Ruxolitinib JAKAVI® (Novartis)
 Nº380
Nº380

Resumen
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. Atendiendo a la condición de enfermedad rara, el medicamento fue designado como huérfano. El nuevo fármaco ha demostrado ser capaz de reducir de forma notable la esplenomegalia en el 29-42% de los pacientes, frente a menos del 0-1% con placebo o con el mejor tratamiento alternativo disponible. Desde el punto de vista de la seguridad, el ruxolitinib se comporta básicamente como un agente mielotóxico, dando lugar a trombocitopenia, anemia, hemorragias y neutropenia, como consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. Aunque se trata de un efecto mielotóxico importante, es previsible y manejable mediante transfusiones y ajustes de la posología del fármaco. En definitiva, se trata de un paso adelante en el tratamiento de una enfermedad rara, aunque su eficacia es modesta y su efecto real sobre la supervivencia global está todavía sujeto a estudio.
MIELOFIBROSIS
Las enfermedades mieloproliferativas crónicas comparten la característica de ser clonales; es decir, se originan en una célula madre de la médula ósea con capacidad de diferenciación pluri- y multipotente. En dichas patologías se produce una proliferación, diferenciación y maduración efectiva de las células en la médula ósea, generando una médula hipercelular y un número incrementado de células maduras en sangre periférica; asimismo, es frecuente encontrar hepatoesplenomegalia debida a hematopoyesis extramedular y/o a secuestro de las células sanguíneas (Novelli, 2011).
Según la clasificación de la Organización Mundial de la Salud, los síndromes mieloproliferativos crónicos incluyen a la leucemia mieloide crónica Filadelfia+ BCR/ABL+, la leucemia neutrofílica crónica, la leucemia eosinofílica crónica/síndrome hipereosinofílico, la policitemia vera, la trombocitemia esencial y la mielofibrosis crónica idiopática, junto con otras enfermedades mieloproliferativas inclasificables.
En concreto, la mielofibrosis crónica idiopática (metaplasia mieloiode, mieloesclerosis) se caracteriza por una intensa fibrosis de la médula ósea, esplenomegalia, hematopoyesis extramedular y leucoeritroblastosis (granulocitos y eritrocitos inmaduros) en sangre periférica y glóbulos rojos en forma de lágrimas. En su fase temprana, se caracteriza por un número elevado de células CD34+ en la médula, mientras que las fases posteriores de fibrosis medular muestran una disminución de células CD34+ en la médula y un aumento de congestión esplénica y hepática con células CD34+. Además, hay formas secundarias de mielofibrosis, que derivan secundariamente de otros trastornos mieloproliferativos, especialmente la policitemia vera y la trombocitemia esencial1.
La mielofibrosis se origina por una mutación somática en una célula madre hematopoyética pluripotente, lo que le confiere a ésta ciertas ventajas proliferativas respecto a los progenitores normales. Esa población celular anormal libera diversas citocinas y factores de crecimiento (PDGF, TGFβ, VEGF, bFGF y calmodulina) en la médula, dando lugar a la producción de fibrosis colágena, osteoesclerosis y angiogénesis, como fenómeno secundario y la colonización de órganos y sitios extramedulares, como el hígado y el bazo, a los que produce un sobrecrecimiento (hepato y esplenomegalia).
Presenta una incidencia de 4-14 casos por millón de habitantes/año, con una mediana de la edad de presentación de 65 años, aunque aproximadamente la tercera parte de los pacientes están asintomáticos al diagnóstico. Se suele diagnosticar entre los 50 y 80 años (menos del 10% son menores de 45 años), pero puede ocurrir a cualquier edad, y afecta tanto a hombres como a mujeres.
Aunque en muchos casos se ignora la etiología de esta enfermedad, en un 50% de los casos está presente una mutación del gen JAK2 y en un 10% una mutación del gen MLP2. La expresión de los genes JAK conduce a la síntesis de la familia de tirosina cinasas Janus (Janus tirosine kinases, JAK; de los que se conocen cuatro tipos, de la JAK1 a la JAK4), un miembro de la superfamilia de los receptores de citocinas. Las JAK parecen ejercer un papel esencial en los procesos de transducción de señales bioquímicas de diversas citocinas; de hecho, la eritropoyetina, la trombopoyetina y el factor de estimulante de colonias de granulocitos y macrófagos (GM-CSF) actúan exclusivamente sobre receptores que utilizan homodímeros de JAK2 en sus procesos de señalización intracelular.
