Alteraciones del eje hipotálamo-hipofisario y de las suprarrenales
 Nº437
Nº437

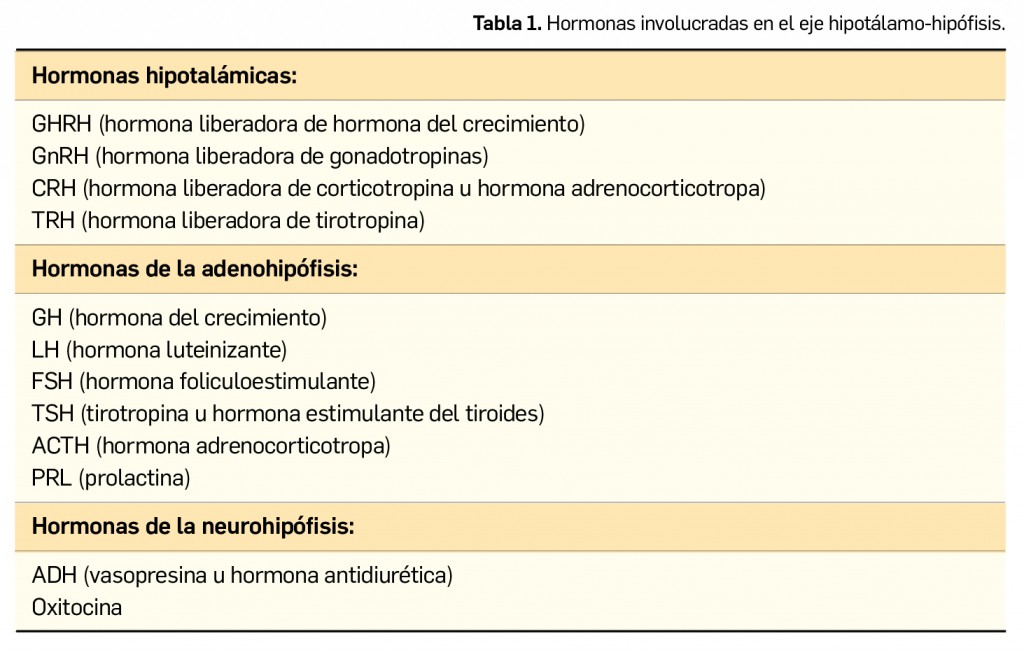
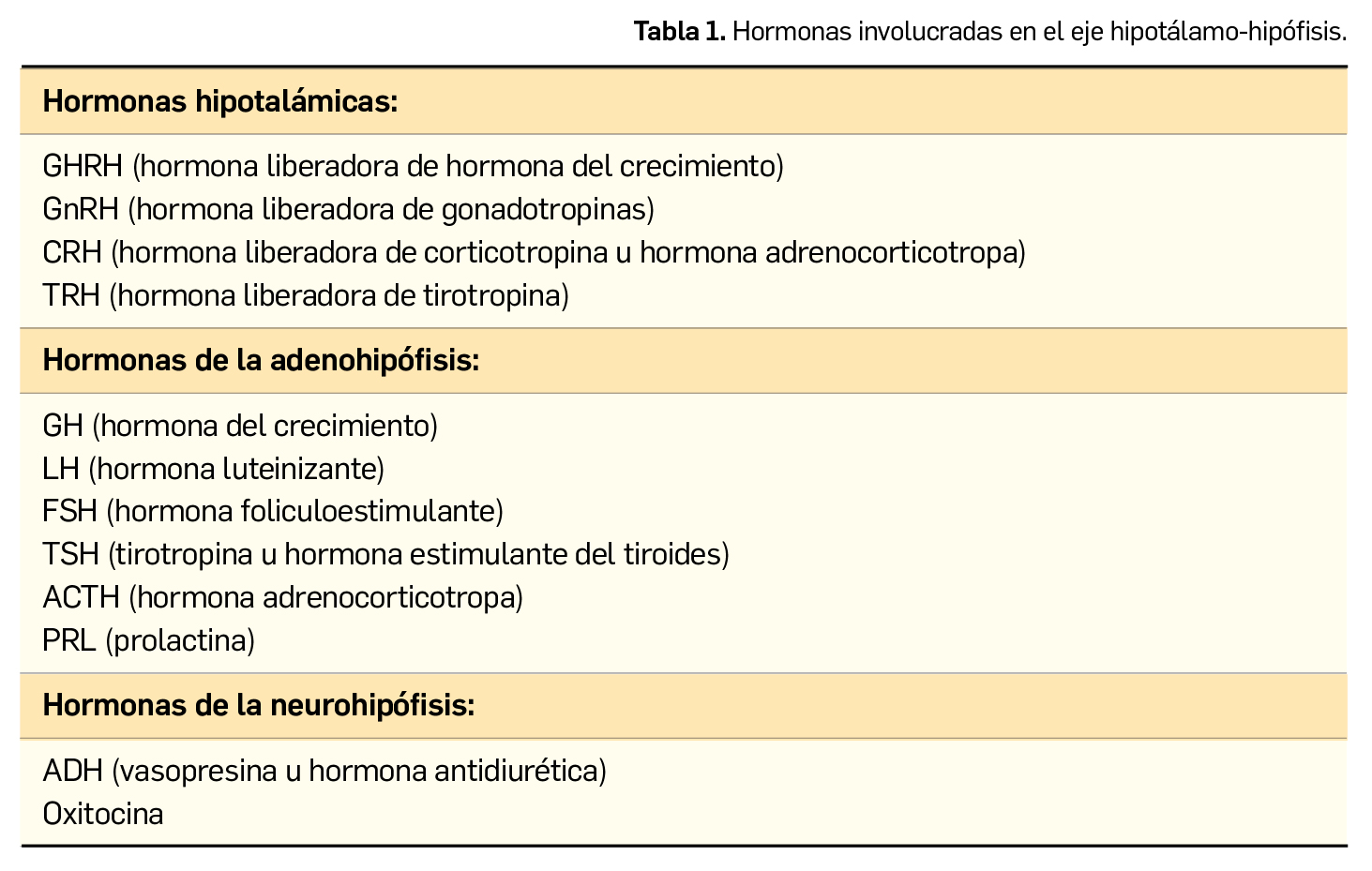
El hipotálamo constituye la principal conexión entre el sistema nervioso y el endocrino. Produce hormonas que van a liberarse en la hipófisis, o bien van a influir para que ésta sintetice las suyas propias, que actuarán a su vez sobre otros sistemas como las gónadas, el tiroides o la corteza suprarrenal (Tabla 1).
Hormona del crecimiento
Hipofunción
Las causas del déficit de hormona del crecimiento (GH) son variadas. Lo más común es que se deba a lesiones que afecten al hipotálamo o a la hipófisis, tales como lesiones compresivas o traumatismos, daño vascular, infecciones, enfermedades autoinmunes o iatrogenia, entre otros. También puede ocurrir de forma congénita.
El déficit de GH tiene su mayor expresión cuando ocurre en la infancia, pues repercute drásticamente sobre la talla impidiendo un crecimiento superior a 3 cm al año. La ausencia total de esta hormona conduce a un verdadero enanismo, en el cual, a pesar de la baja estatura, no existe una desproporción anatómica del crecimiento. Los cartílagos de crecimiento de los huesos largos son los máximos condicionantes de la terapéutica; mientras permanezcan abiertos, una terapia con GH permitirá que los pacientes alcancen una estatura normal o casi normal mientras se haga en el momento y durante el tiempo apropiados. Sin embargo, una vez cerrados, el aporte de GH se traducirá en un crecimiento en grosor y no en longitud.
En el adulto, el déficit de GH también tiene repercusiones importantes, aunque en este caso no sobre la estatura, sino sobre el metabolismo y la composición corporal. Las alteraciones que aparecen como consecuencia (hipertensión, dislipemia, obesidad central, resistencia a la insulina, pérdida de la actividad fibrinolítica del plasma) incrementan el riesgo de sufrir eventos cardiovasculares y se resuelven con la terapia sustitutiva, por lo que también está indicado administrar análogos de GH (somatotropina) o de factor de crecimiento insulínico de tipo 1 (mercasermina) en estos casos.
Hiperfunción (gigantismo y acromegalia)
Se trata de una patología excepcionalmente rara cuya principal causa son los adenomas hipofisarios productores de GH. Tanto el gigantismo como la acromegalia responden a los mismos mecanismos fisiopatológicos, aunque el momento de aparición condiciona que se produzca un cuadro o el otro:
- Gigantismo: en los niños, al no tener aún osificados los cartílagos de crecimiento, un exceso de producción de GH da lugar a un cuadro de gigantismo, presentando un crecimiento estatural excesivo pero armónico. Otros síntomas que acompañan al crecimiento excesivo son: hipogonadismo, neuropatía periférica y debilidad muscular.
