Sistema español de farmacovigilancia de medicamentos: Informe anual de 2025
 Nº492
Nº492

La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha hecho público su informe anual de actividades del Sistema Español de Farmacovigilancia de medicamentos de uso Humano (SEFV-H) en 2025.
- Un incremento anual del 8,9 % en el volumen de notificaciones espontáneas recibidas de casos de reacciones adversas a medicamentos. Total 40 990 casos.
- Las sospechas de reacciones adversas más notificadas fueron de nuevo los trastornos gastrointestinales, los de la piel y tejido subcutáneo y del sistema nervioso.
- Durante 2025, el SEFV-H ha publicado: a) manual de uso de www.notificaRAM.es; b) guía de codificación del SEFV-H; y c) guía de detección de señales del SEFV-H.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha publicado el informe anual de actividades del Sistema Español de Farmacovigilancia de medicamentos de uso Humano (SEFV-H), correspondiente al ejercicio de 2025 (AEMPS, 2026). La AEMPS, como órgano coordinador de las actividades del SEFV-H, según el artículo 7.15 del Real Decreto 1275/2011, reúne datos, analiza e informa de las actividades anuales (BOE, 2011).
En el informe anual se destacan los aspectos más relevantes de los casos de sospechas de reacciones adversas a medicamentos (RAM) y acontecimientos adversos ocurridos tras la vacunación notificados en este período. En el caso de vacunas son los que se conocen como ESAVI, o “Eventos Supuestamente Atribuibles a Vacunación e Inmunización”, que se definen como cualquier ocurrencia médica (cualquier signo desfavorable o involuntario, hallazgo de laboratorio anormal, síntoma o enfermedad) que sigue a la inmunización y que no necesariamente tiene una relación causal con el uso de una vacuna, es decir, se trata de una sospecha que debe ser investigada y aplicar una metodología de análisis de casos para establecer si existe la posible relación causal con la vacuna, o es un hecho casual o coincidente en el tiempo con la inmunización.
En 2025, se recibieron y registraron 40 990 notificaciones de sospechas de RAM y ESAVI en el SEFV-H, lo que supone un aumento del 8,9 % respecto al año anterior. El 48,7 % de ellas fueron comunicadas directamente al SEFV-H a través de los 17 centros autonómicos; otro 51 % procedieron de la industria farmacéutica (recibidas de profesionales sanitarios y pacientes) y el 0,7 % de notificaciones procedían de la revisión de casos publicados en la literatura científica (MLM, del inglés Medical Literature Monitoring). Cabe destacar que un mismo caso puede haber sido comunicado por más de una vía, por ejemplo, se puede notificar un caso por parte del médico de urgencias, por el farmacéutico de cabecera y por el paciente o familiar. Esta posibilidad de “duplicados”, es una de las tareas de los centros autonómicos.
En cuanto a la “gravedad”, de los 40 990 casos recibidos, 13 214 (32,2 %) fueron “graves” (mortal, ingresa o prolonga hospitalización, produce incapacidad, defecto congénito o médicamente relevante), un porcentaje muy similar al observado en 2024 (32,4 %). Entre las notificaciones recibidas por el SEFV-H, el 35,7 % fueron “casos graves”, frente al 28,7 % de las notificaciones remitidas por la industria farmacéutica.
En 2025, la tasa de notificación global se estimó en 41 casos por cada 100 000 habitantes. Las sospechas de RAM y ESAVI más notificadas durante el año se centraron, como el pasado año, en trastornos gastrointestinales, de la piel y del tejido subcutáneo, y del sistema nervioso.
Manual de publicados en 2025
El SEFV-H, compuesto por los 17 centros autonómicos de farmacovigilancia y coordinado por la AEMPS, ha elaborado y publicado en su página web, en 2025, tres documentos de interés:
- El “Manual de uso de NotificaRAM.es”, es una guía práctica para la utilización del formulario electrónico de notificación de sospechas de RAM y ESAVI, dirigida tanto a profesionales sanitarios como a la ciudadanía.
- La “Guía de codificación del SEFV-H” aborda aspectos relacionados con la codificación de casos de sospechas de RAM y ESAVI que pueden generar dudas y para los que se considera necesario establecer criterios consensuados que faciliten el análisis y la evaluación de las notificaciones.
