Pomalidomida imnovid® (Celgene)
 Nº375
Nº375

MIELOMA MÚLTIPLE
El mieloma múltiple o mieloma de células plasmáticas es un agresivo tumor hematológico maligno de linfocitos B, que presenta algunas características similares a la leucemia. Es un tumor infrecuente – de hecho, se le considera una enfermedad rara – que afecta fundamentalmente a la población de edad avanzada, hasta el punto de que la edad media de los pacientes a los que se diagnostica la enfermedad es de 65 años y solo el 2% tienen menos de 40 años en el momento de ser diagnosticados.
En España se registran entre 1.500 y 2.000 casos al año, cifras que alcanzan globalmente en la Unión Europea los 28.000 casos. Su incidencia en España es de 1,5-2,5 por cada 100.000 en menores de 65 años, pero sube a 25-30 a partir de esa edad, afectando a un número ligeramente superior de hombres que de mujeres. Las tasas globales de mortalidad para el mieloma múltiple, ajustadas por edad, son de 3,3 casos por 100.000 en hombres y de 2,2 en mujeres. En general, los países del sur de Europa – España entre ellos– tienen tasas de incidencia y mortalidad mucho más bajas que los del norte; sin embargo, tanto en unos como en otros la mortalidad por esta patología está creciendo. Actualmente, tras la incorporación del bortezomib y de los derivados de la talidomida, así como con el trasplante autólogo de células progenitoras hematopoyéticas (TASPE), la supervivencia relativa ajustada por edad a los 5 años del diagnóstico para este tipo de cánceres es del 40-50% en nuestro país, algo mayor en varones que en mujeres; el pronóstico es mejor si la edad en el momento del diagnóstico es de menos de 45 años, alcanzándose tasas de supervivencia a los 5 años del 65%.
Las células plasmáticas malignas características del mieloma múltiple – como células mielomatosas – se acumulan en la médula ósea y pueden llegar al torrente sanguíneo, provocando una alteración de la función de la médula ósea normal, daños óseos y alteración de la función inmune. Las células mielomatosas pueden también formar tumores localizados, los plasmocitomas, que pueden tener una localización tanto ósea como extraósea; justamente es cuando coexiten varios plasmocitomas, con diversas localizaciones, es cuando se emplea apropiadamente el término mieloma múltiple.
La característica típica de la célula mielomatosa es la producción y liberación al torrente circulatorio de una proteína monoclonal, denominada proteína M, pero también conocida como proteína mielomatosa, para-proteína o proteína en pico (esto último es debido a la determinación mediante electroforesis). Se trata de una inmunoglobulina producida como consecuencia de la aparición de una o más mutaciones en los genes responsables de la producción de inmunoglobulinas en la célula mielomatosa.
La proteína M tiene una secuencia de aminoácidos y una estructura anormales, que provocan la adherencia entre sí y a otros tejidos: células sanguíneas, pared de los vasos sanguíneos u otros componentes de la sangre. Todo ello provoca la disminución del flujo sanguíneo, causando un síndrome de hiperviscocidad. En un 30 % de los casos se produce una mayor cantidad de cadenas livianas que las requeridas para combinar con las cadenas pesadas de las inmunoglobulinas, dando lugar a la proteína de Bence-Jones. Esta tiene un peso molecular de 22,000 daltons y es lo suficientemente pequeña como para ser excretada con la orina y producir un aumento de nivel de proteínas urinarias. Estas proteínas también pueden adherirse entre sí o con otros tejidos (como la inmunoglobulina completa), dando lugar a amiloidosis (depósitos proteicos en cualquier tejido del organismo, como riñón, tejido nervioso o músculo cardíaco) o a la enfermedad de depósito de cadenas livianas (las cadenas livianas se depositan al azar, pero especialmente en los pequeños vasos del ojo o del riñón). Las proteínas monoclonales anormales también tienen pueden provocar un amplio abanico de perturbaciones fisiológicas, al unirse a los factores de la coagulación (dando lugar a trastornos diversos de coagulabilidad) y a otras sustancias circulantes, con diversas consecuencias hormonales y metabólicas.
