 12º Informes de Farmacovigilancia de las Vacunas COVID-19 las novedades y los cambios que, tras la revisión de los datos de seguridad disponibles por parte del Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC), se han incorporado en la información de las fichas técnicas y de los prospectos europeos de las vacunas correspondientes (AEMPS 2021; AEMPS, 2022).
En España, desde el inicio de la pandemia hasta el 17 de enero de 2022, se han comunicado 8.424.503 casos de COVID-19 y 90.993 fallecimientos de infectados confirmados mediante test antes de fallecer (MS, 2022). Hasta el día 9 de enero de 2022, se han administrado un total de 80.109.445 dosis de vacunas frente a la COVID-19, habiéndose registrado 55.455 notificaciones de “acontecimientos adversos” en la base de datos FEDRA. Los acontecimientos notificados con más frecuencia siguen siendo los trastornos generales (fiebre y dolor en la zona de vacunación), del sistema nervioso (cefalea y mareos) y del sistema musculoesquelético (mialgia y artralgia).
Recordemos que se define como acontecimiento adverso “cualquier problema de salud que ocurre después de la vacunación, sin que necesariamente tenga que estar ocasionado por la vacuna”. Estos acontecimientos adversos pueden ser simplemente coincidentes en el tiempo, relacionados con el propio acto de vacunarse (ansiedad, desmayo, error en la administración), relacionados con un potencial problema de calidad de la vacuna, o realmente relacionados con la propia vacuna (como por ejemplo dolor en el lugar de la inyección o fiebre). Por lo tanto, hay que recordar que los acontecimientos adversos que se notifican no significa que estén relacionados con la vacunación como causa-efecto. Por tanto, no son reacciones adversas ni sirven para comparar el perfil de reacciones adversas de las diferentes vacunas.
Se exponen a continuación las recientes conclusiones de la evaluación periódica de los datos de farmacovigilancia de las vacunas COVID-19.
- Miocarditis/ pericarditis asociadas a ▼ComiRNAty®/▼Spikevax®
Desde que se autorizó la comercialización Comirnaty® (BioNTech/Pfizer) en la UE hasta el 1 de diciembre de 2021 se han administrado casi 479 millones de dosis en el Espacio Económico Europeo (EEE). De igual modo, desde que se autorizó la comercialización de Spikevax® (Moderna) en la UE hasta el 1 de diciembre de 2021, se han administrado más de 61,6 millones de dosis de esta vacuna en el EEE.
Tanto la miocarditis como la pericarditis son inflamaciones del corazón que pueden presentar síntomas muy diversos, aunque los más frecuentes son dificultad para respirar, palpitaciones que pueden acompañarse de ritmo cardiaco irregular y dolor en el pecho. Las infecciones víricas, incluyendo la infección con el coronavirus que causa la COVID-19, son causas habituales de miocarditis.
El PRAC ha evaluado datos recientes sobre este riesgo ya conocido para las vacunas de ARN mensajero (Comirnaty® y Spikevax®), incluyendo dos grandes estudios farmacoepidemiológicos realizados en la UE. Uno de los estudios se realizó con datos del Sistema Nacional de Salud Francés (Epi-Phare) y el otro está basado en datos de los registros de países nórdicos. Esta revisión confirma el riesgo de miocarditis y pericarditis que ya está reflejado en la ficha técnica y el prospecto de estas dos vacunas, y se aportan algunos datos adicionales. De acuerdo con los datos revisados, el PRAC ha concluido que el riesgo global de sufrir estas 2 posibles reacciones adversas es muy raro, lo que significa una frecuencia de aparición de, como máximo, una de cada 10.000 personas vacunadas. Además, los datos demuestran que el aumento de riesgo de miocarditis tras la vacunación es mayor en hombres jóvenes.
Tanto la miocarditis como la pericarditis aparecen pocos días después de la vacunación, principalmente en los primeros 14 días, y se observan con mayor frecuencia tras la segunda dosis. Tanto el estudio francés como el nórdico aportan estimadores del número de casos adicionales de miocarditis en varones jóvenes tras la segunda dosis, en comparación con personas de la misma edad y sexo que no han sido vacunadas. En el caso de Comirnaty®, el estudio francés muestra que, en un periodo de 7 días tras la segunda dosis, podría haber alrededor de 3 casos adicionales de miocarditis en 100.000 hombres vacunados de 12 a 29 años, comparados con no vacunados. De acuerdo con el estudio nórdico, en un periodo de 28 días después de la segunda dosis, pueden aparecer 6 casos adicionales de miocarditis en 100.000 hombres vacunados de 16 a 24 años en comparación con no vacunados. Para la vacuna Spikevax®, el estudio francés indica que, en un periodo de 7 días tras la segunda dosis, podría haber alrededor de 13 casos adicionales de miocarditis en 100.000 hombres vacunados de 12 a 29 años en comparación con no vacunados. De acuerdo con el estudio nórdico, en un periodo de 28 días después de la segunda dosis, aparecerían 19 casos adicionales de miocarditis en 100.000 hombres vacunados de 16 a 24 años frente a los no vacunados.
En resumen, los datos disponibles sugieren que el curso de las miocarditis y pericarditis que pueden aparecer tras la administración de estas vacunas no es diferente de las que se presentan independientemente de la vacunación, que habitualmente mejoran con reposo o tratamiento (ver nota de seguridad de la AEMPS MUH(FV) 19/2021).
Hasta el 12 de diciembre de 2021 se han registrado en España 240 notificaciones de miocarditis y/o pericarditis tras la administración de Comirnaty®. A esa misma fecha, se habían administrado en España más de 51 millones de dosis de esta vacuna. En la mayoría de los casos los pacientes estaban en recuperación o se habían recuperado en el momento de la notificación y ocurrieron generalmente en hombres (71%), tras la segunda dosis (63%) y en la primera semana tras recibir la vacuna (61%), siendo más frecuentes en edades jóvenes; del total de casos notificados, tres pacientes cursaron con desenlace mortal, dos de ellos, mayores de 60 años, presentaban causas alternativas para la aparición de miocarditis. En el caso del tercer paciente, también adulto, la información de la que se dispone es limitada para realizar una correcta evaluación sobre su posible relación con la vacuna.
