Teriflunomida AUBAGIO® (Sanofi Aventis)
 Nº380
Nº380

Resumen
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente. Se trata del estereoisómero Z del principal metabolito activo de la leflunomida, utilizada para el tratamiento de la artritis reumatoide y de la artritis psoriásica. Los datos clínicos disponibles muestran un nivel de eficacia modesto aunque clínicamente relevante, con diferencias con respecto al placebo similares a las observadas con otros agentes (glatirámero, interferón beta, etc.), con una reducción en torno al 30% del riesgo de progresión. Por otro lado, la mayor parte de los pacientes que participaron en los ensayos clínicos presentan un bajo nivel de afectación (EDSS 2,0-2,5; sobre un nivel máximo de 10). Desde el punto de vista de la seguridad clínica, la toxicidad no parece ser un problema importante, aunque son comunes los trastornos de naturaleza hepática y digestiva. El mecanismo inmunológico es ciertamente poco específico, pero eso mismo puede achacarse a la mayoría de sus teóricos competidores. Como una potencial – y modesta – ventaja frente a estos, podría mencionarse su cómoda administración oral, una vez al día; pero nada más.
ESCLEROSIS MÚLTIPLE
La esclerosis múltiple es una enfermedad inflamatoria crónica autoinmune del sistema nervioso central, habitualmente de carácter lentamente progresivo, caracterizada por la presencia de múltiples placas diseminadas de desmielinización, distribuidas a lo largo del cerebro y la médula espinal. La enfermedad se caracteriza por múltiples y variados síntomas y signos de disfunción del sistema nervioso central, con remisiones y exacerbaciones recurrentes. En general, los que suelen manifestarse más precozmente son parestesias en una o más extremidades, el tronco o un lado de la cara; debilidad o torpeza de un miembro inferior o la mano; o trastornos visuales; debilidad muscular o fatiga inusual de un miembro, alteraciones leves de la marcha, dificultad en el control vesical, vértigo y trastornos emocionales leves. El curso habitual de la enfermedad se caracteriza por la presencia de remisiones y recaídas, con alguna discapacidad acumulativa, y aunque dicha discapacidad física puede aparecer desde el inicio de la enfermedad, lo más frecuente que comience a manifestarse tras varios años de evolución.
También es denominada como esclerosis en placas o diseminada. Las placas son en realidad lesiones formadas por infiltrados de células, con desmielinización y gliosis (destrucción de células gliales, que constituyen el elemento fundamental del tejido neurológico de sostén). La enfermedad afecta especialmente a adultos jóvenes, en los que produce manifestaciones clínicas muy diversas. Además, tiene un curso variable, observándose frecuentemente brotes y periodos de remisión. Es considerada como la causa de tipo no traumático más frecuente de invalidez o discapacidad neurológica del adulto joven en el mundo occidental.
El comienzo de la enfermedad sucede habitualmente entre los 16 y 50 años de edad, particularmente entre los 20 y los 40, y es dos veces más común en las mujeres, en las cuales suele comenzar más temprano. Es muy rara su aparición antes de los 10 años o después de los 70 años de edad. Se estima que afecta a unos 2,5 millones de personas en todo el mundo, aunque distribuidos de forma irregular; en este sentido, la región de menor prevalencia (<5 casos por 100.000 habitantes) es Asia central, mientras que en la mayoría de los países occidentales, especialmente los más desarrollados (Unión Europea, Estados Unidos, Canadá, Australia, etc.), la prevalencia supera ampliamente los 30 casos por 100.000 habitantes. En la Unión Europea hay aproximadamente 500.000 pacientes, siendo la enfermedad más frecuente en el norte de Europa y menos común en el área mediterránea. En España hay aproximadamente unos 40.000 pacientes1.
La esclerosis múltiple constituye la causa más frecuente de enfermedad por alteración de la mielina en el sistema nervioso central. Se estima que la susceptibilidad de padecer esclerosis múltiple depende de la interrelación con factores ambientales en determinadas áreas geográficas y que parecen operar antes de la pubertad, con una predisposición heredada de tipo multigénico. En este sentido, se ha cuantificado el grado de incremento del riesgo de padecer la enfermedad en función del parentesco familiar. Así, tener un primo afectado supone un 1%; padre o hijo, un 2%; hermano gemelo dicigótico, un 7%; padre y hermano, un 13%; padre y madre, un 20%; padre, madre y un hermano, un 23%; y un hermano gemelo monocigótico, un 30%.
Parece tratarse de una enfermedad de tipo autoinmune, es decir, producida por la reacción inmunológica contra elementos constitutivos del propio organismo, que son detectados – anómalamente – como antígenos. Se ha propuesto que un auténtico antígeno, todavía desconocido pero probablemente de origen externo, actuaría mimetizando proteínas de la mielina de las neuronas. Ese antígeno se presentaría sobre la superficie de los macrófagos en combinación con moléculas de clase 2 del Complejo Mayor de Histocompatibilidad (CMH). La resultante estimulación de los linfocitos T facilitadores (helper, Th1) provocaría la expresión de LFA-1 (lymphocyte function-associated antigen-1)2 y de VLA-4 (Integrin alpha4beta1; Very Late Antigen-4), facilitando la unión de dichos linfocitos T a moléculas de adhesión, como la ICAM-1 ICAM-1 (Intercellular Adhesion Molecule 1, también conocido como CD54 o Cluster of Differentiation 54) y la VCAM-1 (Vascular cell adhesion molecule 1 o CD106, cluster of differentiation), la principal molécula responsable del incremento de la adhesión leucocitaria al endotelio; actuando todos ellos sobre las células endoteliales de los vasos sanguíneos, facilitando su migración a través del endotelio y su penetración en el sistema nervioso central, donde atacarían a las células nerviosas y destruyendo específicamente su capa de mielina.