La Beta 2 microglobulina (β2M) es una pequeña proteína cuyos niveles se encuentran elevados en pacientes con mieloma en actividad, aunque un 10% de los pacientes con mieloma no la producen. Precisamente, el estadiaje internacional del mieloma múltiple se basa en las concentraciones en suero de albúmina y de microglobulina β2: el estadio I se caracteriza por niveles de β2M inferiores a 3,5 mg/L y de albúmina mayores de 3,5 g/L; el II por β2M < 3,5 mg/L y de albúmina < 3,5 g/L, o bien β2M de 3,5 a 5,5 mg/L; finalmente, el estadio III se caracteriza por β2M > 5,5 mg/L (Novelli, 2011). Estos estadios se asocian con valores medianos de supervivencia progresivamente inferiores: I (62 meses), II (44) y III (29).
La etiología del mieloma múltiple es poco conocida. No obstante, se ha asociado el mieloma múltiple con la exposición a radiaciones ionizantes. Igualmente, el riesgo parece ser mayor si existen exposiciones ocupacionales relacionadas con la agricultura, refinerías, industrias del corcho, del metal, del plástico o de la madera, o si se ha trabajado como conductor de camiones. Algunos estudios apuntan también hacia la asociación con algunas substancias químicas como asbesto, benceno, pesticidas o pinturas y disolventes. Se ha sugerido la posible relación con algunas enfermedades autoinmunes, como la artritis reumatoide.
El crecimiento descontrolado de las células mielomatosas tiene como una importante consecuencia la destrucción del esqueleto, la insuficiencia de la médula ósea, hipervolemia e hiperviscosidad sanguíneas, supresión de la producción de inmunoglobulinas normales e insuficiencia renal. Sin embargo la enfermedad puede permanecer asintomática durante muchos años. En la fase sintomática, el dolor óseo es el cuadro de presentación más común. Las células mielomatosas y el aumento del número de osteoclastos parecen ser las responsables de la destrucción ósea. El mecanismo que produce la activación de osteoclásticos es complejo, pero se ha observado que participan diversas citocinas locales, tales como interleucinas (IL-1b, IL-6) y factor de necrosis tumoral alfa (TNFα), quimiocinas como las MIP-1 e las integrinas implicadas en el proceso de adhesión celular.
Los factores endocrinos locales que regulan la formación y remodelación ósea son una auténtica miríada y consisten mayoritariamente en citocinas y factores de crecimiento producidos por células de los sistemas inmune y hematopoyético. Actualmente, se considera que los factores efectores finales que regulan la remodelación ósea forman parte de la superfamilia del Factor de Necrosis Tumoral (TNF) y de la de su receptor. Entre ellos puede citarse el RANKL (Ligando del Receptor del Activador del Factor Nuclear Kappa-B), cuya producción es máxima en las células indiferenciadas del estroma osteoclástico y se reduce a medida que madura el fenotipo osteoblástico. Estimula la diferenciación, supervivencia y fusión de las células precursoras de osteoclastos, activa los osteoclastos maduros y prolonga su vida útil. Como resultado permite la expansión de la masa osteoclástica activa capaz de formar sitios de resorción ósea.
El tejido nervioso es afectado con frecuencia en los pacientes con mieloma, tanto por los efectos directos de los anticuerpos de las proteínas mielomatosas frente a la mielina, o por el depósito de fibrillas proteicas (amiloide). Estos efectos resultan en neuropatías periféricas. Por la susceptibilidad a las infecciones, las infecciones virales de los tejidos nerviosos son muy frecuentes, en especial por varicela zoster y parálisis de Bell. Tanto en el hueso como en los tejidos blandos puede producirse una compresión o desplazamiento de los nervios procedentes de la médula espinal o del tallo cerebral.