Asimismo, en España se han registrado hasta el 12 de diciembre de 2021 un total de 81 notificaciones tras la administración de cerca de 9,5 millones de dosis de Spikevax®. La mayoría de los casos ocurrieron en hombres (90%), tras la segunda dosis (65%) y en la primera semana tras recibir la vacuna (79%), siendo más frecuente en edades jóvenes; la mayoría de los pacientes se recuperaron o estaban recuperándose en el momento de la notificación.
La ficha técnica y el prospecto de estas vacunas se actualizarán para recoger esta nueva información. Se recomienda a las personas que reciban esta vacuna que estén atentas a posibles signos o síntomas de miocarditis y/o pericarditis mencionadas anteriormente, y busquen atención médica inmediata si se presentasen.
- Vasculitis cutánea de pequeños vasos asociada a la vacuna ▼COVID 19 Janssen Vaccine®
Desde que se autorizó su comercialización en la UE hasta el 1 de diciembre de 2021, se han administrado casi 18,1 millones de dosis de esta vacuna en el EEE. Actualmente el uso de esta vacuna en España es irrelevante y, en consecuencia, también los nuevos casos notificados de acontecimientos adversos tras la vacunación. Dado que los nuevos datos respecto a esta vacuna serán anecdóticos, esta información no se incluirá a partir del próximo informe periódico de farmacovigilancia de estas vacunas, excepto que hubiese nueva información relevante.
En relación con los casos de la vasculitis cutánea de pequeños vasos –VCPV– (cursa con inflamación de los vasos sanguíneos de la piel que puede resultar en erupciones cutáneas, manchas rojas puntiagudas o planas debajo de la superficie de la piel o moratones), el PRAC ha concluido que es una posible reacción adversa de COVID-19 Janssen Vaccine®. La VCPV puede ser causada por infecciones, así como algunos medicamentos (incluyendo vacunas). En la mayoría de los casos se resuelve sin necesidad de tratamiento. Esta conclusión se basa en un total de 37 casos notificados mundialmente hasta finales de octubre de 2021. Algunos de ellos fueron considerados como probablemente relacionados con la vacuna al tener una relación temporal cercana a la vacunación y no se identificaron explicaciones alternativas, incluyendo 6 casos verificados por biopsia. Otros casos notificados también tenían una relación temporal con la vacunación, aunque otras causas podrían haber sido responsables. Se ha estimado que alrededor de 36 millones de dosis de esta vacuna se habían administrado en todo el mundo hasta finales de octubre de 2021. En España, hasta el 12 de diciembre de 2021, se habían registrado 4 notificaciones de VCPV tras la administración de esta vacuna; en todas ellas el paciente se había recuperado o estaba en recuperación en el momento de la notificación. Hasta esa fecha, se habían administrado cerca de 2 millones de dosis. La frecuencia de aparición de este posible efecto secundario no se conoce, ya que con los datos de notificación espontánea no se puede estimar adecuadamente.
- Parestesia asociada a la vacuna ▼Spikevax®
Desde que se autorizó su comercialización en la UE hasta el 2 de enero de 2022, se han administrado alrededor de 103 millones de dosis de esta vacuna en el EEE.
La parestesia es un trastorno de la sensibilidad que se manifiesta con sensaciones anormales sin estímulo previo, como el hormigueo. En base a la información de los ensayos clínicos y los casos notificados a nivel mundial, se ha identificado la parestesia como posible reacción adversa que puede aparecer con una frecuencia rara tras la administración de Spikevax®. Previamente ya se han descrito reacciones adversas similares para esta vacuna, como la hipoestesia (disminución de la sensibilidad de la piel).
A fecha de 30 de junio de 2021 se habían identificado 1.425 casos de parestesia a partir del tercer día desde la vacunación con Spikevax®, habiéndose administrado más de 180 millones de dosis de Spikevax® en todo el mundo. En España, hasta el 9 de enero de 2022, se han registrado 158 notificaciones de parestesias tras la administración de Spikevax®; hasta esa fecha se habían administrado cerca de 14,2 millones de dosis de esta vacuna.
- Mielitis transversa asociada a las vacunas ▼Vaxzevria® y ▼COVID Janssen Vaccine®
Desde que se autorizó la comercialización de Vaxzevria® (AstraZeneca/Oxford) en la UE hasta el 2 de enero de 2022 se han administrado alrededor de 69 millones de dosis de esta vacuna en el EEE. Igualmente, se han administrado alrededor de 18,7 millones de dosis de COVID Janssen Vaccine en el EEE desde que se autorizó su comercialización en la UE hasta el 2 de enero de 2022.
La mielitis transversa es un raro trastorno neurológico que se caracteriza por una inflamación de uno o ambos lados de la médula espinal y que a menudo daña la capa de mielina que cubre las fibras nerviosas. Puede manifestarse por debilidad en brazos y piernas, síntomas sensoriales (como hormigueo, entumecimiento, dolor o sensación de pérdida del dolor) o problemas funcionales de la vejiga o intestino, como la necesidad de orinar con más frecuencia, incontinencia urinaria, dificultad para orinar y estreñimiento.
En base a la revisión de casos de mielitis transversa notificados tras la administración de Vaxzevria® y los datos publicados en la literatura científica, el PRAC ha concluido que no se puede descartar una relación causal de mielitis transversa tras su administración, si bien el número de casos identificados es muy bajo. Se han analizado, en total, 25 casos, habiéndose estimado una exposición global de 1.400 millones de dosis. En España, hasta el 9 de enero de 2022, se ha registrado una sola notificación que cumple con los criterios diagnósticos de mielitis transversa tras la administración de Vaxzevria®. El paciente se encontraba en recuperación en el momento de la notificación. En ese periodo, se habían administrado cerca de 9,8 millones de dosis de esta vacuna.