- Finalmente, la “Guía para la detección de señales del SEFV-H” que describe la metodología utilizada por el SEFV-H para la identificación de señales, entendidas como posibles nuevos riesgos de los medicamentos o cambios en los ya conocidos.
El motivo de la publicación de estos documentos es fomentar la notificación por parte de los profesionales sanitarios y la ciudadanía, y compartir criterios y aspectos de interés para su correcta codificación y posterior análisis.
"Señales" en el SEFV-H
Toda esta información reunida contribuye a mejorar la detección de señales de farmacovigilancia y a reforzar la vigilancia continua de la seguridad de los medicamentos. Entre los 19 971 casos notificados directamente al SEFV-H, un 12,8 % de ellos describía reacciones adversas “desconocidas”, para el medicamento que se consideró sospechoso, tomando como referencia su ficha técnica. Además, las notificaciones que fueron a la vez “graves” y “desconocidas” (no estaban descritas en su ficha técnica) representaron un 5,7 %. El análisis de estos casos resulta especialmente relevante para la generación de señales de farmacovigilancia, entendidas como potenciales nuevas reacciones adversas que requieren investigación adicional. Así, durante 2025, el Comité Técnico del SEFV-H ha identificado y validado nuevas “señales” que se han presentado en el comité europeo de FV, el PRAC (del inglés, Pharmacovigilance Risk Assessment Committee), en sus reuniones mensuales. Por ejemplo, las “señales” identificadas en el SEFV-H, a partir de las notificaciones espontáneas realizadas por los profesionales sanitarios y la ciudadanía españoles, se describen en la siguiente tabla (Tabla 2).
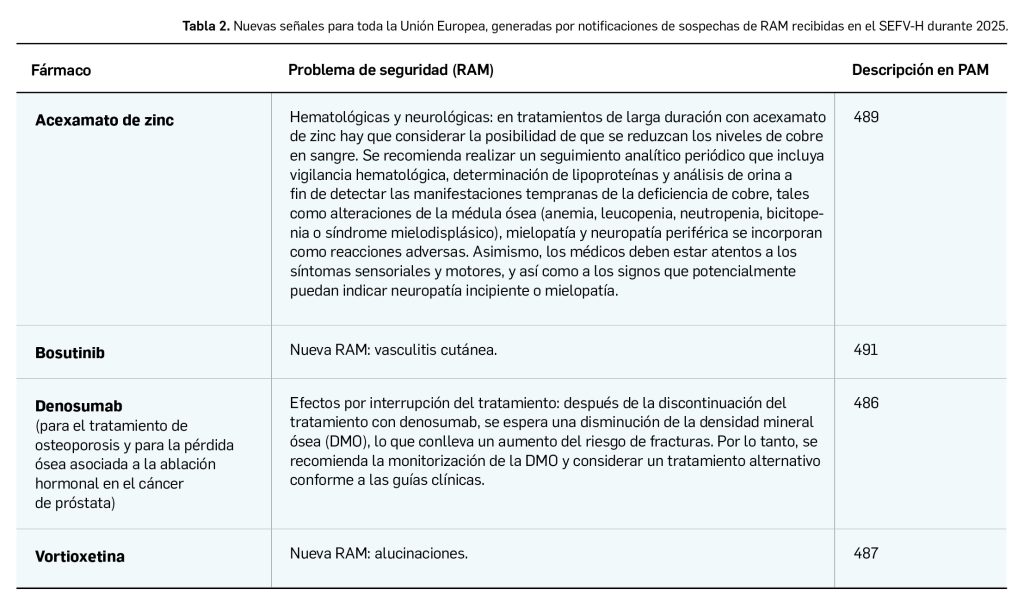
La lectura de los datos anteriores (Tabla 2) motiva enviar un agradecimiento amplio por la participación y colaboración de los profesionales sanitarios españoles, en particular, y de los ciudadanos en general, en la notificación espontánea de sospechas de RAM. EL SEFV-H EN EL ENTORNO EUROPEO
El SEFV-H en el entorno europeo
Todo esto sucede en el contexto del marco europeo, ya que el SEFV-H, coordinado por la AEMPS, es una parte de la Red Europea de Regulación de Medicamentos, EMRN (del inglés, European Medicines Regulatory Network). Tal como la EMA refleja en su Informe anual de 2025, recientemente publicado (EMA, 2026), el volumen de notificaciones en la base de datos europea EudraVigilance (EV), se ha incrementado un 0,5 % comparado con 2024, el número de notificaciones recibidas (ICSR, del inglés Individual Case Safety Report), hasta un total de 1,8 millones de ICSR, tanto de los países del espacio económico europeo (EEE) como de países de fuera del EEE. Las notificadas desde el EEE se incrementaron un 1 %, y disminuyó un 5 % la notificación de casos de “RAM no graves” (EMA, 2026).
