SEVeM, un sistema contra los medicamentos falsificados
 Nº489
Nº489

La entrada de medicamentos falsificados en la cadena de suministro legal es una amenaza grave para la salud pública. Para combatirla, la Unión Europea estableció un marco normativo robusto que exige la implementación de sistemas de verificación en todos los Estados miembros.
Este marco normativo de la Directiva 2011/62/UE (conocida como Directiva sobre Medicamentos) fue desarrollado a través del Reglamento Delegado (UE) 2016/161, estableciendo así la obligatoriedad, desde el 9 de febrero de 2019, de incluir dos dispositivos de seguridad en el embalaje exterior de la mayoría de los medicamentos de prescripción (y algunos sin receta con riesgo de falsificación).
Estos dispositivos de seguridad, que son elementos fundamentales que hacen posible la verificación, consisten en un número de identificador único (IU) y un dispositivo contra manipulaciones (DCM).
Para que la verificación del IU funcione a nivel europeo, se creó una estructura supranacional, una organización que coordina el sistema a nivel europeo: European Medicines Verification Organisation (EMVO). A su vez, cada Estado miembro debe tener su propio sistema nacional conectado con el nodo europeo (EU Hub); en España se creó el Sistema Español de Verificación de Medicamentos (SEVeM).
El repositorio nacional de SEVeM está conectado al EU Hub de EMVO para recibir la información de los fabricantes, y también para permitir verificaciones transfronterizas si fueran necesarias. A su vez, los diferentes agentes del sistema se conectan al repositorio nacional. Actualmente, esa conexión es directa en el caso de los laboratorios titulares de autorización de comercialización o importadores paralelos, los almacenes de distribución, los servicios de farmacia de los hospitales y/o clínicas privadas y las farmacias militares. Los servicios de farmacia de hospitales del Sistema Nacional de Salud aún no están conectados directamente con el sistema y se conectarán, previsiblemente, a través del nodo SNSFarma. Las farmacias comunitarias se conectan a través de Nodofarma Verificación.
Cuando se realiza una verificación/desactivación y el sistema no identifica correctamente el IU, se genera una alerta. Se está implementando un sistema diseñado para investigar y resolver estas alertas, involucrando a todos los agentes que interactúan con el sistema: desde el titular del medicamento hasta la farmacia y, si procede, a SEVeM y a las autoridades sanitarias. Su objetivo es facilitar la gestión de las diferentes alertas generadas en el sistema, haciendo posible registrar las investigaciones que permiten diferenciar errores técnicos o de procedimiento de posibles casos de falsificación.
A nivel global, el sistema de verificación aporta: integridad de la cadena legal (desde el fabricante, hasta el paciente), trazabilidad mejorada, lucha contra el fraude, cumplimiento normativo europeo, datos para la farmacovigilancia y gestión de la facturación.
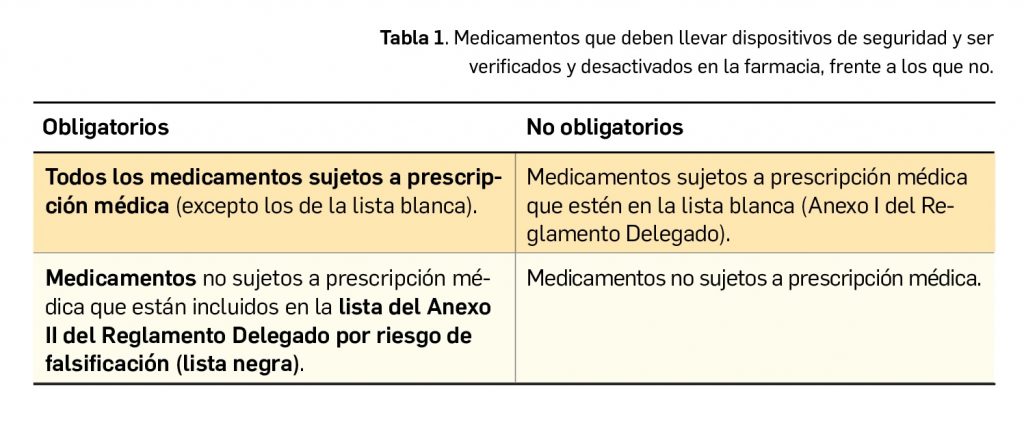
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) publica y actualiza la información sobre qué medicamentos comercializados en España requieren llevar los dispositivos (Nomenclátor de prescripción) (Tabla 1).
La notificación de sospechas por parte de las personas autorizadas para dispensar es una obligación legal y un acto de responsabilidad fundamental para proteger la salud pública. La notificación se realiza ante la sospecha fundada, no es necesario tener la certeza absoluta de que es una falsificación. El objetivo es alertar a las autoridades para que inicien la investigación pertinente.
Se deben adoptar las siguientes medidas ante una sospecha:
- NO DISPENSAR: el medicamento sospechoso no debe llegar al paciente.
- SEPARAR Y CUSTODIAR: mantenerlo en cuarentena, identificado y a disposición de las autoridades sanitarias.
- NOTIFICAR INMEDIATAMENTE: comunicar a la autoridad competente en un plazo de 24 horas laborables desde la confirmación de dicha sospecha.
La notificación debe dirigirse a la Autoridad Sanitaria Competente de la Comunidad Autónoma donde esté ubicada la farmacia comunitaria o servicio de farmacia, o al Ministerio de Defensa en el caso de sus farmacias/hospitales.
La notificación debe ser lo más completa posible e incluir, como mínimo, la siguiente información: datos del notificante, datos del medicamento sospechoso, datos del identificador único, datos del suministro (si se dispone), motivo de la sospecha, información adicional y fotografías.
Debe notificarse siempre que exista sospecha de que:
- El envase ha sido manipulado (el DCM está roto o muestra signos de manipulación).
- El medicamento puede no ser auténtico tras la verificación del IU en SEVeM, o se detectan anomalías graves en el embalaje o etiquetado no atribuibles a un simple defecto de calidad.
Mientras la Comunidad Autónoma obtiene del sistema el informe de pista de auditoría del envase sospechoso y realiza las actuaciones oportunas (informando a la AEMPS), las farmacias llevarán a cabo las siguientes acciones:
- Mantenerse disponibles y colaborativas con la autoridad sanitaria competente o la AEMPS en la investigación.
- Mantener en cuarentena y custodiado el medicamento hasta recibir instrucciones de la autoridad sanitaria.
- Documentar todas las acciones realizadas en el sistema de gestión de alertas o en los registros propios de la farmacia.