La predisposición a las infecciones es quizá el rasgo más característico de los pacientes con mieloma junto con la enfermedad ósea, debido a la inhibición de las funciones inmunes normales: producción deficitaria de anticuerpos normales, daño de la función de los linfocitos T y activación anómala de la función monocito/macrófago. Los pacientes con mieloma son particularmente susceptibles a las infecciones virales y a las infecciones con bacterias encapsuladas como el neumococo.
El tratamiento se basa fundamentalmente en la supresión de las células mielomatosas mediante quimio y/o radioterapia, asociado eventualmente a trasplante de células progenitoras hematopoyéticas periféricas. El fármaco antineoplásico más comúnmente utilizado es el melfalán, que continúa siendo el mejor agente único para el tratamiento del mieloma múltiple. La mayoría de los pacientes responden, particularmente cuando se lo utiliza combinado con prednisona. La asociación melfalán/prednisona (protocolo MP) es utilizada frecuentemente, dando lugar a respuestas objetivas en el 60% de los pacientes, manifestada por un 50% mejora en los niveles de proteína M, en los recuentos sanguíneos y en otros resultados bioquímicos, además de la mejoría de varios síntomas de la enfermedad, como el dolor óseo y la fatiga. La ciclofosfamida puede reemplazar el melfalán ya que tiene una actividad antimielomatosa similar, aunque está menos experimentada. Asimismo, en pacientes con mieloma múltiple, los resultados alcanzados con bendamustina son, como mínimo, equiparables a los obtenidos con melfalán (Cuéllar, 2011); incluso, la tasa de respuestas completas es sustancialmente mayor con bendamustina y el porcentaje de pacientes supervivientes a los cinco años también (29% vs. 19%).
Aunque se han desarrollado combinaciones quimioterápicas más complejas con respuestas similares, o incluso ligeramente mejores, no está claro que sean de primera elección. De hecho, el protocolo MP sigue considerándose como el estándar terapéutico de inicio, dejando las combinaciones más complejas como una segunda línea para aquellos pacientes que no alcanzan una respuesta satisfactoria.
Diferentes líneas de investigación han facilitado la incorporación de otros agentes antineoplásicos, de carácter más selectivo, como es el caso del bortezomib, un inhibidor del proteasoma 26S, autorizado en 2004 para el tratamiento de los pacientes con mieloma múltiple que han recibido previamente al menos dos tratamientos y que presentan progresión de la enfermedad demostrada con el último de estos tratamientos. Los datos clínicos disponibles indican unas tasas de respuesta del orden del 35% para respuestas completas y parciales, con tiempos de progresión de la enfermedad de 9 a 13 meses para estos pacientes respondedores, bastante superiores a los descritos en la bibliografía para los pacientes con mieloma múltiple refractario o recidivante (alrededor de tres meses).
Algunos estudios premiliminares mostraron que la talidomida es capaz de producir una significativa respuesta en pacientes con mieloma, con tasas de respuesta del orden del 25% en pacientes con mieloma recidivante/refractario, en su mayoría, después de un doble trasplante. El problema que plantea este tratamiento es el de la toxicidad. De hecho, la talidomida comenzó a ser investigada tan solo cuatro años después de su retirada por su desgraciada implicación en numerosos y graves casos de dismorfogénesis fetal, calculándose que entre 1956 y 1962 más de 10.000 niños nacieron con importantes deformidades, fundamentalmente focomelia. Sin embargo, sus interesantes propiedades inmunomoduladoras atrajeron el interés de los científicos y algunos años después se había comprobado su potencial utilidad en múltiples patologías de tipo dermatológico, infeccioso y autoinmune. Asimismo, se comprobó su potencial antiangiogénico, siendo ensayada en diversos modelos de cáncer.