En base a la revisión de casos de mielitis transversa notificados tras la administración de la vacuna de Janssen y los datos publicados en la literatura científica, el PRAC ha concluido que, igual que para Vaxzevria®, no se puede descartar una relación causal de mielitis transversa tras la administración de esta vacuna, aunque el número de casos identificados es muy bajo. En total se analizaron 13 casos notificados con esta vacuna, habiéndose estimado una exposición global de 33,5 millones de dosis. En España, hasta el 9 de enero de 2022, no se ha registrado ninguna notificación de mielitis transversa tras la administración de COVID-19 Janssen Vaccine®, habiéndose administrado hasta esa fecha cerca de 2 millones de dosis.
Se recomienda a las personas vacunadas buscar atención médica inmediata si tras recibir una de estas dos vacunas experimentan problemas sensoriales, debilidad o mal funcionamiento de la vejiga o del intestino, con incontinencia urinaria o estreñimiento.
Colchicina y riesgo de sobredosis mortal: medidas en Nueva Zelanda
La agencia de medicamentos de Nueva Zelanda ha elaborado una nota informativa alertando del riesgo de sobredosis mortales con colchicina (Colchicina Seid®, Colchimax®), fármaco de primera línea en el tratamiento de los ataques agudos de gota, entre otras indicaciones. Algo similar ya ocurrió en España en 2010, y motivó ciertas medidas reguladoras por parte de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS).
La agencia de medicamentos de Nueva Zelanda, MedSafe ha informado sobre una advertencia recordando al público el alto riesgo de muerte por sobredosis de colchicina y que no existen tratamientos efectivos disponibles para la intoxicación grave por colchicina (MedSafe, 2021).
La colchicina es un fármaco indicado para el tratamiento de la gota aguda cuando los medicamentos antiinflamatorios no esteroideos están contraindicados, son ineficaces o no se toleran. Se trata de un principio activo de estrecho margen terapéutico que, en sobredosis, es muy tóxico, con gran variabilidad en cuanto a la dosis letal, entre 0,5 y 0,8 mg/kg de peso. Con la separación bien definida entre las dosis terapéuticas y tóxicas, algunas guías clínicas pueden hacer referencia a esquemas de dosificación no aprobados para la colchicina en Nueva Zelanda.
Desde enero de 2016 hasta enero de 2021 el Centro Nacional de Intoxicaciones (NPC) neozelandés recibió 56 casos relacionados con intoxicación por colchicina. Las principales razones del envenenamiento fueron el comportamiento exploratorio de niños, el error terapéutico y la autointoxicación intencionada. Los profesionales de la salud deben comunicar a los pacientes la importancia de guardar los medicamentos fuera de la vista y del alcance de los niños y asegurarse de que los pacientes sepan cuándo y cómo tomar la colchicina.
En España, en agosto de 2010 se comunicaron varios casos a través de los sistemas de prevención de errores de medicación, del ISMP-España y del Sistema Español de Farmacovigilancia, junto con casos publicados en revistas en los años anteriores (AEMPS, 2010). Entonces, la AEMPS tomó varias medidas reguladoras:
- Revisar la información de las fichas técnicas y de los prospectos de los medicamentos con colchicina (Colchicina Seid® y Colchimax®, este último en asociación con dicicloverina), actualizando la posología en las indicaciones ya establecidas para la presentación de 1 mg por comprimido, como son: ataques agudos por inicio del tratamiento con movilizadores del ácido úrico, y enfermedad periódica (fiebre mediterránea familiar). El tratamiento del ataque agudo de gota se inicia con la administración de 1 mg de colchicina al primer signo de la crisis. Si el alivio del dolor no se consigue se puede administrar 0,5 a 1 mg de colchicina pasadas 1-2 h tras la primera toma. No se debe administrar más de 2 mg en 24 h, pero sí se puede continuar la administración hasta 4 días seguidos, sin superar la dosis total acumulada de 6 mg durante los 4 días.
- Incluir una advertencia en el apartado de posología de todos los medicamentos con colchicina: “La colchicina tiene un margen terapéutico estrecho y en caso de sobredosis es extremadamente tóxica. No se debe sobrepasar la dosis indicada a continuación, en ninguna ocasión, ya que puede resultar mortal.”
- Tramitar la autorización de una nueva presentación comercial de 0,5 mg por comprimido (Colchicina Seid 0,5 mg comprimidos) para facilitar la posología en el tratamiento en adultos de la pericarditis aguda y pericarditis recurrente en combinación con ácido acetilsalicílico u otros antiinflamatorios no esteroideos (AINE).
- Actualizar toda la información del fármaco relativa a interacciones, dado que cuando se asocia la colchicina con principios activos que son metabolizados o interaccionan con el sistema del citocromo P450, en particular con el isoenzima CYP3A4, o con la glicoproteína P, se pueden producir sobredosis relativas de colchicina con la dosificación adecuada; por ejemplo, incrementos plasmáticos de colchicina –hasta niveles tóxicos– al simultanear tratamientos con inhibidores de CYP3A4 o de la glicoproteína P, ya que su uso simultáneo elevará los niveles plasmáticos de colchicina e incrementará su toxicidad: antibióticos macrólidos como claritromicina, telitromicina, eritromicina, josamicina; con ketoconazol, itraconazol, fluconazol; indinavir, atazanavir, nelfinavir, ritonavir, saquinavir, amprenavir, fosamprenavir; diltiazem, verapamilo, zumo de pomelo y otros cítricos amargos. Tampoco debe utilizarse junto con ciclosporina. En casos necesarios se ajustará la dosis de colchicina.
- Recordar que solo se debe utilizar bajo prescripción médica, explicando claramente al paciente la pauta posológica con el fin de evitar errores en la dosificación.