Los 40 990 casos españoles recibidos en el SEFV-H se integraron en el total acumulado de 17 347 873 casos individuales que hay en EV, en el módulo de posautorización, EV-PM (del inglés EudraVigilance-Postmarketing Module), a finales de 2025.
"Señales" en la Unión Europea
Teniendo en cuenta que el objetivo principal de los sistemas de farmacovigilancia es la identificación de “señales”, en el informe de la EMA se detallan los datos siguientes relativos (EMA, 2026):
El equipo de la EMA, para la gestión de señales, revisaron un total de 1 201 señales potenciales de seguridad (es decir, pares de fármaco y RAM obtenidos mediante el análisis de la base de datos EudraVigilance, la literatura médica o la información recibida de otras autoridades reguladoras), relacionadas con 995 principios activos de medicamentos de registro centralizado europeo, lo que representa un 4 % menos que en 2024.
Un total de 60 señales se remitieron al PRAC para su análisis inicial, priorización y evaluación en 2025. De estas 60, 33 fueron validadas por la EMA y 27 por las autoridades nacionales competentes durante la vigilancia continua de la seguridad mediante el análisis de informes de monitorización de reacciones adversas, casos de ICSR, literatura médica y otros datos de seguridad.
Veintiséis de las señales evaluadas (43 %) dieron lugar a una recomendación para actualizar la información del medicamento para pacientes (prospectos) y profesionales sanitarios (fichas técnicas), proporcionando así una guía actualizada sobre el uso seguro y eficaz de los medicamentos afectados. Para 12 señales (20 %), se consideró suficiente continuar con la vigilancia rutinaria de la seguridad del medicamento. La evaluación de 21 señales (35 %) seguía en curso a finales de 2025, incluidas 13 mediante un procedimiento de seguimiento de señales y 8 como parte de los próximos PSUR. Una señal condujo a un procedimiento de arbitraje (referral) con la vacuna frente al virus Chikungunya, y otra resultó en una DHPC, la de clozapina, dado que el resultado fue la actualización de la información (ficha técnica y prospecto) y la DHPC enviada (EMA, 2026).
El año 2025 también ha marcado un hito en materia de farmacovigilancia: finalizó el programa piloto iniciado en febrero de 2018, sobre identificación de nuevas señales en farmacovigilancia, realizado por los laboratorios titulares de la autorización de comercialización (TAC), a partir de una “lista piloto” de fármacos y combinaciones de sustancias activas.
El 22 de julio de 2025, la Comisión Europea publicó el Reglamento de Ejecución (UE) 2025/1466. Este reglamento modifica el Reglamento de Ejecución (UE) n.º 520/2012 relativo a la realización de actividades de farmacovigilancia, previsto en el Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo y en la Directiva 2001/83/CE del Parlamento Europeo y del Consejo. Entre otras actualizaciones, este reglamento modifica los requisitos en lo que respecta a la monitorización de la base de datos EudraVigilance y elimina la obligación de que los TAC presenten señales validadas a la EMA y a las autoridades nacionales competentes como notificaciones de señales independientes.
Como consecuencia de las modificaciones del Reglamento de Ejecución 520/2012, ha finalizado el programa piloto iniciado en febrero de 2018 para que los TAC monitorizaran EudraVigilance para determinados principios activos. La EMA emitió una comunicación y un anuncio para indicar la finalización del programa piloto.