La destrucción de la mielina se debe a tres mecanismos complementarios:
- liberación de Factor de Necrosis Tumoral alfa (TNFα) por los linfocitos Th1
- liberación de TNFα y de radicales libres de oxígeno, óxido nítrico y proteasas por parte de macrófagos activados
- activación de la cascada del complemento mediante anticuerpos
Una de las causas propuestas es la infección por un virus latente (posiblemente herpesvirus o retrovirus humanos, como el HTLV-1, que causa la paraparesia espástica tropical) en la cual la activación vírica y su expresión desencadenan una respuesta inmune secundaria. Se considera, no obstante, que es más probable que infecciones comunes (tales como las producidas por virus del sarampión u otros similares) a una determinada edad desencadenen en personas susceptibles mecanismos de mimetismo molecular por los que presentarían una sensibilización cruzada contra la mielina. Por otro lado, la incidencia familiar y la asociación con ciertos alotipos HLA justifican la relativa susceptibilidad genética observada.
La esclerosis múltiple es, por consiguiente, una enfermedad básicamente desmielinizante, que conduce a la muerte neuronal desde las etapas iniciales de la enfermedad. Probablemente, esta pérdida neuronal es lo que contribuye decisivamente a la creciente discapacidad a la que se enfrentan los pacientes con esclerosis múltiple. Se caracteriza por la presencia de placas de desmielinización con destrucción de la oligodendroglía e inflamación perivenular diseminadas en el sistema nervioso central, especialmente en la sustancia blanca, con cierta especificidad por las columnas laterales y posteriores de la médula espinal, los nervios ópticos y las áreas periventriculares. También se encuentran afectadas las vías del mesencéfalo, la protuberancia y el cerebelo, al igual que la sustancia gris cerebral y medular.
El término de esclerosis se debe a la sustitución de la mielina normal por una proliferación de astrocitos. El infiltrado rodea a los pequeños vasos venosos – perivenular – y está formado por linfocitos T CD8+, CD4+, células fagocitarias, linfocitos B y células plasmáticas (estas últimas en menor número), y puede haber edema. A medida que las vainas de mielina van degenerando, se hace más apreciable la proliferación de células fagocitarias (macrófagos/microglia) y de astrocitos. Los cuerpos neuronales y los axones no suelen estar afectados, sobre todo en las lesiones recientes, pero posteriormente puede haber destrucción axonal, principalmente en las vías largas, y gliosis fibrosa que produce esclerosis de los haces de fibras, lo que justifica la irreversibilidad observada de algunos síntomas.
La principal consecuencia de la destrucción de las vainas mielínicas es la alteración de la conducción de los impulsos nerviosos transmitidos por las fibras desmielinizadas. La velocidad de transmisión se hace más lenta y los estímulos no se transmiten correctamente o incluso no lo hacen en modo alguno. El grado de anormalidad de la conducción puede variar dependiendo de circunstancias como la temperatura corporal, el ejercicio o la composición iónica del espacio extracelular. La sintomatología de la enfermedad depende de este fenómeno. En el caso concreto de la temperatura tiene consecuencias clínicas importantes, ya que la mayoría de los pacientes experimentan un empeoramiento de sus síntomas al aumentar la temperatura corporal. Se distingue actualmente cuatro variedades o formas clínicas de esclerosis múltiple:
- Remitente-recurrente (RR). Es el tipo más frecuente y afecta a más del 80% de las personas con esclerosis múltiple. En las fases iniciales puede no haber síntomas, a veces incluso durante varios años; sin embargo, a pesar de la ausencia de síntomas, sí se van produciendo lesiones inflamatorias en el sistema nervioso central. Los brotes son imprevisibles y pueden aparecer síntomas en cualquier momento – nuevos o ya conocidos – que pueden durar desde algunos días a varias semanas, desapareciendo posteriormente. Entre las recidivas no parece haber progresión de la enfermedad.
- Progresiva secundaria (SP). El grado de discapacidad persiste o incluso empeora entre brotes. Puede aparecer después de una fase recurrente-remitente y se considera una forma avanzada de la esclerosis múltiple. Entre un 30% y un 50% de los pacientes que sufren inicialmente la forma recurrente-remitente desarrolla la forma secundaria progresiva, habitualmente entre los 35 y los 45 años. Se caracteriza por una progresión continua con o sin recidivas ocasionales, remisiones poco importantes y fases de estabilidad.
- Progresiva primaria (PP). Afecta al 10% de todos los pacientes. Se caracteriza por la ausencia de brotes definidos, pero hay un comienzo lento y un empeoramiento constante de los síntomas sin un periodo intermedio de remisión. No hay episodios de recidiva ni periodos de remisión, sólo fases de estabilidad ocasionales y mejorías pasajeras poco importantes.
- Progresiva recidivante (PR). Es la forma más atípica, con progresión desde el comienzo, aunque a diferencia de aquellos con la forma progresiva primaria (PP), hay brotes agudos claros, con o sin recuperación completa. Los períodos entre brotes se caracterizan por una progresión continua.