- Acromegalia: a partir de la pubertad, al estar cerrados los cartílagos de conjunción, no existe la posibilidad de crecimiento longitudinal, de tal manera que el exceso de GH origina un aumento de tejidos blandos inicialmente, y de las porciones distales (“acras”) del esqueleto después (nariz, manos, pies, etc.). Estos pacientes presentan un característico aspecto facial tosco, prognatismo y maloclusión dentaria. Se presentan alteraciones respiratorias (síndrome de apnea obstructiva del sueño, agravamiento de la voz) por aumento de los tejidos de las estructuras laríngeas, y debilidad muscular y dolor asociado por compresión nerviosa (síndrome del túnel carpiano), además de otras alteraciones cutáneas, hormonales, cardiovasculares (hipertensión, disfunción ventricular), etc.
Prolactina
Hipoprolactinemia
Aparece como consecuencia de causas de hipopituitarismo (tumores que destruyan la hipófisis, granulomas, traumatismos, lesiones iatrogénicas, destrucción isquémica, etc.). El síntoma más evidente es la dificultad para la lactación posparto. No es necesario su tratamiento específico, pero sí la sustitución de otras hormonas hipofisarias.
Hiperprolactinemia
Se define como hiperprolactinemia niveles sanguíneos de esta hormona de > 20 µg/l en mujeres y > 15 µg/l en hombres, en una muestra extraída por la mañana y en ayunas. Los valores de referencia pueden diferir sensiblemente según las técnicas de laboratorio empleadas.
Causas:
- Iatrogénica: es la causa más frecuente de hiperprolactinemia. La producen fármacos antagonistas de receptores de dopamina D2 en la vía tuberoinfundibular, sobre todo antipsicóticos y otros como sulpirida, metoclopramida, domperidona, etc.
- Prolactinomas: son los tumores funcionantes más frecuentes de la hipófisis.
- Hipotiroidismo: a veces se acompaña de hiperprolactinemia, probablemente por un aumento de la actividad de la hormona liberadora de tirotropina (TRH), que estimula la producción de prolactina.
- Mastectomías, traumatismos torácicos: se acompañan en ocasiones de hiperprolactinemia, probablemente por estimulación de los nervios aferentes de la mama relacionados con el reflejo de succión.
Los síntomas más comunes de la hiperprolactinemia en mujeres son las alteraciones menstruales (amenorrea u oligomenorrea) que deben su origen a la inhibición de LH, y la galactorrea. Son más frecuentes la disminución de la líbido y la disfunción sexual en mujeres que en varones; en estos últimos es muy habitual la aparición de ginecomastia.
En aquellos casos en los que la causa sea iatrogénica, la solución consiste en ajustar, sustituir o retirar la medicación cuando sea posible. Si la hiperprolactinemia es patológica puede tratarse con agonistas D2 (cabergolina, bromocriptina) a dosis inferiores a las utilizadas en la terapia antiparkinsoniana.
Gonadotropinas
La sintomatología de las alteraciones del eje hipotálamo-hipófiso-ovárico incluye desde irregularidades del ciclo menstrual, con menstruaciones más o menos frecuentes, hasta su ausencia total, lo que se denomina amenorrea. Cuando la alteración es prepuberal, aunque se pueden desarrollar parcialmente los caracteres sexuales secundarios, por lo general éstos están retrasados en el tiempo y no llegan a aparecer menstruaciones. Se denomina entonces al cuadro amenorrea primaria. Cuando la alteración ocurre después del desarrollo puberal, las reglas, que hasta ese momento ocurrían de manera más o menos regular, desaparecen, en cuyo caso se habla de amenorrea secundaria. La mayoría de las amenorreas primarias (60%) tienen un origen congénito y se deben a errores del desarrollo gonadal o de los genitales.