En respuesta a la activación del receptor de citocinas, estas tirosina cinasas (que no forman parte propiamente de dicho receptor) son rápidamente fosforiladas dando lugar a una activación celular en cascada y supone la activación de varias vías de transducción de señales, que determinan la aparición de cambios en la expresión genética que promueven la progresión y maduración celular.
En realidad, la mutación de JAK2 no parece ser la causa primaria de la enfermedad sino más bien una mutación somática secundaria. Aunque la mutación JAK2V617F3 parece ser la más común (aproximadamente, el 65%) de las asociadas a la mielofibrosis primaria, se han identificado otras mutaciones que activan anormalmente al JAK2, como la MPLW515L/Kin del receptor de MPL. Sea como fuere, los altos niveles de citocinas encontradcos en los pacientes con mielofibrosis, particularmente de interleucina 6 (IL-6) parece ser los responsables del estado hipercatabólico y de los síntomas constitucionales de la enfermedad (pérdida de peso, fatiga, etc.). Muchas de estas citoxinas utilizan tirosina cinasas de tipo JAK1 en su mecanismo señalizador celular.
Las principales causas de muerte en los pacientes con mielofibrosis son la insuficiencia medular evolutiva, la transformación a leucemia no linfoblástica aguda, las infecciones oportunistas, las complicaciones trombohemorágicas, la insuficiencia cardíaca y la hipertensión portal. Los factores pronósticos más relevantes son:
- Edad de 65 años o más.
- Anemia (hemoglobina <10 g/dl).
- Síntomas inespecíficos: fiebre, sudores nocturnos o pérdida de peso.
- Leucocitosis (>25 × 109/l).
- ≥1% de blastocitos circulantes.
Los pacientes sin ninguna característica adversa, excepto la edad, tienen una mediana de supervivencia de más de 10 a 15 años (mediana de 135 meses: 11,25 años), pero la presencia de uno de los mencionados factores adversos la reduce a 95 meses (7,9 años), dos factores a 48 meses (4 años) y tres o más a 27 meses (2,25 años). Las anomalías cariotípicas también pueden afectar el pronóstico. En este sentido, algunas series retrospectivas han permitido observar que las deleciones 13q y 20q y la trisomía 9 se han correlacionado con una mayor supervivencia y ninguna transformación leucémica, en comparación con el peor pronóstico de la trisomía 8, un cariotipo complejo, -7/7q-, i(17q), inv(3), -5/5q-, 12p- o el reordenamiento 11q23.
Los pacientes asintomáticos de riesgo bajo solo requieren un seguimiento clínico, sin ninguna intervención terapéutica, que solo estaría justificada en caso de aparición de anemia sintomática, leucocitosis marcada, sudores nocturnos abundantes, pérdida de peso, fiebre o esplenomegalia sintomática.
El trasplante alogénico de células madre hematopoyéticas es el único tratamiento curativo de la mielofibrosis, pero su aplicabilidad está limitada por la avanzada edad de la mayoría de los pacientes y por la existencia de diversas condiciones comórbidas (Odenike, 2013)
Hasta ahora, el único tratamiento mielosupresor específicamente indicado en España (y algunos otros países de la Unión Europea, como Francia, Italia o Suecia) en la mielofibrosis. El ruxolitinib, atendiendo a su condición de inhibidor de JAK1 y JAK2, ha sido autorizado para reducir la esplenomegalia y los síntomas debilitantes de la pérdida de peso, la fatiga y los sudores nocturnos de los pacientes de mielofibrosis primaria, mielofibrosis posterior a trombocitopenia esencial o mielofibrosis posterior a policitemia vera, tanto positivos como negativos para JAK2.
ACCIÓN Y MECANISMO
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia.
Las JAK parecen ejercer un papel esencial en los procesos de transducción de señales bioquímicas de citocinas hematopoyéticas como la eritropoyetina, la trombopoyetina y el factor de estimulante de colonias de granulocitos y macrófagos (GM-CSF). En respuesta a la activación del receptor de citocinas, estas tirosina cinasas (que no forman parte propiamente de dicho receptor) son rápidamente fosforiladas dando lugar a una activación celular en cascada y supone la activación de varias vías de transducción de señales, que determinan la aparición de cambios en la expresión genética que promueven la progresión y maduración celular. El ruxolitinib impide dichos procesos de fosforilación al competir directamente con el ATP, impidiendo de esta manera los efectos proliferativos de dichas citocinas.