Información de seguridad procedente de la evaluación periódica de los datos de farmacovigilancia que decide el Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC)
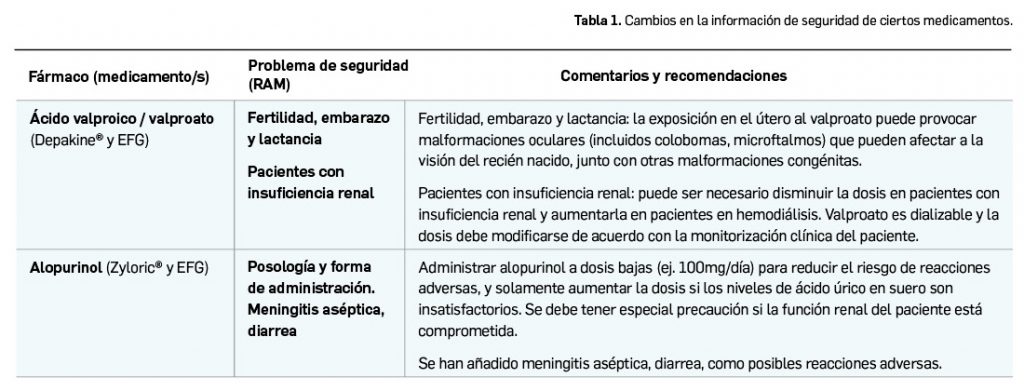
El Comité europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en la información autorizada de las fichas técnicas y de los prospectos de los medicamentos europeos por motivos de seguridad. Una vez que se revisan y evalúan los datos de los informes periódicos de seguridad (IPS; en inglés PSUR), de forma colaboradora entre todas las 27 agencias nacionales, se presentan los cambios y se acuerdan en las reuniones mensuales del PRAC. A continuación, se muestran los últimos cambios de información de seguridad acordados.
El Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en las fichas técnicas y los prospectos de los siguientes medicamentos, siendo los más importantes los que se describen en la Tabla 1, según informa la AEMPS en sus Boletines de Seguridad de Medicamentos de Uso humano de los meses de agosto y septiembre, y del mes de octubre de 2021.
Las fichas técnicas y prospectos de los medicamentos pueden consultarse en la web de la AEMPS, dentro de la sección CIMA: Centro de Información Online de Medicamentos.
Esta información que se incorpora a las fichas técnicas y prospectos de los medicamentos, supone una actualización permanente, por lo que es necesario consultar sus datos y la fecha de la actualización, al final del texto de las fichas técnicas y prospectos, cuando se consultan en la web de la AEMPS (sección CIMA: Centro de Información Online de Medicamentos).
Información importante
El Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (SEFV-H) se basa en el programa de notificación espontánea de un profesional sanitario (médico, odontólogo, farmacéutico, enfermero, otros) o de un ciudadano, de una sospecha de relación entre un medicamento (incluidos vacunas, sueros, gases medicinales, fórmulas magistrales, plantas medicinales) y un síntoma o signo adverso (reacción adversa, RAM) que manifieste el paciente o familiar (programa de tarjeta amarilla). El Real Decreto 577/2013 de Farmacovigilancia de medicamentos de uso humano (BOE núm. 179, de 27 de julio de 2013) entró en vigor el 28 de julio de 2013. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) coordina el SEFV-H. A su vez se integra en el Sistema Europeo de Farmacovigilancia que desde 1995 coordina la Agencia Europea de Medicamentos (EMA), y participa desde 1984 en el Programa Internacional de Farmacovigilancia de la OMS, junto con más de 130 países.
¿Qué notificar? Se deben notificar las sospechas de RAM:
Con medicamentos autorizados, incluidas las de aquellos que se hayan utilizado en condiciones diferentes a las autorizadas o con medicamentos extranjeros importados con autorización de la AEMPS.
Principalmente las RAM ‘graves’ (mortales, o que amenacen la vida, prolonguen o provoquen una hospitalización, causen incapacidad o sean médicamente importantes y las trasmisiones de un agente infeccioso a través de un medicamento) o RAM ‘inesperadas’ de cualquier medicamento
Con medicamentos de ‘seguimiento adicional’ (durante sus primeros 5 años desde la autorización, identificados con un triángulo negro invertido (▼) a la izquierda del nombre del medicamento en el material informativo, en el prospecto y en la ficha técnica);
ver la lista mensual de los medicamentos con “triángulo negro” en la web de la AEMPS: https://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/seguimiento_adicional.htm#lista_europea
Las que sean consecuencia de ‘errores de medicación’, que ocasionen daño en el paciente,
Las originadas por ‘interacciones’ con medicamentos, plantas medicinales, incluso alimentos (zumo de pomelo, ahumados, crucíferas, etc).
¿Cómo notificar?
No olvide notificar cualquier sospecha de RAM a su Centro Autonómico o Regional de Farmacovigilancia mediante las ‘tarjetas amarillas’. Consulte en este directorio su Centro Autonómico de Farmacovigilancia correspondiente.
MÉTODO electrónico: desde el 15 de enero de 2013 se puede notificar a través del sitio web https://www.notificaRAM.es/, y el sistema electrónico hace llegar a su centro correspondiente la notificación de sospecha de RAM. Sirve para profesionales sanitarios y para ciudadanos, en formularios diferentes. La nueva legislación europea de farmacovigilancia establece esta posibilidad para facilitar la notificación de las sospechas de RAM por la población en general.
¿Dónde conseguir tarjetas amarillas?
Consultando a su Centro correspondiente del SEFV-H. Podrá encontrar el directorio de Centros en las primeras páginas del “Catálogo de Medicamentos” y en las páginas de Internet http://www.portalfarma.com y http://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/docs/dir_serfv.pdf.
¿Dónde consultar las fichas técnicas y prospectos de los medicamentos?
En la página web de la AEMPS http://www.aemps.gob.es, seleccionando ”CIMA: Centro de Información on-line de Medicamentos de la AEMPS, Humanos”, se pueden consultar por nombre comercial o por sus principios activos. También están disponibles en la base de datos BOT PLUS.
NOTA: la mención de marcas comerciales en el texto solo tiene fines de identificación, y en absoluto se les debe asignar directamente lo descrito en el texto.