A pesar de haber sido autorizada como medicamento huérfano, incluso para el mieloma múltiple – en 2006 – su toxicidad siempre ha sido un factor limitante de todo su potencial farmacológico. De ahí, la necesidad de fármacos que mantuvieran sus propiedades o incluso las ampliaran, pero limitando su notable perfil toxicológico. Por ello, se desarrollaron derivados como la lenalidomida, que no solo mantenían las propiedades de la talidomida sobre diversos biomarcadores implicados en diversas patologías, sino que incluso las ampliaban, mejorando los resultados clínicos en mieloma múltiple, con un perfil toxicológico algo más benigno.
Un aspecto confuso del tratamiento del mieloma ha sido el descubrimiento de que una disminución de los niveles de la proteína mielomatosa en el suero y/o en la orina no se traslada necesariamente a una remisión o a un aumento de la supervivencia. Dado que ningún tratamiento actual erradica todas las células mielomatosas, las características de aquellas residuales tras la quimioterapia inicial son de particular importancia. En este sentido, unas pocas células agresivas residuales pueden causar más problemas que una gran cantidad de células inactivas.
El empleo de altas dosis de quimioterapia (AD) asociado con el trasplante autólogo de células progenitoras hematopoyéticas (TASPE) ha demostrado mejorar tanto las tasas de respuesta como las expectativas de vida en los pacientes con mieloma. Se han alcanzado tasas de remisión completa que oscilan entre un 25% y un 75%. No obstante, el 90% de los pacientes recaen, con tiempos medios de recaída de 18 a 24 meses. La expectativa global de vida en estos pacientes es de aproximadamente 4 a 5 años.
La radioterapia es una importante de tratamiento del mieloma. Para los pacientes con importante destrucción ósea, intenso dolor y/o compresión nerviosa o de la médula espinal, la radioterapia local puede ser muy efectiva. La mayor desventaja es que daña en forma permanente las células progenitoras hematopoyéticas normales de la médula ósea en el área tratada.
Al margen del tratamiento específico, los pacientes con mieloma requieren una serie de tratamientos de soporte, destinados a paliar o a suprimir las principales complicaciones orgánicas del mieloma. En este sentido, la epoetina o sus análogos son administraodos para mejorar los niveles de hemoglobina en los pacientes que tienen anemia persistente. Los bisfosfonatos son empleados como coadyuvantes en los pacientes con mieloma que tienen problemas óseos. Igualmente, el uso de altas dosis de gammaglobulina puede ser requerido en pacientes con infecciones agudas y severas recurrentes. Por otro lado, los factores de crecimiento de colonias, como el GM-CSF, pueden ayudar a mejorar el recuento de glóbulos blancos en un esfuerzo por evitar complicaciones infecciosas.
Las recaídas que ocurren tras 1-3 años de haberse producido una primera remisión, son un problema frecuente en el mieloma. Aunque el inteferón alfa o la prednisona en el tratamiento de mantenimiento pueden ser útiles para prolongar el período de remisión inicial, la recaída que sobreviene inevitablemente, requiere una reinducción con quimioterapia. Si ocurre una recaída después de una remisión de por lo menos 6 meses a un año, la primera estrategia es reutilizar la terapéutica que ha producido la remisión la primera vez. Aproximadamente el 50% de los pacientes alcanzarán la remisión. Esto ocurre especialmente en los pacientes en remisión de más de un año, tras la inducción inicial. Si la remisión ha durado menos de 6 meses, se deben utilizar algunas terapias alternativas. Éste también es el caso para la recaída que ha ocurrido después de la segunda o tercera vez que se utilizó el esquema inicial. Si la recaída es sistémica, el uso de la dexametasona, sola o asociada a un derivado de talidomida puede ser útil para controlar la enfermedad durante algún tiempo.
ACCIÓN Y MECANISMO
La pomalidomida es un agente antitumoral análogo de la talidomida, con propiedades inmunomoduladoras y antiangiogénicas. Ha sido autorizada para el tratamiento en combinación con dexametasona de pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
La pomalidomida induce la apoptosis a través de la caspasa-8, inhibe la angiogénesis, modula la secreción de diversas citocinas y obstaculiza las interacciones entre estroma y células de mieloma. La proteína cereblon (CRBN) ha sido identificada como diana farmacológica para la talidomida y sus análogos, asociándose una baja expresión de CRBN con la resistencia a estos fármacos en mieloma. La CRBN interacciona con el factor 4 regulador de interferón, que desempeña un papel crucial en la supervivencia de las células de mieloma (Jurczyszyn, 2014).