ASPECTOS MOLECULARES
El ruxolitinib está estrechamente relacionado farmacológicamente con otros miembros de la serie de inhibidores de tirosina cinasas. Se han desarrollado modelos moleculares de relación estructura-actividad, en los que las interacciones estéricas y electrostáticas han demostrado ser las más determinantes para el grado de inhibición y el tipo de tirosina cinasa susceptible.
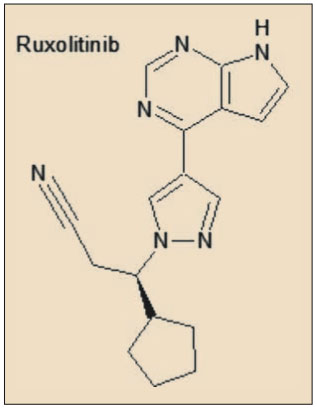
En concreto, la estructura del ruxolitinib emula la del ATP (trifosfato de adenosina); de hecho, se trata de un análogo isostérico de la adenosina. En tal calidad, el ruxolitinib actúa como un antagonista competitivo del ATP específicamente en la JAK1 y JAK2, lo que conduce a un bloqueo del proceso de la fosforilación en la cadena de señalización bioquímica intracelular de determinadas citocinas hematopoyéticas.
La mutación genética más comúnmente registrada en pacientes con mielofibrosis afecta a la estructura de la JAK2 (JAK2V617F), pero lo hace en la región externa al bolsillo de unión al ATP del enzima, por lo que los inhibidores de la cinasa JAK2 competitivos del ATP – como el ruxolinitib – no son capaces de distinguir entre las enzimas JAK2 normales y las mutadas. Por ello, este tipo de fármacos producen una mielosupresión no selectiva que, si bien facilita el control la hiperproliferación de las células hematopoyéticas en mielofibrosis y en otras patologías mieloproliferativas (pero también es responsable de efectos adversos mielosupresores), pueden no ser capaces en eliminar los clones mutantes.
EFICACIA Y SEGURIDAD CLÍNICAS
Le eficacia y la seguridad clínica del ruxolitinib en la indicación autorizada han sido contrastadas mediante dos ensayos clínicos principales aleatorizados de fase III; el primero de ellos (COMFORT-I; 309 pacientes) es un estudio doblemente ciego y controlado con placebo, mientras que el segundo (COMFORT-II, 209 pacientes) es un estudio abierto, comparando el tratamiento con ruxolitinib con el mejor tratamiento disponible (seleccionado por el investigador en función de las circunstancias específicas de cada paciente). Las pautas posológicas de ruxolitinib fueron ajustadas en función del recuento plaquetario basal de los pacientes, con dosis orales de partida de 15 mg/12 h (100.000-200.000 plaquetas/µL) o 20 mg/12 h (>200.000 plaquetas/µL), con incrementos de 5 mg/12 h cada 4 semanas de tratamiento, de acuerdo a la evolución del recuento plaquetario y de la respuesta al tratamiento (según el volumen palpable del bazo).
La mediana de edad de los pacientes en ambos estudios es de 66 y 70 años (35-91); entre un 51% y un 58% eran varones. Presentaban mielofibrosis primaria en un 45-55% de los casos, secundaria a policitemia vera en un 27-33% y secundaria a trombocitemia esencial en un 14-23%. Un 40-47% tenían un grado 3 de fibrosis, un 33-41% de grado 2 y un 4-14% de grado 1. Tenían un tamaño mediano de bazo palpable bajo el margen costal de 14-16 cm, mutación de JAK2 en un 67-80% y habían sido tratados previamente con hidroxiurea en un 57-75%.
En el estudio COMFORT-I (Verstovsek, 2012) la duración del tratamiento fue de 24 semanas y la variable primaria de eficacia fue la proporción de pacientes con una reducción del volumen esplénico de al menos un 35% al final de las 24 semanas de tratamiento, determinado por técnicas de imagen (resonancia magnética). Entre las variables secundarias se incluyeron la durabilidad de la respuesta, la variación en las puntuaciones de los síntomas (MFSAF, Formulario de Evaluación de los Síntomas de Mielofibrosis, v2.0) y la supervivencia global de los pacientes.