Nueva vacuna frente al herpes zóster
De cara al futuro, la prevención del herpes zóster supone un reto interesante. Por una parte, porque aunque no representa una alta mortalidad, la carga de enfermedad que conlleva puede ser un problema de salud pública y social, de cara al aumento de la edad media de la población que se espera para las próximas décadas. Además, ese aumento de la esperanza de vida va inexorablemente asociado a un mayor número de personas con comorbilidades e inmunodepresión, factores que a su vez las hacen más susceptibles a la infección. Por otro lado, porque una de sus principales complicaciones, la neuralgia postherpética (NPH), va asociada, como se verá más adelante, a una merma de la calidad de vida. La NPH representa un riesgo importante para la autonomía del paciente, acelerando en muchos casos su discapacidad y dependencia.
Estamos al comienzo de la década que la Organización Mundial de la Salud denomina como década del envejecimiento saludable (2021-2030). Por esto, estamos virando de un calendario donde las vacunas infantiles eran la clave, hacia un calendario a lo largo de la vida, donde la vacunación del adulto está cobrando un peso cada vez mayor.
El virus varicela-zóster
El virus varicela-zóster (VVZ) forma parte de la familia Herpesviridae. El mismo virus produce dos manifestaciones clínicas distintas: varicela y herpes zóster (HZ). La varicela es la infección primaria. Transcurrida esta, se acantona en los ganglios nerviosos dorsales o craneales, donde puede permanecer de forma latente durante años. La reactivación del virus es lo que se conoce como HZ.
La sintomatología inicial del HZ suele ser picor, hormigueo, o dolor intenso o punzante en el dermatoma correspondiente al ganglio donde ha estado acantonado el VVZ. Posteriormente aparecen vesículas que tardan entre 2 y 4 semanas en desaparecer y se acompañan con dolor en la zona. En el cas0 del HZ, los dermatomas que más frecuentemente son afectados son los inervados por los ganglios sensoriales espinales de T1 a L2 (> 50% de los casos) y por la rama oftálmica del ganglio trigémino (entre el 8% y el 15% de los casos). Las molestias derivadas pueden durar años.
Como ya se ha mencionado, la principal complicación del HZ es la NPH, un dolor de tipo neuropático que puede durar más de 3 meses después de la remisión de los síntomas iniciales, y que viene asociado frecuentemente con una merma importante de la autonomía y de la calidad de vida del paciente. Son muy frecuentes también las consultas que llegan a las farmacias de pacientes con medicación para la NPH, en las que se observa que dicha medicación puede no ser segura y/o efectiva.
La incidencia anual en Europa de HZ está entre 2 y 4,6 casos por cada 1.000 habitantes. Como más del 90% de la población adulta ha estado en contacto con el VVZ, potencialmente pueden desarrollar HZ. El aumento de incidencia se acentúa a medida que vamos cumpliendo años o bien ante determinadas condiciones de riesgo o inmunocompromiso. También se ha observado que el sexo (femenino) es factor de riesgo independiente de la edad.
Los individuos vacunados en la infancia también pueden desarrollar HZ, aunque se ha visto que disminuye un 69% el riesgo cuando las personas se vacunan con menos de 10 años y con las 2 dosis administradas. Para estudiar si la vacunación sistemática a los niños frente a la varicela pudiera aumentar los casos de HZ, es muy interesante estudiar los datos de Navarra, donde se vacuna de varicela con dos dosis, a los 15 meses y a los 3 años, a todos los niños desde el año 2007. Tras 14 años de vacunación en estas condiciones, podemos concluir que no hay un incremento significativo en la incidencia de HZ en la población adulta. En la gráfica de incidencia en función de la edad se puede observar claramente un punto de inflexión a partir de los 50 años, en el que los casos comienzan a aumentar con una pendiente acentuada. Se estima que el riesgo de padecer HZ a partir de 85 años es de más del 50%.
Con respecto a las hospitalizaciones, el 80,2% de las mismas por HZ y el 91,4% por NPH ocurren en mayores de 50 años.
Vacunas autorizadas frente a HZ
A fecha de publicación de este artículo, en España existen dos vacunas comercializadas frente al HZ, Zostavax® (ZLV), del laboratorio MSD, autorizado por la Comisión Europea en mayo de 2006, y Shingrix® (HZ/su), de GSK, autorizado en Europa desde marzo de 2018. Las dos vacunas están indicadas para prevenir el HZ y la NPH en personas de 50 años o más.
Zostavax® contiene virus vivos atenuados de la varicela-zóster de la cepa Oka/Merck, concretamente más de 19.400 Unidades formadoras de placa (UFP) producidos en células diploides humanas (MRC-5). Estos virus también están presentes en la vacuna de la varicela Varivax®, pero con una concentración 14 veces superior en el caso de Zostavax®. La pauta es una única dosis y, al tratarse de virus vivos, está contraindicada en pacientes inmunodeprimidos.
Shingrix® es una vacuna producida por técnicas de recombinación de ADN de subunidad adyuvada. Contiene glicoproteína E del virus de la varicela zóster producida en células de Ovario de Hámster Chino (OHC) mediante tecnología del ADN recombinante como componente antigénico viral (50 µg). Está adyuvada con AS01B que contiene: extracto de la planta Quillaja saponaria Molina, fracción 21 (QS-21), 3-O-desacil-4’-monofosforil lípido A (MPL) de Salmonella minnesota y la glicoproteína antes mencionada (gE). La vacuna está diseñada para inducir respuestas inmunes humorales y celulares antígeno-específicas en individuos con inmunidad preexistente frente al VVZ. Al tratarse de una vacuna inactivada que a priori genera menos respuesta, se añade el adyuvante con el sistema AS01B, que aumenta la respuesta inmunitaria y la persistencia en el tiempo. La pauta son dos dosis, pudiéndose administrar la segunda entre los 2 y los 6 meses de la primera, aunque también existe una pauta acelerada que permite un mes entre dosis. Dicho esto, lo recomendable es separar las dosis 2 meses, buscando la inmunización completa lo antes posible ya que mejora la efectividad de la vacuna, como se verá en el siguiente epígrafe. Esta vacuna aporta una importante novedad: puede administrarse en pacientes en situación de inmunocompromiso a partir de los 18 años de edad.