Existe una gran controversia sobre la existencia real de una quinta forma de esclerosis múltiple, la benigna (B), que se caracterizaría por tener tan solo una recidiva inicial y, posiblemente, solo un brote adicional y una recuperación completa entre estos episodios, pudiendo transcurrir hasta 20 años hasta que se produzca una segunda recidiva, por lo que el proceso únicamente progresa de forma limitada. Para algunos especialistas, la forma benigna sería en realidad un cuadro recurrente-remitente (RR) sintomáticamente muy leve y con discapacidad mínima. No obstante, estos pacientes acaban progresando en su mayoría y experimentan deterioro cognitivo. Se estima que constituyen aproximadamente el 15% de todos los casos diagnosticados de esclerosis múltiple.
La forma sintomática de la enfermedad incluye una amplia variedad de síntomas y signos, entre los que resultan más frecuentes los mentales (apatía, alteración del juicio o inatención, etc.), los de pares craneales (especialmente oculares y acústicos), los motores (incremento de los reflejos tendinosos profundos, la combinación de espasticidad y ataxia cerebelosa puede llegar a ser totalmente incapacitante; además, las lesiones hemisféricas pueden producir hemiplejia), los sensitivos (parestesias, entumecimiento y embotamiento de la sensibilidad) y los autónomos (urgencia urinaria, dificultad para la micción, retención urinaria parcial o incontinencia leve, estreñimiento, etc.).
En general, a los 5 años de la aparición de los primeros síntomas, algo más del 50% de los pacientes tiene algún tipo de afectación leve, en otro 40 % hay afectación moderada y en menos de un 10 % es grave; un 70% de los pacientes están en condiciones de trabajar habitualmente. No obstante, a los 15 años solo el 25-30% de los pacientes continúa con una afectación leve y un 50% requieren ayuda para caminar. A los 20 años, un 35% continúa en condiciones de trabajar y un 20% ha muerto como consecuencia de las complicaciones.
Más del 60% de los pacientes con esclerosis múltiple evidencia un deterioro de la movilidad, que aparece en todos los tipos de esclerosis, incluso en etapas tempranas; en este sentido, en España, un 42% de los pacientes con una antigüedad de diagnóstico de ≤5 años reportan dificultades en la marcha y un 53% pérdida de equilibrio. De hecho, la mayoría de los pacientes con valores ≥4 en la escala EDSS3 tienen problemas para caminar y dos de cada tres consideran que su vida familiar se ve significativamente afectada por sus problemas de movilidad (Arroyo, 2013).
La esclerosis múltiple produce globalmente una reducción media de unos 9 años sobre la duración de vida en los varones y hasta de 14 en las mujeres. La esperanza de vida es de unos 25 años tras el comienzo de la enfermedad, aunque con notables variaciones interindividuales. En este sentido, la supervivencia depende sobre todo del grado de incapacidad existen en los paciente: sólo el 7 % de los enfermos que caminan han fallecido a los 10 años, mientras que asciende al 49 % para los que apenas se mantienen en pie y al 84 % para los encamados de forma permanente.
El pronóstico depende fundamentalmente del número de ataques, siendo un signo de mal pronóstico la existencia de una elevada frecuencia de recaídas durante los primeros años de enfermedad (la frecuencia media de ataques en los primeros años es de uno anual). Igualmente, el tipo de ataques es relevante para el pronóstico, ya que los síntomas primarios de tipo motriz, ataxia o problemas bulbares se asocian con peores pronósticos, mientras que si son de tipo visual, el pronóstico es más favorable.
Por el momento, no existe ningún tratamiento curativo de la enfermedad y sus objetivos consisten en reducir la gravedad y la frecuencia de las recaídas, limitar la discapacidad persistente, aliviar los síntomas y promover la reparación tisular. El tratamiento de elección para las recaídas agudas son los corticosteroides, atendiendo al carácter inflamatorio e inmunológico de la esclerosis múltiple. Reducen la intensidad y la duración de la recaída, probablemente reduciendo el edema, pero no afecta a la progresión de la discapacidad. El régimen preferible consiste en el empleo de metilprednisolona IV en dosis elevadas (1 g durante tres días), aunque si se opta por la vía oral, la pauta recomendada es una dosis inicial de 60 mg de prednisona por día, durante una semana, reduciéndola posteriormente a lo largo de tres semanas. No obstante, en la forma recidivante-remitente la tendencia es que la eficacia de los corticosteroides disminuya con el tiempo. Por otro lado, en los cuadros agudos graves resistentes a corticosteroides, se opta por un cambio de plasma en días alternos, medida que produce excelentes resultados en más del 40% de los pacientes afectados.
La terapia modificadora de la enfermedad se emplea únicamente en las formas recidivante-remitente y en la secundariamente progresiva, pero no en la progresiva primaria. En la forma recidivante-remitente, este tratamiento busca reducir la frecuencia e intensidad de los ataques y prevenir la acumulación de discapacidad asociada con la transición a la forma secundariamente progresiva. Hasta el momento las terapias inmunomoduladoras indicadas en la esclerosis múltiple son los interferones beta, el acetato de glatirámero, el fingolimod, el natalizumab y los agentes citotóxicos, como azatioprina o ciclofosfamida.
El interferón beta 1b consiste en una leve variación molecular del interferón beta humano, difiriendo en un aminoácido y no está glucosilado. En pacientes con forma recidivante-remitente, la administración de dosis subcutáneas de 8 MU cada dos días reduce en un 34% la tasa de recaídas, pero no parece reducir de forma significativa la acumulación de discapacidad. Por su parte, en pacientes con esclerosis múltiple secundariamente progresiva el tratamiento de dos años ha demostrado un significativo incremento del tiempo hasta la progresión de la enfermedad, así como otros importantes beneficios, tales como el retardar la necesidad de utilizar silla de ruegas, reducir el consumo de corticosteroides y el número de hospitalizaciones. Por su parte, el interferón beta 1a es estructuralmente idéntico a la citocina humana, tanto en la secuencia de aminoácidos como en los restos glucídicos. Reduce en un tercio la tasa de recaídas, prolongando una media de cinco meses el tiempo transcurrido hasta la primera recaída. También incrementa de forma significativa el periodo transcurrido hasta la progresión sostenida de la enfermedad.