El fármaco incrementa la proliferación de linfocitos T (ranto CD4 como CD8) y de células NK (natural killers), así como la expresión de diversas moléculas coestimuladoras, como CD28. Inhibe la producción de diversas citocinas proinflamatorias (Factor de Necrosis Tumoral α [TNFα] y las interleucinas IL-1β, IL-6 e IL-12), incrementando la de otras otras citocinas de carácter antiinflamatorio (IL-10). Aumenta, asimismo, la producción de interferón gamma (IFNγ) e IL-2.
La pomalidomida ha mostrado inhibir el crecimiento tumoral de células resistentes a otros fármacos antineoplásicos, incluyendo la lenalidomida.
ASPECTOS MOLECULARES
La pomalidomida es un análogo estructural de la talidomida y de la pomalidomida, aunque más potente que estos últimos. Químicamente, corresponde a la (RS)4-amino-2-(2,6-dioxopiperidin-3-il)isoindol-1,3-diona. Este tipo de fármacos está emparentados química y farmacológicamente con los antiepilépticos e hipnóticos de tipo ureídico (hidantoínas, barbitúricos, etc.).
La talidomida tiene dos enantiómeros (fruto del carbono asimétrico en la posición 3 del anillo de piperidindiona, que conecta con el anillo indólico). El enatiómero (S) es intensamente teratógeno en seres humanos, mientras que el (R) es el responsable de sus acciones farmacológicas hipnótico-sedantes. El problema no se resuelve utilizando solo el enantiómero (R), ya que ambos enantiómeros sufren fácilmente procesos de interconversión en condiciones fisiológicas, por lo que su
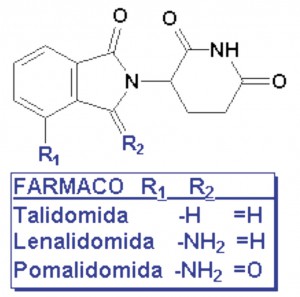
uso por mujeres embarazadas (especialmente durante el primer trimestre) representa un peligro cierto, como lo atestigua tristemente el amplio y devastador historial de malformaciones congénitas que ha dejado especialmente en Europa.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de la pomalidomida han sido adecuadamente contrastadas para la indicación autorizada mediante un ensayo clínico principal de fase III (MM-03; San Miguel, 2013), multicéntrico, multinacional (Australia, Canadá, Estados Unidos, Europa y Rusia, abierto y aleatorizado, en el que se compararó la administración de un régimen de pomalidomida (oral: 4 mg/día, días 1 a 21 de cada ciclo de 28) asociada con dexametasona en dosis bajas (oral: 40 mg, días 1, 8, 15 y 22, de cada ciclo de 28), frente a dexametasona sola en dosis altas (oral: 40 mg, días 1 a 4, 9 a 12 y 17 a 20, de cada ciclo de 28). Se seleccionaron a 455 pacientes diagnosticados de melanoma múltiple con enfermedad mensurable (proteína M sérica ≥ 500 mg/dL o en orina ≥ 200 mg/24 h) que fuese refractaria o refractaria-recidivante tras dos tratamientos al menos, incluyendo a lenalidomida y bortezomib. Los pacientes tenían una mediana de edad de 64 años (35-87), siendo el 46% de ellos mayores de 65 años y el 8% mayores de 75; el 60% eran varones, el 79% de raza caucásica y en cuanto a los antecedentes terepéuticos, el 92% era refractario a lenalidomida, el 78% a bortezomib y el 72% a ambos.