Eficacia y efectividad de ambas vacunas
Para estimar la eficacia de la vacuna ZLV se realizaron dos estudios. En el primero, llamado SPS, participaron 38.546 adultos de 60 años y mayores que habían pasado la varicela. Se concluyó que la eficacia de la vacuna fue de un 51,3% en la prevención del HZ y de un 66,5% en la prevención de la NPH. También se observó que la eficacia de la vacuna disminuye considerablemente a medida que aumenta la edad de vacunación. En el segundo estudio, denominado ZESP, participaron 22.439 pacientes de entre 50 y 59 años que también habían padecido la varicela. Aquí la eficacia frente a HZ fue del 70%. En un ensayo de extensión posautorización del estudio SPS se hizo un seguimiento de los pacientes vacunados y su protección a lo largo del tiempo, y los resultados obtenidos permiten concluir que la eficacia de la vacuna disminuye a lo largo de los años, más con referencia al HZ que a la NPH.
Con respecto a la efectividad, en 2013 se introdujo la vacunación frente a HZ en Reino Unido a los 70 años con captación gradual de las personas entre 71 y 79 años. La efectividad observada frente a la incidencia de HZ rondó entre el 62% y 64%, mientras que frente a la NPH estuvo entre el 81% y el 88%. Estos porcentajes fueron similares a los obtenidos en otros países. También se ha observado que, igual que ocurre con la eficacia, la efectividad disminuye con el tiempo. En este sentido, se observó en los estudios que esta efectividad era similar en los distintos grupos de edad analizados.
En el caso de HZ/su, también se han desarrollado dos estudios principales: ZOE-50 y ZOE-70. En el primer estudio, ZOE-50, participaron 16.145 personas mayores de 50 años con contacto previo con VVZ. La eficacia global de la vacuna frente a HZ fue del 97,2%. Por su parte, en ZOE-70, en el que participaron 14.816 pacientes mayores de 70 años, se obtuvo una eficacia del 89,8%. El análisis agrupado de los dos estudios reveló una eficacia del 91,3%-91,4% frente a HZ y del 88,8% frente a NPH en personas de 70 años y mayores. Además, este segundo estudio valoró el impacto de las complicaciones relacionadas con la enfermedad y concluyó que producía una reducción del 93,7% en mayores de 50 años y de un 91,6% en mayores de 70.
Con respecto a la efectividad, Sun y colaboradores observaron un 85,5% de efectividad en pacientes con una edad media de 65 años, mientras que Izurieta y colaboradores observaron una efectividad del 70,1% con una edad media de 74. Estos autores confirmaron también que un retraso en la segunda dosis no interfiere en la eficacia vacunal, pero se señala que es importante completar la pauta de vacunación, ya que la efectividad de una dosis es del 56,9%.
Como ya hemos comentado en el apartado anterior, esta vacuna aporta la importante novedad de poderse administrar en personas en situación de inmunocompromiso. Un estudio llevado a cabo en receptores de trasplantes autólogos de células madres hematopoyéticas resultó en una eficacia frente a HZ de 68,2% y frente a NPH de 89,3%. Por otro lado, en personas con hemopatías malignas se obtuvo una eficacia frente a HZ de 87,2%. Con respecto a la efectividad, el estudio de Izurieta y colaboradores antes mencionado muestra cómo se comporta HZ/su en pacientes con inmunocompromiso, en quienes se reporta una efectividad vacunal del 64,1%.
Estos resultados son esperanzadores ya que se observa que en estos pacientes la vacuna produce no solo inmunidad humoral, sino lo que es más importante, inmunidad mediada por células incluso cuando la vacuna se administra entre ciclos de quimioterapia, situación en la que el paciente sufre mayor estado de inmunocompromiso.
Se ha realizado un estudio, entre abril de 2016 y 2018, de seguimiento en sujetos vacunados 10 años antes en República Checa, Alemania y Suecia, donde se puso de manifiesto la persistencia de respuesta inmunitaria tanto humoral como celular. En base a estos datos y aplicando un modelo matemático, se ha estimado la persistencia a largo plazo, llegando a la conclusión que la respuesta inmunitaria en pacientes de 60 años o más persiste, al menos, 10 años y los modelos matemáticos predicen que esta inmunidad podría mantenerse incluso durante 20 años o más.
Podemos concluir, por tanto, que la eficacia de la vacuna HZ/su frente a HZ y NPH es muy superior a la obtenida por la vacuna ZVL. La eficacia y efectividad de la vacuna ZVL disminuye de manera pronunciada a lo largo de los años, siendo también menor a mayor edad, mientras que la de la vacuna HZ/su no es dependiente de la edad. Además, la vacuna HZ/su reduce de forma significativa la incidencia de HZ y de NPH en pacientes inmunodeprimidos.
Seguridad de las vacunas frente a HZ
En los ensayos clínicos ZEST y SPS se evaluó la seguridad de la vacuna ZVL, que se caracterizó por las siguientes reacciones adversas más frecuentes: dolor en el lugar de inyección, seguido de dolor de cabeza, erupción cutánea y dolor en una extremidad. Estas reacciones fueron de intensidad leve, mientras que las notificadas como graves fueron similares entre el grupo control y el vacunado. Los eventos adversos fueron más frecuentes en los pacientes de entre 50 y 60 años, y también se notificaron con más frecuencia en las administraciones subcutáneas frente a las intramusculares. Al tratarse de una vacuna de virus vivo, fue de especial interés analizar las reacciones de tipo herpetiforme que pudieran ser consecuencia de la misma vacuna, y se observó que la frecuencia de este tipo de exantemas fue similar en el grupo placebo y en el grupo control. Con esta vacuna podemos analizar lo recogido en farmacovigilancia en vida real: se han registrado casos de exantemas de tipo herpetiforme y de enfermedad diseminada en los que efectivamente se aisló la cepa vacunal, aunque con frecuencia extremadamente baja. Tanto en estos casos como en los de muerte asociada con varicela o con HZ, ocurrieron en personas inmunocomprometidas, lo que refuerza la contraindicación de la vacuna en este tipo de pacientes.