El glatirámero es una mezcla de péptidos sintéticos formados por copolímeros de ácido L-glutámico, L-alanina, L-tirosina y L-lisina, parcialmente acetilados. No se conoce su mecanismo de acción, aunque se ha sugerido que podría actuar como un péptido que mimetiza a la proteína base de la mielina, provocando un efecto inductor de los linfocitos T supresores, deficitarios en la esclerosis múltiple, e inhibiendo el efecto de los antígenos anti-mielina del sistema nervioso central, al inhibir e efecto de los linfocitos T autorreactivos. El acetato de glatirámero actúa sobre las células dendríticas, que tienen una intensa capacidad presentadora de antígenos, orquestando las respuestas Th1 y Th2. Es capaz de reducir en un 30% el número de recaídas, y la discapacidad resultante, en los pacientes con esclerosis múltiple de tipo remitente-recidivante. Sin embargo, no hay evidencia de que este tratamiento tenga efectos beneficiosos sobre la duración o gravedad de la recaída. Tampoco hay datos clínicos significativos en pacientes afectados con formas progresivas de la enfermedad.
El fingolimod es, previa transformación en el fosfato correspondiente, un modulador del receptor 1 de la esfingosina-1-fosfato (S1P), localizado en la superficie de los linfocitos y al que se une con alta afinidad, actuando como un antagonista funcional al inducir su desacoplamiento o internalización. Este proceso hace a los linfocitos insensibles al S1P, bloqueando así la señal bioquímica que induce la salida de los linfocitos desde los órganos linfoides y, en consecuencia, provoca una redistribución linfocitaria4. Como consecuencia de ésta, se reduce la infiltración de los linfocitos al sistema nervioso central, y con ello reduce el riesgo de provocar inflamación y lesiones en el tejido nervioso en los pacientes con esclerosis múltiple. El fingolimod ha sido autorizado en monoterapia para el tratamiento modificador del curso de la enfermedad en la esclerosis múltiple remitente recurrente muy activa para pacientes tratados con interferón beta o aquellos con una evolución rápida. El tratamiento crónico con fingolimod da lugar a una reducción del recuento de linfocitos, especialmente de linfocitos T y B que circulan a través de los órganos linfoides. Sin embargo, los linfocitos que circulan en sangre periférica – implicados principalmente en la defensa inmunológica periférica – no son afectados significativamente por el fármaco. En este sentido, el fingolimod reduce levemente (20%) los niveles de neutrófilos y prácticamente no afecta a los de monocitos.
El natalizumab es un anticuerpo monoclonal que inhibe selectivamente las moléculas de adhesión, uniéndose a la subunidad a4 de las integrinas humanas, evitando la penetración de los leucocitos al sistema nervioso central inflamado, facilitando con ello la reducción de la inflamación y de las lesiones neurológicas asociadas a la esclerosis múltiple. En este sentido, es capaz de reducir el número recaídas en pacientes con esclerosis múltiple recidivante-remitente, así como el riesgo de progresión de la discapacidad. Se emplea en monoterapia en la forma remitente recidivante muy activa que no responda adecuadamente al interferón beta o en esclerosis múltiple remitente recidivante grave de evolución rápida. Presenta una pauta posológica cómoda, consistente una única administración al mes. Sin embargo, se ha mencionado la persistencia de anticuerpos inactivadores en el 6% de los pacientes, que anulan su actividad e inducen cuadros de hipersensibilidad. Aún más importante es que su uso ha sido asociado con la aparición – aunque excepcionalmente infrecuente: 1 caso por cada 2.000 pacientes tratados – de leucoencefalopatía multifocal (LMP), una enfermedad subaguda progresiva del SNC causada por la reactivación del virus JC, predominantemente en pacientes inmunodeprimidos y que suele provocar una discapacidad grave o la muerte. La sintomatología de la LMP es muy similar a un brote de esclerosis múltiple y el riesgo de desarrollar la enfermedad parece aumentar a partir de los dos años de tratamiento.
Entre los tratamientos inmunosupresores, los fármacos más utilizados son la ciclofosfamida y la azatioprina. El fundamento de su aplicación la disminución de las células en rápida proliferación, entre ellas las linfoides responsables de la destrucción de la mielina del SNC. Sin embargo, el uso de fármacos inmunosupresores en las formas progresivas más graves no ha mostrado un beneficio uniforme y tienen notables riesgos tóxicos. La ciclofosfamida puede ser beneficiosa en pacientes de menos de 40 años de edad, pero produce efectos tóxicos graves y su uso parece contar cada vez con menos partidarios. La azatioprina, por su parte, se ha utilizado mucho más que la anterior, porque sus efectos tóxicos son menos acusados y el manejo clínico es más sencillo; administrada sola o junto con corticoides orales en dosis bajas, ha demostrado una eficacia modesta en algunos aspectos clínicos como la rapidez de progresión o el número de recaídas, pero no en la discapacidad. Este leve beneficio es el principal motivo por justifica que continúe siendo un fármaco usado en pacientes con múltiples brotes o en rápida progresión. Otros fármacos inmunosupresores ensayados y con resultados más o menos decepcionantes (por su escasa eficacia o por su inaceptable toxicidad) han sido la ciclosporina, la mioxantrona, el clorambucilo, la cladribina y el metotrexato.