Pubertad precoz
Cuando el desarrollo puberal, tanto en la mujer como en el varón, ocurre mucho antes de lo que les corresponde, que son 12-13 años en la mujer y 14-15 en el varón, hablamos de pubertad precoz. La fisiopatología supone la puesta en marcha del eje hipotálamo-hipófiso-gonadal a una edad en la que, en condiciones normales, debería estar todavía inactivo. Es un fenómeno mucho más frecuentemente en la mujer que en el hombre. También puede tener un origen tumoral a nivel del hipotálamo. Comienzan a desarrollarse las mamas (telarca), y aparece vello en el pubis y bajo las axilas (pubarca). Finalmente, en las mujeres aparece la menstruación. En los varones aparece el bigote y la barba, produciéndose además un aumento del desarrollo muscular. En ambos sexos se acelera la velocidad de crecimiento, pero como la secreción gonadal en varones y mujeres determina un cierre precoz de los cartílagos de crecimiento, la estatura final suele ser corta.
Se denomina pubertad precoz verdadera cuando la activación de eje hipotálamo-hipófisis-ovárico ocurre antes de los 9 años, produciéndose crecimiento folicular y, de forma alternante, niveles elevados de estrógenos. Aparece toda la sintomatología mencionada antes, como consecuencia del funcionamiento del eje. Es muy importante hacer un diagnóstico diferencial entre la pubertad precoz verdadera y la pseudopubertad precoz, que puede ocurrir por un contacto del niño o niña con hormonas sexuales de orígenes variados.
El tratamiento de elección en la pubertad precoz verdadera lo constituyen los análogos de la hormona liberadora de gonadotropinas (GnRH). Su administración conduce a la regresión de los caracteres sexuales secundarios y evitan el adelantamiento de la edad ósea, impidiendo el consiguiente acortamiento de la talla final. A los 12-13 años se suspende el tratamiento permitiendo el inicio normal de la pubertad.
Amenorrea hipoganadotropa
A efectos prácticos, se clasifican en un mismo grupo todas las alteraciones ováricas que cursan con gonadotropinas bajas, independientemente de su origen. Salvo casos extremadamente puntuales, la mayoría de estas alteraciones son de origen hipotalámico por afectarse la síntesis o la secreción de GnRH.
La mayoría de los casos de alteración hipotálamo-hipofisaria son debidos a trastornos funcionales, entre los que destacan, por su frecuencia, los factores psicológicos y los nutricionales (pérdida de peso, anorexia).
También cabe destacar la amenorrea que aparece en mujeres deportistas o bailarinas como consecuencia de un exceso de actividad física con la consiguiente liberación de opiáceos endógenos y/o de estimulación del eje hipotálamo-hipófiso-suprarrenal. El tratamiento con gonadotropinas puede ser de ayuda en estos casos, una vez restablecido el peso normal.
Vasopresina
Diabetis insípida
Aparece cuando la vasopresina no puede actuar. Se clasifica en diabetes insípida de origen central, cuando se debe a falta de hormona antidiurética, o nefrógena, cuando se debe a falta de respuesta del túbulo renal a la vasopresina.
La diabetes insípida de origen central generalmente se debe a traumatismos que lesionan la conexión hipotálamo-hipofisaria (traumatismos craneales en accidentes de tráfico), lo que interrumpe el transporte de la hormona por los axones neuronales. En otras ocasiones, puede tratarse de un tumor hipotalámico que destruya los núcleos donde se sintetiza. La falta de hormona en ambos casos da lugar a incapacidad renal para concentrar la orina, lo que causa diuresis de hasta 18 litros diarios de una orina muy diluida. Hay que hacer el diagnóstico diferencial con la diabetes mellitus, donde la diuresis por falta de insulina es de tipo osmolar, por acompañamiento de agua en la eliminación de glucosa por el riñón, al saturarse el transporte máximo de glucosa en los túbulos renales.
El tratamiento se realiza con desmopresina, un análogo de vasopresina de acción más prolongada, que facilita por tanto su aplicación en terapéutica. Se administra por vía intranasal (10-40 µg/día en dosis única o dividida en 2 o 3 dosis). Los análogos de vasopresina presentan otras indicaciones terapéuticas, como el tratamiento de la enuresis nocturna (desmopresina), enfermedad de Von Willebrand (desmopresina) y hemorragia por varices esofágicas (terlipresina). Las diferencias en sus utilidades terapéuticas radican en las acciones farmacológicas sobre receptores V1 (efectos vasoconstrictores) y V2 (control de la diuresis).