En los ensayos clínicos ZOE-50 y ZOE-70 se analizó también la seguridad de la vacuna HZ/su. La reacción adversa más frecuente fue dolor en el lugar de la inyección, seguida de dolor muscular, fatiga y dolor de cabeza. La mayoría de los síntomas se describieron como de intensidad moderada y su duración estuvo entre 2 y 3 días. Las reacciones adversas fueron similares en la primera y la segunda dosis. No se identificaron diferencias en referencia a reacciones adversas graves, fatales o de tipo inmune entre el grupo placebo y el grupo que recibió la vacuna. Por otra parte, cinco estudios que analizaron la reactogenicidad en pacientes mayores de 18 años con situación de inmunocompromiso (receptores de trasplante de células autólogas, VIH, tumores sólidos, cáncer hematológico y trasplante renal) recogieron resultados similares.
Con respecto a la coadministración con otras vacunas, las vacunas frente a HZ se pueden administrar junto con la de gripe estacional, vacuna antineumocócica polisacárida conjugada 23-valente y difteria-tétanos-tosferina acelular sin que se hayan observado diferencias en la respuesta inmunitaria o hayan alterado el perfil de seguridad. Al igual que en otros casos de coadministración en vacunas, cuando esto se lleve a cabo se hará en lugares anatómicos diferentes y con distintas inyecciones.
Un meta-análisis que compara la seguridad de las dos vacunas concluye que HZ/su se asocia con una mayor reactogenicidad tanto local como sistémica en comparación con la vacuna ZLV, teniendo en cuenta que la manera de medir las reacciones adversas y la definición de las mismas fue diferente en cada una de las vacunas que aquí analizamos. En cualquier caso, ambas vacunas son seguras, y las reacciones adversas graves en los dos casos fueron similares en el grupo placebo y en el de vacunados.
Recomendaciones de vacunación frente al herpes zóster en España
En 2018 se revisó la vacunación en grupos de riesgo y se recomendó la utilización de HZ/su en personas con trasplante de progenitores hematopoyéticos, de órgano sólido, pacientes con VIH y en tratamiento con fármacos anti-JAK, aunque no pudo llevarse a cabo por falta de existencias de las vacunas. Ya el pasado año se modificaron las recomendaciones teniendo en cuenta, ahora sí, la disponibilidad de la vacuna. Se incluyeron, por tanto, nuevos grupos: hemopatías malignas y tumores sólidos en tratamiento con quimioterapia. Además de vacunar a estos grupos de riesgo antes mencionados, la recomendación clave de cara al envejecimiento saludable apuesta por vacunar a la cohorte de 65 años a partir de este año 2022, vacunando, además, siempre que haya dosis disponibles, al menos a otra cohorte cada año empezando por la que cumple 80 años, y bajando así hasta llegar a la primera cohorte que se vacunó los 65 años.
Calendarios de vacunaciones del adolescente de la SEMA
La Sociedad Española de Medicina de la Adolescencia (SEMA) ha elaborado un documento de consenso titulado “Calendario de Vacunaciones del Adolescente”, donde se expone el calendario de vacunaciones del Consejo Interterritorial del Sistema Nacional de Salud y se detalla el que la Asociación Española de Pediatría (AEP), a través de su Comité Asesor de Vacunas (CAV-AEP), recomienda para este grupo de edad.
Actualmente, el calendario del adolescente, dentro del calendario común de vacunaciones a lo largo de toda la vida del Consejo Interterritorial de Salud, recoge las siguientes vacunas: vacuna antimeningocócica tetravalente ACWY a los 12 años (que sustituye desde 2019 a la antigua antimeningocócica C), vacuna de la varicela con dos dosis a los individuos que no hayan pasado la enfermedad ni estén correctamente vacunados, vacuna frente al virus del papiloma humano (VPH) a las niñas a los 12 años, vacuna de tétanos y difteria (Td) a los 14 años, y vacuna frente al SARS-CoV-2 si no han sido vacunados. Además, se ofrece rescate de estas y otras vacunas a niños que lleguen a la adolescencia con el calendario vacunal incompleto, grupos de riesgo y determinadas situaciones.
La Asociación Española de Pediatría, por su parte, incluye, además, las siguientes recomendaciones: vacuna difteria, tétanos y tosferina acelular (Tdpa) a los 12-14 años, en vez de solo TD (esta vacuna ya se administra en el Principado de Asturias) y vacuna VPH también en chicos, y preferentemente a los 12 años (antes del debut de las relaciones sexuales). Además, se propone ir introduciendo otras recomendaciones, como ya se hace en otros países o regiones: vacuna frente a la hepatitis A a los 10 años (se administra en Cataluña, Ceuta y Melilla), vacuna antigripal tetravalente en campaña, y vacuna antimeningocócica B a los 14-18 años con pauta de dos dosis.
La vacunación del adolescente representa un reto por las características propias de la adolescencia: acuden menos a la consulta que los niños, y por tanto las coberturas son menores; a partir de los 14-16 años se produce el tránsito de la pediatría a la atención primaria; viajan mucho por intercambios, formación o turismo; y actúan como si no existiesen los riesgos. Como profesionales, la formación por tanto es prioritaria, ya que es clave aprovechar cada visita. No se puede dejar pasar oportunidades para hablar de vacunas o vacunar a los adolescentes por falsas contraindicaciones o falta de conocimiento. También es muy importante explorar las preocupaciones y dudas tanto de padres como adolescentes.