Desde el punto de vista de los tratamientos sintomáticos, el fármaco más relevante es la fampridina, un bloqueante de canales iónicos de potasio (K+) dependientes del voltaje que limita la fuga de iones potasio a través de dichos canales en los axones desmielinizados de los pacientes con esclerosis múltiple, prolongando la repolarización e intensificando el potencial de acción en las neuronas afectadas y, con ello, mejorando algunas de las funciones neurológicas perturbadas en estos pacientes, particularmente la marcha en pacientes adultos. No obstante, sus efectos clínicos son modestos y solo son observados en un tercio de la población susceptible de su uso; por otro lado, presenta un perfil toxicológico nada desdeñable, todo lo cual limita su potencial terapéutico (Cuéllar, 2013).
ACCIÓN Y MECANISMO
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente.
La etapa limitante en la biosíntesis de bases nucleicas de tipo pirimidínico es la formación del N-carbamilaspartato, partiendo del carbamilfosfato y del aminoácido aspartato y gracias al enzima aspartato-transcarbamilasa. El núcleo pirimidínico se genera a partir del N-carbamilfosfato, formándose el dihidroorotato, el cual, mediante la DHODH, da lugar a orotato y, tras incorporar ribosa-fosfato, forma orotidilato, un precursor del uridilato (UMP), el cual a su vez puede ser convertido en los nucleótidos citidilato (en el ARN) y timidilato (en el ADN).
Los requerimientos de nucleósidos y de sus correspondientes bases nucleicas para la síntesis de ADN y ARN en los procesos de proliferación celular pueden ser cumplimentados mediante la administración exógena (fundamentalmente, con la dieta) o por la síntesis de novo a partir de precursores bioquímicos. Sin embargo, la capacidad para reutilizar o reconvertir nucleótidos a partir de restos de ácidos nucleicos propios, permite que el organismo humano pueda prescindir prácticamente de la obtención de bases purínicas y pirimidínicas a partir de la dieta. De hecho, la vía de la reutilización constituye la principal fuente de nucleótidos para la síntesis de ADN, ARN y cofactores enzimáticos (AMP, ATP, GMP, etc). Esto es así para la mayoría de las células humanas, pero no tanto para las células con alta capacidad de división (en función de las demandas orgánicas), como ocurre con los linfocitos B y T activados.
Por este motivo, el bloqueo de la síntesis de novo de pirimidinas en los linfocitos T y B es capaz de interferir con la síntesis proteica y de ARN en estas células, activando las moléculas sensoras que bloquean la progresión del ciclo celular en la fase G1 (crecimiento). Esto se manifiesta como un efecto fundamentalmente citostático. Este efecto es capaz de bloquear la acción combinada de los linfocitos T y B activados.
La esclerosis múltiple es considerada como una enfermedad de tipo autoinmune, posiblemente relacionado con la existencia de un antígeno – aún sin identificar – que se presentaría sobre la superficie de los macrófagos en combinación con moléculas de clase 2 del Complejo Mayor de Histocompatibilidad (CMH). La resultante estimulación de los linfocitos T facilitadores (Th1) provocaría la expresión de LFA-1 y de VLA-4, facilitando la unión de dichos linfocitos T a moléculas de adhesión, como la ICAM-1 y la VCAM-1, sobre las células endoteliales de los vasos sanguíneos, facilitando su migración a través del endotelio y su penetración en el sistema nervioso central, donde atacarían a las células nerviosas y destruyendo específicamente su capa de mielina. Esta destrucción de la mielina se debe a tres mecanismos complementarios: liberación de Factor de Necrosis Tumoral alfa (TNFa) por los linfocitos Th1, así como del propio TNFa y de radicales libres de oxígeno y óxido nítrico, y proteasas por parte de macrófagos activados y la activación de la cascada del complemento mediante anticuerpos.
En este sentido, además de la inhibición de la proliferación de linfocitos B y T activados, y de la interferencia de la adhesión de estos a las células endoteliales, la teriflunomida disminuye la síntesis de citocinas inmunosupresoras, como el Factor de Crecimiento Transformante beta (TGF-β). Asimismo, inhibe diversas isoenzimas tirosina cinasa, el Factor Nuclear potenciador de las cadenas ligeras Kappa de las células B activadas (NF-kB) y metaloproteasas (MMP). También parece desarrollar efectos antiinflamatorios específicos, al reducir la liberación de histamina e inhibir la inducción de la ciclooxigenasa 2 (COX 2).