El 41,9% (IC95% 34,1 a 50,1) de los pacientes tratados con ruxolitinib alcanzaron la variable primaria, en comparación con el 0,7% (IC95% 0 a 3,6) con placebo. Asimismo, el porcentaje de pacientes que experimentaron una reducción de al menos un 50% en la puntuación de síntomas al cabo de 24 semanas fue del 45,9% vs. 5,3% (p<0,0001), con una variación media de -8,6 puntos (desde un nivel basal de 18,0) vs. +3,2 (desde 16,5). La supervivencia global fue del 91,6% vs. 84,3%, con una reducción del riesgo de muerte del 50% (HR= 0,50; IC95% 0,25 a 0,98; p= 0,04). La tasa de descontinuación del tratamiento durante el estudio debido a eventos adversos fue del 11,0% con ruxolitinib vs. 10,6% placebo.
Tras un seguimiento medio de dos años (Verstovsek, 2013) de los pacientes que habían formado parte de la rama de tratamiento con ruxolitinib en el estudio anterior, 100 de los 155 inicialmente asignados al mismo mantenían el tratamiento (65%), mientras que todos aquellos asignados inicialmente a placebo comenzaron a ser tratados con el fármaco o suspendieron definitivamente su participación en el estudio, dentro de los tres meses posteriores al análisis primario. Los resultados mostraron una reducción media del volumen esplénico del 31,&% a la semana 24 y del 34,9% a la semana 96, manteniéndose igualmente los parámetros de calidad de vida de los pacientes. La supervivencia global al final del periodo fue claramente mejor con ruxolitinib, con una reducción del 42% del riesgo de muerte (HR= 0,58; IC95% 0,36 a 0,95; p= 0,03).
Por su parte, en el estudio COMFORT-II la duración del tratamiento fue de 48 seamanas, siendo la variable primaria de eficacia la proporción de pacientes con una reducción del volumen esplénico de al menos un 35% en las primeras 48 semanas de tratamiento, observándose que un 28,5% (IC95% 21,3 a 36,6) de los pacientes tratados con ruxolitinib habían alcanzado la variable primaria, en comparación con el 0% (IC95% 0 a 5,0) con el mejor tratamiento alternativo; a las 24 semanas, los valores correspondientes habían sido del 31,9% vs. 0%.
Un análisis sectorializado de los datos (Guglielmelli, 2014) ha mostrado que las respuesta sobre los síntomas y la esplenomegalia en el estudio COMFORT-II, así como el riesgo de desarrollar anaemia y trombocitopenia por el ruxolitinib fue similar entre los pacientes con diferentes perfiles mutacionales. Además, se apreció una reducción del riesgo de muerte entre aquellos portadores de mutaciones asociadas a peor pronóstico (ASXL1, EZH2, SRSF2, IDH1/2), con una reducción del 43% (HR= 0,57; IC95% 0,30 a 1,08) vs. mejor tratamiento, aunque no se llegó a alcanzar la significación estadística.
Tras un seguimiento medio de tres años (151 semanas) de los pacientes que inicialmente habían sido tratados con ruxolitinib en el estudio COMFORT-II y que habían mantenido el tratamiento posteriormente (45% al final de este periodo de seguimiento), se observó que la reducción se la esplenomegalia se había mantenido durante al menos 144 semanas, con la probabilidad del 50% (IC95% 36 a 63) entre los pacientes que habían obtenido algún grado de respuesta (Cervantes, 2013). La tasa de mortalidad de los pacientes originalmente aleatoriamente asignados a ruxolitiniv fue un 52% inferior a la de aquellos asignados al mejor tratamiento alternativo (HR= 0,48; IC95% 0,28 a 0,85; p= 0,009).
Desde el punto de vista de la seguridad, el perfil toxicológico del ruxolitinib se caracteriza fundamentalmente por la mielotoxicidad, manifestada comúnmente por trombocitopenia, anemia y neutropenia, consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. La incidencia de eventos adversos asociados al tratamiento con ruxolitinib en los ensayos clínicos principales es elevada, particularmente anemia (82%), trombocitopenia (70%), hemoorragias (33%) y neutropenia (16%); fueron también muy frecuentes las alteraciones de los valores de transaminasas (19-27%), hipercolesterolemia (17%), vértigo (15%), cefalea (14%), infecciones del tracto urinario (12%) y aumento de peso (10%). Considerando específicamente aquellos valorados como graves o muy graves (grados 3-4), los más comunes fueron anemia (43%), trombocitopenia (11%), neutropenia (6,6%), hemorragia (4,7%), aumento de niveles de transaminasas (1,3%) e infecciones del tracto urinario (1,0%).