Como variable primaria de eficacia se estableció mediana de la supervivencia libre de progresión tumoral (tiempo transcurrido desde la primera dosis hasta la constatación de progresión de la enfermedad o muerte del paciente); como variables secundarias se determinaron, entre otras, la supervivencia global (tiempo hasta la muerte del paciente, por cualquier causa), la tasa de respuesta objetiva (parcial y completa), tiempo hasta la progresión tumoral y la duración de la respuesta.
Los resultados obtenidos mostraron unos valores medianos de supervivencia libre de progresión tumoral de 15,7 semanas (IC95% 13,0 a 20,1) vs. 8,0 (IC95% 7,0 a 9,0), con una reducción de la tasa de riesgo del 55% (HR: 0,45; IC95% 0,35 a 0,59; p< 0,0001). La supervivencia global no pudo ser establecida completamente por la insuficiente duración del seguimiento (IC95% 48,5 semanas a no determinado) para la combinación de pomalidomida-dexametasona, pero fue significativamente mayor que con dexametasona en altas dosis (HR: 0,53; IC95% 0,37 a 0,74; p< 0,0001), que fue de 34,0 semanas (IC95% 23,4 a 39,9); por su parte, las tasas de respuesta objetiva fueron del 15,2% vs. 3,3%. No se observaron diferencias significativas (P= 0,565) en el tiempo hasta respuesta (en los respondedores): 8,1 (IC95% 4,0 a 24,2) vs. 7,9 (IC95% 4,1 a 24,1).
En cuanto a la seguridad, la pomalidomida asociada a la dexametasona en dosis menores mostró un perfil toxicológico complejo, con eventos adversos frecuentes y graves, aunque con una incidencia similar a la de la dexametasona en dosis altas; en este sentido, el 78% vs. 76% de los pacientes mostraron eventos adversos de grado 3-4. Entre ellos, la incidencia de eventos adversos hematológicos fue especialmente marcada: neutropenia (42% vs. 15%), anemia (27% vs. 29%) y trombocitopenia (21% vs. 24%); otros eventos adversos graves fueron neumonía (9,0% vs. 7,4%), dolor óseo (6,3% vs. 2,7%), fatiga (4,7% vs. 4,7%), disnea (4,7% vs. 4,7%) y fiebre (3,0% vs. 2,7%); las alteraciones bioquímicas graves fueron más comunes con la dexametasona en altas dosis: hiperglucemia (3,0% vs. 6,7%) e hipercalcemia (3,7% vs. 5,4%). El 5% vs. 4% de los pacientes con al menos un evento adverso relacionado con el tratamiento suspendieron el tratamiento por este motivo. La incidencia de eventos adversos con deslace fatal fue del 5% vs. 4%.
ASPECTOS INNOVADORES
La pomalidomida es un agente antitumoral análogo de talidomida y lenalidomida, con propiedades inmunomoduladoras y antiangiogénicas. Ha sido autorizada para el tratamiento en combinación con dexametasona de pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento. Dada la condición de enfermedad rara que tiene el mieloma múltiple, la pomalidomida fue designada como medicamento huérfano.
El ensayo clínico pivotal en el que se fundamenta su autorización por la EMA y la FDA es metodológicamente consistente y sus conclusiones estadística y clínicamente robustas, lo que resulta especialmente interesante atendiendo a la condición de medicamento huérfano. Sea como fuere, se ha establecido una diferencia estadísticamente significativa a favor de la combinación pomalidomida-dexametasona vs. dexametasona en altas dosis de casi dos meses (15,7 vs. 8,0 semanas) para la supervivencia libre de progresión tumoral, y mejoras también significativas en la supervivencia global. Aunque los datos numéricos no son obviamente espectaculares, una mejora de estas características debe valorarse favorablemente teniendo en cuenta que se ha conseguido en pacientes con refractarios o refractarios y recidivantes tras al menos dos tratamientos previos, en los que se incluía bortezomib y lenalidomida, considerados como piezas clave en el tratamiento del mieloma múltiple.