Aumento de casos de enfermedad meningocócica grupo B en Inglaterra
La enfermedad meningocócica se manifiesta generalmente con meningitis, septicemia o una combinación de las dos. Los bebés y mayores son los que más riesgo tienen frente a la infección, seguidos de adolescentes y adultos jóvenes, que son los portadores y transmisores principales. El uso de mascarillas, la distancia social y los confinamientos que han tenido lugar durante la pandemia por COVID-19 han hecho que la circulación natural y transmisión de virus y bacterias haya descendido. Pero la otra cara de la moneda es que esta disminución también produce un descenso de la inmunidad comunitaria, haciendo que aumente el número de personas susceptibles. Es lo que los expertos llaman “deuda inmunológica”.
La incidencia de la enfermedad meningocócica invasiva en Inglaterra disminuyó de 1,93/100.000 personas (1.016 casos) en 2010/11 hasta 0,95/100.000 (530 casos) en 2018/19 y 0,74/100.000 en 2019/20 (419 casos). Durante el cierre nacional por la pandemia (abril-agosto de 2020), la incidencia fue un 75% más baja que durante abril-agosto de 2019. En julio de 2021 se retiraron las medidas de contención de la COVID-19 y colegios, institutos y universidades inglesas volvieron a abrir sus puertas. Entre septiembre y noviembre de 2021, los casos de enfermedad meningocócica invasiva grupo B aumentaron en adolescentes y adultos jóvenes superando incluso los niveles previos a la pandemia.
El serogrupo y la distribución por edad de estos casos sugiere que los programas de vacunación meningocócica mantienen tasas bajas de enfermedad de los grupos C/W/Y (los adolescentes ingleses se vacunan frente a estos grupos a los 13-14 años); sin embargo, la baja inmunidad contra las cepas del grupo B y la alta transmisión de meningococo entre adolescentes y adultos jóvenes han dado como resultado el regreso de la enfermedad del grupo B, particularmente en estudiantes universitarios. Es, por tanto, probable que veamos esta y otras infecciones en nuestros pacientes después de la pandemia, así que debemos estar preparados.
Obesidad y sobrepeso: semaglutida mejora los resultados de liraglutida
Un reciente ensayo de fase 3 es el primer estudio comparativo entre estos dos análogos de GLP-1 para el tratamiento del sobrepeso o la obesidad y ha revelado mejoras notables con el primero: a la semana 68 redujo el peso en un -16% frente a un -6% con liraglutida. La probabilidad de que los pacientes alcanzaran reducciones de peso de ≥ 10% y de hasta ≥ 20% fue al menos 6 veces mayor con semaglutida, si bien la incidencia de eventos adversos gastrointestinales se mantuvo en un nivel similar. Su mejor pauta posológica lo convierte en un interesante candidato para el tratamiento de estas patologías.
Cuando se habla de fármacos antidiabéticos, se debe tener presente que, además de su efecto hipoglucemiante, se trata de fármacos que ejercen otras acciones farmacológicas secundarias beneficiosas, por ejemplo, sobre el sistema cardiovascular. Si de forma específica nos centramos en los análogos del péptido similar al glucagón o GLP-1 (grupo del que forman parte dulaglutida, exenatida o lixisenatida), también se ha probado que liraglutida es eficaz en la reducción del peso.
De hecho, este fármaco está autorizado, además de su indicación en diabetes mellitus tipo 2, para el tratamiento –en combinación con una dieta baja en calorías y un aumento de la actividad física– de la obesidad (IMC ≥ 30 kg/m²) o bien del sobrepeso (IMC entre 27-30 kg/m²) asociado a al menos una comorbilidad relacionada con el peso (prediabetes o diabetes mellitus tipo 2, hipertensión, dislipidemia o apnea obstructiva del sueño).
El último fármaco en incorporarse a este grupo ha sido semaglutida (comercializado en España en 2019). Se divulgan ahora los resultados de un ensayo clínico multicéntrico, comparativo y abierto, de fase 3 y de 68 semanas de duración, que ha investigado su eficacia y seguridad cuando se usa para el control del peso en pacientes obesos o con sobrepeso. El estudio incluyó a un total de 388 pacientes no diabéticos con IMC ≥ 30 kg/m2 o bien ≥ 27 kg/m2 con al menos una comorbilidad relacionada con el peso (media de edad de 49 años, 78% mujeres, peso promedio de 105 kg e IMC promedio de 38 kg/m2), quienes fueron aleatorizados (3:1:3:1) a recibir 2,4 mg de semaglutida una vez a la semana (n= 126) o 3 mg de liraglutida diarios (n= 127), en ambos casos por vía subcutánea, con un grupo placebo equivalente (n= 85 en los dos grupos), y siempre en combinación con dieta y actividad física. El 94% de los pacientes completó el estudio, y el 80% cumplió el tratamiento durante todo el periodo de estudio.
Los resultados obtenidos al finalizar apuntan a una reducción de peso significativamente mayor con semaglutida que con liraglutida. Así, el cambio medio en el peso corporal (variable primaria) fue de -15,8% con semaglutida frente a -6,4% con liraglutida (diferencia de -9,4 puntos porcentuales; p< 0,001); el cambio de peso medio con placebo fue de solo -1,9%, posible reflejo de que en todos los brazos del estudio se recomendaron cambios dietéticos y de actividad física. Frente a liraglutida, los pacientes tratados con semaglutida tenían una probabilidad notablemente incrementada (p< 0,001) de alcanzar reducciones de peso de ≥ 10% (71% vs. 26%; OR= 6,3), de ≥ 15% (56% vs. 12%; OR= 7,9) y de ≥ 20% (39% vs. 6%; OR= 8,2), que se consideraron entre las variables secundarias confirmatorias. Además, la proporción de participantes que discontinuó el tratamiento por cualquier motivo fue menor en el brazo de semaglutida (14% vs. 28%). En términos de seguridad, cabe destacar que la incidencia de eventos adversos gastrointestinales fue similar con ambos análogos de GLP-1 (84% vs. 83%).
En definitiva, parece previsible que en un futuro a corto plazo se pueda autorizar la semaglutida para el control del peso en asociación a cambios de dieta y una mayor actividad física, teniendo un régimen posológico más favorable que liraglutida.