Aproximadamente el 60% de los pacientes tratados con ruxolitinib tuvieron reducciones en su poslogía y cerca del 30% suspendieron el tratamiento, aunque la mayoría lo hizo por motivos diferentes a efectos adversos. Aunque muy comunes y eventualmente graves, las citopenias pudieron ser manejadas adecuadamente mediante ajuste posológico y transfusiones sanguíneas.
ASPECTOS INNOVADORES
El ruxolitinib es un inhibidor selectivo de las tirosina cinasas de tipo Janus JAK1 y JAK2, que participan en la señalización intracelular de diversas citocinas hepatopoyéticas implicadas en la proliferación de células hematológicas. Ha sido autorizado para el tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. Atendiendo a la condición de enfermedad rara, el medicamento fue designado como huérfano.
El nuevo fármaco ha demostrado ser capaz de reducir de forma notable la esplenomegalia en el 29-42% de los pacientes, frente a menos del 0-1% con placebo o con el mejor tratamiento alternativo disponible. Una reducción del 35% del volumen del bazo determinado mediante técnicas de imagen (resonancia magnética nuclear o tomografía computarizada) viene a equivaler a una reducción del 50% en el tamaño palpable externamente, lo que es considerado internacionalmente como un criterio objetivo de mejoría clínica, que se asocia con una mejora sustancial de los síntomas abdominales y del riesgo en caso de requerirse una esplenotomía. Por otro lado, es preciso indicar que la mayoría (57-75%) de los pacientes incluidos en los estudios habían sido tratados previamente con hidroxiurea, el único tratamiento actualmente autorizado de la mielofibrosis.
Los pacientes que más se benefician del tratamiento con ruxolitinib son los portadores de la mutación JAK2v617F+, con tasas de eficacia del 33-48% vs. 0-1%; una mutación que está presente, en término medio, en el 65% de los pacientes con mielofibrosis. Sin embargo, también es significativa la respuesta en pacientes no portadores de dicha mutación, con tasas de respuesta que siguen estando significativamente por encima (14-28% vs. 0-1%) de las obtenidas con placebo o con tratamientos alternativos (Becker, 2014).
Los datos procedentes de estudios de seguimiento de los estudios clínicos pivotales muestran que el tratamiento continuado es capaz de mantener la respuesta durante periodos de 1 a 3 años, si bien es cierto que en este tipo de estudios se produce la pérdida del seguimiento, por motivos diversos, de un buen número de los pacientes inicialmente incluidos, lo que limita la robustez de los resultados. No obstante, parece apreciarse un incremento de la supervivencia global de los pacientes tratados, que es preciso confirmar con nuevos estudios de seguimiento que incluyan un mayor número de pacientes y periodos más prolongados de observación.
Desde el punto de vista de la seguridad, el ruxolitinib se comporta básicamente como un agente mielotóxico, dando lugar a trombocitopenia, anemia, hemorragias y neutropenia, como consecuencia directa del mismo mecanismo mielosupresor responsable de su uso terapéutico. Aunque se trata de un efecto mielotóxico importante, es previsible y manejable mediante transfusiones y ajustes de la posología del fármaco.
El efecto mielotóxico del ruxolitinib no depende del estatus mutacional JAK2V617F, ya que esta mutación afecta a la estructura de la región por fuera del bolsillo de unión al ATP del enzima, por lo que los inhibidores de la cinasa JAK2 competitivos del ATP – como el ruxolitinib – no pueden distinguir entre las enzimas JAK2 normales y las mutadas. Por ello, este tipo de fármacos producen una mielosupresión no selectiva que, si bien es útil para controlar la hiperproliferación de las células hematopoyéticas en mielofibrosis y en otras patologías mieloproliferativas, pueden no ser capaces de eliminar los clones mutantes.
Aunque es evidente que el ruxolitinib forma parte del ya muy amplio grupo de inhibidores de tirosina cinasas (TKI, tyrosine-kinase inhibitors) disponibles, es preciso matizar que se trata de un fármaco muy peculiar, dada su acción altamente selectiva sobre tirosina cinasas de tipo Janus (JAK), las cuales están estrechamente ligadas a los procesos de señalización celular hematopoyética, lo que determina marcadamente tanto su perfil farmacológico como el toxicológico, agrupados por el efecto mielosupresor del fármaco; en este sentido – pero solo en éste – cabría hablar de cabeza de serie. En cualquier caso, se ha convertido en un referente para el desarrollo de nuevos fármacos más selectivos y eficaces en esta indicación, ofreciendo asimismo la posibilidad de combinar varios mecanismos con un objetivo terapéutico común, permitiendo diluir las consecuencias toxicológicas (Rosenthal, 2014).