ASPECTOS MOLECULARES
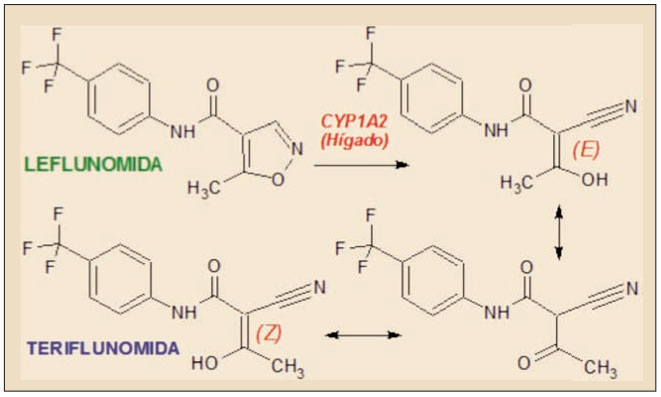
La teriflunomida es el estereoisómero Z del principal metabolito activo de la leflunomida, un agente inmunomodulador autorizado para el tratamiento de la artritis reumatoide y de la artritis psoriásica. La leflunomida es transformada en el hígado humano mayoritariamente (70%) en el metabolito M1 principalmente a través del citocromo P450 (CYP), particularmente por el isoenzima CYP1A2, mediante la apertura del anillo isoxazólico y dando lugar a una mezcla interconvertible de estereoisómeros enólicos (E y Z) y de una cetona, de los que el estereoisómero enólico Z es el más estable y, por tanto, el predominante.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínica de la teriflunomida han sido adecuadamente contrastadas en las indicaciones autorizadas mediante tres ensayos clínicos principales de fase 3, multinacionales, aleatorizados, doblemente ciegos, con diseño de grupos paralelos y controlados con placebo o con comparador activo (interferón beta 1a). Dichos ensayos clínicos fueron llevados a cabo sobre pacientes con esclerosis múltiple mayoritariamente (>91%) remitente-recurrente y moderadamente afectados, con una puntuación no superior a 5,5 (sobre un máximo de 10) en la escala de discapacidad de Kurtzke o EDSS5 (Expanded Disability Status Scale, Escala Expandida del Estado de la Discapacidad), que mide el estado funcional de los pacientes con esclerosis múltiple, aunque la mediana fue de 2,0 a 2,5; los pacientes experimentaron los primeros síntomas entre 4 y 7 años antes y habían sido diagnosticados hace entre uno y cuatro años. Asimismo, los pacientes debían haber experimentado al menos un recaída en el último año o dos recaídas en los dos últimos años.
En todos los estudios se empleó teriflunomida por vía oral, tanto en dosis de 7 como de 14 mg/24 h, durante un periodo no inferior a 48 semanas (en caso contrario, se valoró como suspensión prematura del tratamiento) y, en su caso, el correspondiente placebo. En el estudio comparativo con interferón beta 1a, éste se empleó con dosis de 44 µg tres veces por semana6, por vía subcutánea. Como variable primaria de eficacia se midió en los estudios controlados con placebo la tasa anualizada de recaídas confirmadas, mientras que entre las variables secundarias se determinó la mediana de tiempo trascurrido hasta progresión de la discapacidad (aumento persistente durante al menos 12 semanas de al menos un punto en la escala EDSS). En el estudio comparativo con interferón beta, se determinó la mediana de tiempo hasta fallo terapéutico (definido como la aparición de la primera recaída confirmada o la suspensión del tratamiento por cualquier causa, lo que sucediese en primer lugar). Otros parámetros determinados fueron la variación experimentada en la escala de impacto de fatiga (Fatigue Impact Scale, FIS) y el cuestionario de satisfacción del tratamiento (Treatment Satisfaction Questionnaire for Medication, TSQM).
Dos de los estudios fueron controlados con placebo y tuvieron un diseño similar. El primero de ellos (TEMSO; O'Connor, 2011) se llevó a cabo sobre 1.088 pacientes con una mediana de edad de 38 años, 72% mujeres, 98% de raza caucásica, mediana de 2,5 en la escala EDSS (un 23% tenía >3,5) y un 27% habían recibido tratamiento previamente. El tratamiento se prolongó durante 108 semanas.
Los resultados mostraron una tasa anualizada de recaídas de 0,37 (IC95% 0,32 a 0,43) con teriflunomida 7 mg, de 0,37 (IC95% 0,31 a 0,44) con teriflunomida 14 mg y de 0,54 (IC95% 0,47 a 0,62) con placebo; esto supone una reducción del riesgo de recaída con respecto al placebo del 31%, tanto con la dosis de 7 mg (HR= 0,69; IC95% 0,56 a 0,84; p= 0,0002) como con la de 14 mg (HR= 0,69; IC95% 0,55 a 0,85; p= 0,0005). Por su parte, el porcentaje de pacientes con progresión de la enfermedad fue del 18,6% con teriflunomida 7 mg, del 17,3% con teriflunomida 14 mg y del 23,7% con placebo, lo que implica una reducción del riesgo de progresión de la enfermedad del 24% con la dosis de 7 mg (HR= 0,76; IC95% 0,56 a 1,05; p= 0,0835, no significativa) y del 30% con la de 14 mg (HR= 0,70; IC95% 0,51 a 0,97; p= 0,0279).
En un análisis secundario (O'Connor, 2013), los pacientes tratatados con las dosis de 7 y de mg de teriflunomida mostraron una reducción de la tasa anualizada de recaídas con secuelas neurológicas del 32% y del 36% respectivamente (ambas, estadísticamente significativas). También se apreció una reducción estadísticamente significativa en la tasa de recaídas que finalizaron en la hospitalización del paciente (36% y 59%, respectivamente para las dosis de 7 y 14 mg), así como en la de pacientes que requirieron la administración IV de corticosteroides para tratar las recaídas (29% y 34%).
El segundo estudio pivotal (TOWER; Confavreux, 2014) fue realizado en 1.169 pacientes con una mediana de 38 años, 71% mujeres, 82% de raza caucásica, mediana de 2,5 en la escala EDSS (un 24% tenía >3,5) y un 33% habían recibido tratamiento previamente (16% con interferón beta y 12% con glatirámero).