Síndrome de secreción inadecuada de ADH
Se denomina así a la liberación mantenida de ADH en ausencia de sus estímulos habituales. Esta situación es la causa más frecuente de hiponatremia y su diagnóstico exige descartar otras situaciones que cursan con disminución de la volemia eficaz. El tratamiento se basa en el uso de diuréticos acuaréticos (antagonistas de receptores de vasopresina V2) que producen eliminación de agua sin depleción de electrolitos y, con ello, corrigen la hiponatremia. Tolvaptán es el único comercializado actualmente en España (Samsca®).
Glucocorticoides
Síndrome de Cushing
Dentro de los síndromes de Cushing espontáneos o no iatrogénicos, los más frecuentes son los debidos a tumores hipofisarios productores de ACTH y es lo que se denomina enfermedad de Cushing. La ACTH, secretada en exceso, estimula la esteroidogénesis de las zonas fascicular y reticular de la glándula suprarrenal, produciendo un aumento de la secreción de cortisol, andrógenos y desoxicorticosterona (DOC) junto con hiperplasia suprarrenal bilateral. Pasireótida (Signifor®) disminuye la producción de ACTH de un tumor hipofisario si no puede llevarse a cabo su extirpación quirúrgica o bien no resulta eficaz.
El tratamiento del síndrome de Cushing dependerá de su etiología. En el caso de un Cushing iatrogénico se procederá a la retirada paulatina del medicamento glucocorticoide. El tratamiento de los síndromes de Cushing de origen hipofisario es la hipofisectomía. En los de origen suprarrenal, la extirpación de la glándula afectada. El tratamiento farmacológico se utiliza en pacientes con enfermedad de Cushing que no se curan tras la cirugía o radioterapia, así como en aquellos que rehúsan este tipo de terapias.
Aldosterona
Hiperaldosteronismo
- Hiperaldosteronismo primario o síndrome de Conn: se define así a un grupo de procesos caracterizados por un exceso crónico en la producción de aldosterona, que son independientes de forma total o parcial del sistema renina-angiotensina, y que suelen cursar con hipertensión arterial y, frecuentemente, con alteraciones metabólicas. La causa más común de la hipersecreción de aldosterona, aunque rara, es la existencia de un tumor de la corteza suprarrenal, en cuyo caso el tratamiento es quirúrgico.
- Hiperaldosteronismo secundario: se define así a la hipersecreción de aldosterona que aparece como resultado de ciertas enfermedades o en respuesta a estímulos fisiológicos, en ausencia de adenoma o hiperplasia suprarrenal. El aumento de aldosterona suele deberse a la activación del sistema renina-angiotensina-aldosterona. El exceso de aldosterona estimula, en principio, la reabsorción de sodio en los túbulos distales y colectores y, secundariamente, de agua, lo que da lugar a hipertensión, por aumento del volumen plasmático.
En aquellos casos en los que no esté recomendada la cirugía, el tratamiento de elección del hiperaldosteronismo primario son los antagonistas de los receptores de aldosterona (espironolactona y eplerenona). En el caso de que se trate de un hiperaldosteronismo secundario se pueden utilizar tanto los inhibidores de la enzima de conversión de angiotensina (IECA), como enalapril o ramipril, como los antagonistas del receptor de angiotensina AT1 (valsartán) y los antagonistas del receptor de aldosterona.
Enfermedad de Addison (hipoaldosteronismo)
Se trata de una insuficiencia primaria crónica que da lugar a una carencia de glucocorticoides y mineralocorticoides, por lo que es una enfermedad mortal si no se trata adecuadamente. La tuberculosis supuso durante mucho tiempo una de las principales causas de destrucción de la glándula, que gracias al control actual de las enfermedades infectocontagiosas, ha ido disminuyendo en incidencia.
La carencia de mineralocorticoides disminuye la retención renal de sodio, lo que da lugar a hiponatremia e hiperpotasemia y a un aumento de la secreción de renina y angiotensina II. La hipotensión y la hiperpotasemia se manifiestan con astenia, alteraciones musculares y tendencia a la acidosis. La falta de glucocorticoides es responsable de la característica hiperpigmentación de la piel de estos pacientes, que les confiere un característico color aceitunado que es especialmente visible en la mucosa bucal.
El tratamiento de sustitución de los glucocorticoides que faltan deberá realizarse con hidrocortisona (Hidroaltesona®), que es el preparado más parecido al cortisol endógeno.


{kind=link}
{kind=link}
{kind=link}
{kind=link}