En definitiva, se trata de un paso adelante en el tratamiento de una enfermedad rara, aunque su eficacia es modesta y su efecto real sobre la supervivencia global está todavía sujeto a estudio.
|
VALORACIÓN |
|
|---|---|
|
Ruxolitinib ► JAKAVI® (Novartis) |
|
|
Grupo Terapéutico (ATC): L01XE. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Citostáticos: inhibidores directos de la proteína cinasa. |
|
|
Indicaciones autorizadas: Tratamiento de la esplenomegalia u otros síntomas en pacientes adultos con mielofibrosis primaria (mielofibrosis idiopática crónica), secundaria a policitemia vera o secundaria a trombocitemia. |
|
|
VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. |
♣ ♣ |
|
Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar y puede resultar útil en algunos cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
⇑ |
|
Novedad molecular: Incorpora un mecanismo de acción innovador frente al de los tratamientos previamente disponibles para la misma indicación terapéutica. |
⇑ |
|
FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA |
|||
|---|---|---|---|
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Imatinib |
Glivec |
Novartis |
2002 |
|
Erlotinib |
Tarceva |
Roche |
2006 |
|
Sunitinib |
Sutent |
Pfizer |
2007 |
|
Dasatinib |
Sprycel |
Bristol Myers Squibb |
2007 |
|
Sorafenib |
Nexavar |
Bayer |
2007 |
|
Lapatinib |
Tyverb |
Glaxo |
2008 |
|
Nilotinib |
Tasigna |
Novartis |
2008 |
|
Gefitinib |
Iressa |
AstraZeneca |
2010 |
|
Pazopanib |
Votrient |
Glaxo |
2011 |
|
Crizotinib |
Xalkori |
Pfizer |
2014 |
|
Vemurafenib |
Zelboraf |
Roche |
2014 |
|
Dabrafenib |
Tafinlar |
GlaxoSmithKline |
2014 |
|
Axitinib |
Inlyta |
Pfizer |
2014 |
|
Afatinib |
Giotrif |
Boehringer Ingelheim |
2014 |
|
Ruxolitinib |
Jakavi |
Novartis |
2015 |
|
Vandetanib |
Caprelsa |
AstraZeneca |
2015 |
BIBLIOGRAFÍA
Bibliografía
- Becker H, Engelhardt M, von Bubnoff N, Wäsch R. Ruxolitinib. Recent Results Cancer Res. 2014; 201: 249-57. doi: 10.1007/978-3-642-54490-3_16.
- Cervantes F, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Sirulnik A, Stalbovskaya, et al; COMFORT-II investigators. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013; 122(25): 4047-53. doi: 10.1182/blood-2013-02-485888.
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- European Medicines Agency (EMA). Ruxolitinib®. European Public Assessment Report (EPAR). EMA/445881/2012; EMEA/H/C/002464. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002464/WC500133226.pdf
- Guglielmelli P, Biamonte F, Rotunno G, Artusi V, Artuso L, Bernardis I, et al; COMFORT-II Investigators; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative (AGIMM) Investigators. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. 2014; 123(14): 2157-60. doi: 10.1182/blood-2013-11-536557.
- Novelli Canales S, García Cadenas I. Terapéutica farmacológica de los cánceres hematológicos. En: Terapéutica farmacológica de los trastornos neoplásicos e inmunológicos. Consejo General de Colegios Oficiales de Farmacéuticos. Madrid; 2011. pp. 189-220.
- Odenike O. Beyond JAK inhibitor therapy in myelofibrosis. Hematology Am Soc Hematol Educ Program. 2013; 2013: 545-52. doi: 10.1182/asheducation-2013.1.545.
- Rosenthal A, Mesa RA. Janus kinase inhibitors for the treatment of myeloproliferative neoplasms. Expert Opin Pharmacother. 2014; 15(9): 1265-76. doi: 10.1517/14656566.2014.913024.
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366(9): 799-807. doi: 10.1056/NEJMoa1110557.
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013; 98(12): 1865-71. doi: 10.3324/haematol.2013.092155.
1 El riesgo de desarrollar mielofibrosis en los siguientes diez años secundariamente a una policitemia vera es del 10% y menos del 4% en el caso de la trombocitemia esencial.
2 MLP (myeloproliferative leukemia protein) es el receptor de trombopoyetina, también denominado CD110.
3 Responsable de la sustitución de fenilalanina por valina en la posición 617 de la cinasa JAK2.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