Se ha publicado un documento específico de preguntas y respuestas para ayudar a los titulares de autorizaciones de comercialización a adaptarse a estas nuevas obligaciones, proporcionándoles orientación práctica. En 2026, el Módulo IX de las Buenas Prácticas de Farmacovigilancia, GVP (del inglés, Good PharmacoVigilance Practices) sobre Gestión de Señales se actualizará para garantizar su conformidad con el nuevo marco legal del nuevo Reglamento de Ejecución (EMA, 2026).
Todos estos cambios se llevarán a cabo en el marco de los acuerdos de acceso de los laboratorios TAC a sus datos en la base de datos EudraVigilance, el conocido como “Eudravigilance Access Policy (version 5)”. Así como el marco normativo europeo de protección de datos personales, en consonancia con las medidas acordadas tras la auditoría del European Data Protection Supervisor (EDPS), y que la EMA publicó en julio de 2025 como el Anexo II del GVP-Módulo VI: Enmascaramiento de datos personales en los informes individuales de seguridad de casos presentados a EudraVigilance (EMA & HMA, 2025). Este anexo al Módulo VI de las GVP (Buenas Prácticas de Farmacovigilancia) se elaboró en colaboración con la Red de la UE y el Grupo de Trabajo de Expertos de EudraVigilance. En conclusión, nuevos pasos en el trabajo colaborativo europeo entre las agencias reguladoras y los titulares de autorización de comercialización (EMA, 2026).
Recomendaciones para los profesionales sanitarios
Como recomendación, después de agradecer la participación de los profesionales sanitarios y del resto de ciudadanos en las actividades del SEFV-H, debemos recordar la iniciativa de la AEMPS, la campaña denominada “Cómo notificar las sospechas de reacciones adversas de los medicamentos”, con el objetivo de fomentar la participación activa de profesionales sanitarios y ciudadanía en la notificación de sospechas de reacciones adversas a los medicamentos, del pasado 27 de noviembre de 2024, que se muestra en la web. Todo ello se puede resumir en el lema, “Tu notificación importa”. Agradecimiento por la colaboración (AEMPS, 2024).
Bibliografía
-
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). La AEMPS publica el Informe Anual del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano. Referencia: MUH (FV), 01/2026. 2026. Disponible en: https://www.aemps.gob.es/informa/la-aemps-publica-el-informe-anual-del-sistema-espanol-de-farmacovigilancia-de-medicamentos-de-uso-humano-3/ (consultado 26 de marzo de 2026).
-
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Campaña para incentivar la notificación de sospechas de reacciones adversas a los medicamentos. 2024. Disponible en: https://www.aemps.gob.es/informa/campana-para-incentivar-la-notificacion-de-sospechas-de-reacciones-adversas-a-los-medicamentos/ (consultado 27 de marzo de 2026).
-
- Boletín Oficial del Estado (BOE). Real Decreto 1275/2011, de 16 de septiembre, por el que se crea la Agencia estatal “Agencia Española de Medicamentos y Productos Sanitarios” y se aprueba su Estatuto. BOE núm. 229, de 23 de septiembre de 2011. Disponible en: https://www.boe.es/diario_boe/txt.php?id=BOE-A-2011-15044 (consultado 26 de marzo de 2026).
-
- European Medicines Agency (EMA) & Heads of Medicines Agencies (HMA). Guideline on good pharmacovigilance practices (GVP). Module VI Addendum II–Masking of personal data in individual case safety reports submitted to EudraVigilance. Ref.: EMA/178902/2025. 2025. Disponible en https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-good-pharmacovigilance-practices-gvp-module-vi-addendum-ii-masking-personal-data-individual-case-safety-reports-submitted-eudravigilance_en.pdf (consultado 27 de marzo de 2026).
-
- European Medicines Agency (EMA). 2025 Annual Report on EudraVigilance for the European Parliament, the Council and the Commission. Reporting period: 1 January to 31 December 2025. EMA/371010/2025- Noted. Management Board meeting of 12 March 2026. 2026. Disponible en: https://www.ema.europa.eu/en/documents/report/2025-annual-report-eudravigilance-european-parliament-council-commission_en.pdf (consultado 27 de marzo de 2026).