Se registró una tasa anualizada de recaídas de 0,39 (IC95% 0,33 a 0,46) con teriflunomida 7 mg, de 0,32 (IC95% 0,27 a 0,38) con teriflunomida 14 mg y de 0,50 (IC95% 0,43 a 0,58) con placebo; todo lo cual implica una reducción del riesgo de recaída con respecto al placebo del 22% con la dosis de 7 mg (HR= 0,78; IC95% 0,63 a 0,96; p= 0,0183) y del 36% con la de 14 mg (HR= 0,64; IC95% 0,51 a 0,79; p= 0,0001). Los correspondientes porcentajes de pacientes con progresión de la enfermedad fueron del 16,0% con teriflunomida 7 mg, del 11,9% con teriflunomida 14 mg y del 16,8% con placebo, lo que implica una reducción del riesgo de progresión de la enfermedad del 4% con la dosis de 7 mg (HR= 0,96; IC95% 0,68 a 1,35; p= 0,7620, no significativa) y del 31% con la de 14 mg (HR= 0,69; IC95% 0,47 a 1,00; p= 0,0442).
El estudio comparativo con interferón beta 1a (TENERE; Vermersch, 2014) incluyó a 324 pacientes con una mediana de 35 años, 68% mujeres, 100% de raza caucásica, mediana de 2,0 en la escala EDSS (un 12% tenía >3,5) y un 19% habían recibido tratamiento previamente. Los datos registrados indicaron que el porcentaje de pacientes que habían experimentado fallo terapéutico fue del 48,6% con 7 mg de teriflunomida, del 37,8% con 14 mg y del 42,3% con interferón beta, siendo la probabilidad de fallo a las 96 semanas del 59% (HR= 0,59; IC95% 0,46 a 0,71) con teriflunomida 7 mg, del 41% (HR= 0,41; IC95% 0,31 a 0,51) con teriflunomida 14 mg y del 44% (HR= 0,44; IC95% 0,34 a 0,54) con interferón beta; en este sentido, no encontrándose superioridad de ninguna de las dosis de teriflunomida vs. interferón beta 1a (OR= 1,12; IC95% 0,75 a 1,67; p= 0,5190, no significativa, para la dosis de 7 mg; OR= 0,86; IC95% 0,56 a 1,31; p= 0,5953, no significativa, para la de 14 mg).Las puntuaciones en el índice de fatiga (FIS) indicaron una fatiga más frecuente con interferón beta, aunque las diferencias fueron solo estadísticamente significativas con teriflunomida 7 mg. También las puntuaciones en el cuestionario de satisfacción del tratamiento (TSQM) fueron más favorables para la teriflunomida.
Desde el punto de vista de la seguridad clínica, los eventos adversos más frecuentemente asociados al tratamiento con teriflunomida son de carácter hepático, digestivo e inmunológico: alteraciones de las pruebas funcionales hepáticas (17,8% con la dosis de 14 mg de teriflunomida; 17,0% con la de 7 mg y 10,5% con placebo), diarrea (17,3%; 14,5% y 8,3%), náusea/vómitos (16,6%; 11,7% y 9,0%), alopecia (14,7%; 11,4% y 4,3%), parestesia/disestesia (14,7%; 12,1% y 10,2%), neutropenia (4,6%; 2,3% y 0,7%), infecciones virales (6,5%; 4,4% y 1,9%), hipertensión (4,6%; 3,5% y 1,9%), metrorragia (3,1%; 1,2% y 0,5%), dolor muscular (3,1%; 4,0% y 1,4%). La tasa de eventos adversos graves fue del 16,1% (14 mg), 12,8% (7 mg) y 7,6% (placebo), provocando directamente la suspensión del tratamiento en un 11,8%, 9,1% y 7,6%, respectivamente.
ASPECTOS INNOVADORES
La teriflunomida es un agente inmunosupresor selectivo, que actúa fundamentalmente como un inhibidor del enzima mitocondrial dihidroorotato deshidrogenasa (DHODH), que cataliza un paso clave en la síntesis de novo de bases nucleicas pirimidínicas, esencial para la proliferación de los linfocitos B y T activados, implicados en la etiología autoinmune de la esclerosis múltiple. Ha sido autorizada para el tratamiento de pacientes adultos con la forma remitente-recurrente. Se trata del estereoisómero Z del principal metabolito activo de la leflunomida, utilizada para el tratamiento de la artritis reumatoide y de la artritis psoriásica.
El meta-análisis de los datos agregados de los dos estudios controlados con placebo muestra una tasa anualizada de recaídas de 0,354 para la teriflunomida 14 mg y de 0,534 para placebo (P< 0,0001), siendo los respectivos porcentajes de pacientes con progresión sostenida del 17,9% vs. 25,2%, lo que supone una reducción del riesgo del 30,5%. Por lo que respecta a la comparación con interferón beta 1a, el ensayo pivotal correspondiente no ha sido capaz de mostrar superioridad con ninguna de las dos dosis de teriflunomida ensayadas. No se dispone de ningún otro estudio directamente comparativo con otros medicamentos utilizados en la esclerosis múltiple.
Desde el punto de vista de la seguridad clínica, la toxicidad no parece ser un problema importante, aunque son comunes los trastornos de naturaleza hepática y digestiva. Hasta un 12% de los pacientes tratados con la dosis de 14 mg suspendieron el tratamiento por eventos adversos, frente a un 9% con placebo. El perfil toxicológico no parece presentar diferencias con por su precursor farmacológico, la leflunomida, por lo que no son previsibles grandes sorpresas en este campo.
Los datos clínicos registrados solo corresponden a la forma recidivante-recurrente – por otro lado, la más común – y muestran un nivel de eficacia modesto aunque clínicamente relevante, con diferencias con respecto al placebo similares a las observadas con otros agentes (glatirámero, interferón beta, etc.), con una reducción en torno al 30% del riesgo de progresión. Por otro lado, la mayor parte de los pacientes que participaron en los ensayos clínicos presentan un bajo nivel de afectación (EDSS 2,0-2,5; sobre un nivel máximo de 10).
El mecanismo inmunológico es ciertamente poco específico, pero eso mismo puede achacarse a la mayoría de sus teóricos competidores. Como una potencial – y modesta – ventaja frente a estos, podría mencionarse su cómoda administración oral, una vez al día; pero nada más.
|
VALORACIÓN |
|
|---|---|
|
Teriflunomida ► AUBAGIO® (Sanofi Aventis) |
|
|
Grupo Terapéutico (ATC): L04AX. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores selectivos. |
|
|
Indicaciones autorizadas: Tratamiento de pacientes adultos con esclerosis múltiple remitente recurrente. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad físico-química: Vía de administración más cómoda para el paciente. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA* |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Leflunomida |
Arava |
Sanofi Aventis |
2000 |
|
Teriflunomida |
Aubagio |
Sanofi Aventis |
2014 |
* Se indican únicamente los primeros registros autorizados con ese principio activo.
BIBLIOGRAFÍA
Bibliografía
- Arroyo R. Los problemas de movilidad en los pacientes con esclerosis múltiple. Eficacia de Fampyra y beneficio para el paciente. Hospital Clínico San Carlos; Madrid, 17 de septiembre de 2013.
- Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, Wolinsky JS, Bagulho T, Delhay JL, Dukovic D, Truffinet P, Kappos L); TOWER Trial Group. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014; 13(3): 247-56. doi: 10.1016/S1474-4422(13)70308-9.
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Fampridina (Pampyra) en esclerosis múltiple. Panorama Actual Med. 2013; 37(367): 258-63.
- European Medicines Agency (EMA). Aubagio®. European Public Assessment Report (EPAR). EMA/529790/2013; EMEA/H/C/002514. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002514/WC500148684.pdf
- O’Connor PW, Lublin FD, Wolinsky JS, Confavreux C, Comi G, Freedman MS, Olsson TP, Miller AE, Dive-Pouletty C, Bégo-Le-Bagousse G, Kappos L. Teriflunomide reduces relapse-related neurological sequelae, hospitalizations and steroid use. J Neurol. 2013; 260(10): 2472-80. doi: 10.1007/s00415-013-6979-y.
- O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, Benzerdjeb H, Truffinet P, Wang L, Miller A, Freedman MS; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011; 365(14): 1293-303. doi: 10.1056/NEJMoa1014656.
- Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, Olsson TP, Benamor M, Bauer D, Truffinet P, Church M, Miller AE, Wolinsky JS, Freedman MS, O’Connor P; TENERE Trial Group. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014; 20(6): 705-16. doi: 10.1177/1352458513507821.
1 Federación Española para la Lucha contra la Esclerosis múltiple. http://www.esclerosismultiple.com/esclerosis_multiple/index.php
2 LFA-1 pertenece a la superfamilia de las integrinas y participa en la adhesión firme de los leucocitos al endotelio de las zonas inflamadas durante el proceso de extravasación. Así mismo, está involucrada en la facilitación de la adhesión o la migración de las células T vírgenes y de memoria a los órganos linfoides secundarios.
3 Expanded Disability Status Scale; con valores de discapacidad progresiva de 0 (ausencia de discapacidad) a 10 (discapacidad absoluta).
4 Algunos datos experimentales sugieren que el efecto del fingolimod modulando los receptores de S1P podría no limitarse a los linfocitos, sino que podrían afectar también a dichos receptores presentes sobre los astrocitos que rodean la vaina mielínica axónica, favoreciendo la aparición de efectos neuroprotectores y/o reparadores
5 La EDSS evalúa la discapacidad de acuerdo a ocho sistemas o escalas funcionales (EF): piramidal, cerebelo, tronco del encéfalo, sensibilidad, vejiga, intestino, visión y funciones mentales. La puntuación global va entre 0 (examen neurológico normal, con valoración 0 en todas las escalas funcionales) y 10 (muerte relacionada directamente con la esclerosis múltiple). Por ejemplo, una puntuación de 2,0 supone una mínima incapacidad, con un EF grado 2, otros 0 o 1; 2,5 implica lo mismo, pero con dos EF de grado 2. Una puntuación de 4,0 supone un paciente completamente ambulante sin asistencia, autosuficiente, capacitado durante 12 horas diarias a pesar de una relativamente grave incapacidad consistente en una escala funcional grado 4 (otros 0 o 1) o combinaciones de grados menores excediendo los límites de los grados previos, capaz de caminar sin ayuda o descanso uno 500 metros. Por su parte, una puntuación de 5,5 implica que el paciente es capaz de caminar 100 metros sin ayuda ni descanso, pero con incapacidad suficientemente grave como para impedir las actividades del día entero. Con una puntuación de 8,0 el paciente está esencialmente relegado a la cama o silla de ruedas o en silla, pero capaz de permanecer fuera de la cama gran parte del día, siendo capaz de realizar muchas de las funciones de su cuidado personal; generalmente conserva el uso efectivo de los brazos.
6 Las dos primeras semanas se utilizaron dosis de 8 µg y las dos siguientes de 22 µg; a partir de entonces se mantuvieron los 44 µg a lo largo de todo el estudio.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