Su perfil toxicológico – sin olvidar la teratogenicidad heredada de la talidomida – es importante, aunque es preciso relativizarlo en el contexto de la enfermedad neoplásica avanzada en la que se utiliza y, además, considerando que la dexametasona que acompaña a la pomalidomida en su indicación contribuye en buena manera a dicho perfil toxicológico. En cualquier caso, es particularmente importante la toxicidad hematológica (neutropenia, especialmente), aunque comparativamente la pomalidomida produce menos neuropatía que la talidomida y menos tromboembolismo que la lenalidomida.
Los datos tanto in vitro como clínicos muestran que la pomalidomida presenta una actividad antimielomatosa mayor que la talidomida y la lenalidomida, con un perfil toxicológico similar o incluso algo más favorable. La importancia de este hecho viene reflejada por la circunstancia de que tanto la FDA como la EMA utilizaron un sistema acelerado de autorización, al considerar que la pomalidomida podría constituirse en referencia terapéutica en estos cuadros avanzadas y multirresistentes de mieloma, tradicionalmente con muy mal pronóstico (Offidani, 2014). En este sentido, a pesar de las mejoras incorporadas en el tratamiento del mieloma, especialmente con el bortezomib y la lenalidomida (tras la talidomida), la mayoría de los casos acaban por hacerse resistentes y, por ello, la llegada de la pomalidomida puede considerarse un cierto avance en este campo, al haber mostrado eficacia sobre dichos cuadros resistentes (Mark, 2014). Así y todo, la magnitud global de la respuesta – y el lado oscuro de su toxicidad – no permite considerar a la pomalidomida como un avance sustancial en este campo, aunque sí viene a aportar alguna mejora en relación a sus antecedentes (talidomida y lenalidomida).
BIBLIOGRAFIA
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Bendamustina (Levact®) en leukemia linfocítica crónica, linfoma no-Hodgkin y mieloma múltiple. Panorama Actual Med. 2011; 35(344): 499-513.
- European Medicines Agency (EMA). Imnovid®. European Public Assessment Report (EPAR). EMA/540982/2013; EMEA/H/C/002682. http://www.ema.europa.eu/.
- Jurczyszyn A, Legieć W, Helbig G, Hus M, Kyrcz-Krzemień S, Skotnicki AB. New drugs in multiple myeloma - role of carfilzomib and pomalidomide. Contemp Oncol (Pozn). 2014; 18(1): 17-21. DOI: 10.5114/wo.2014.40175
- Mark TM, Coleman M, Niesvizky R. Preclinical and clinical results with pomalidomide in the treatment of relapsed/refractory multiple myeloma. Leuk Res. 2014; 38(5): 517-24. doi: 10.1016/j.leukres.2014.02.008.
- Novelli Canales S, García Cadenas I. Terapéutica farmacológica de los cánceres hematológicos. En: Terapéutica farmacológica de los trastornos neoplásicos e inmunológicos. Consejo General de Colegios Oficiales de Farmacéuticos. Madrid; 2011. p. 189-220.
- Offidani M, Corvatta L, Caraffa P, Leoni P, Pautasso C, Larocca A, Palumbo A. Pomalidomide for the treatment of relapsed-refractory multiple myeloma: a review of biological and clinical data. Expert Rev Anticancer Ther. 2014; 14(5): 499-510. doi: 10.1586/14737140.2014.906904.
- San Miguel J, Weisel K, Moreau P, Lacy M, Song K, Delforge M, Karlin L, Goldschmidt H, Banos A, Oriol A, Alegre A, Chen C, Cavo M, Garderet L, Ivanova V, Martinez-Lopez J, Belch A, Palumbo A, Schey S, Sonneveld P, Yu X, Sternas L, Jacques C, Zaki M, Dimopoulos M. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013; 14(11): 1055-66. doi: 10.1016/S1470-2045(13)70380-2.
| VALORACIÓN | |
|---|---|
| POMALIDOMIDA IMNOVID®(Celgene) | |
| Grupo Terapéutico (ATC): L04AX. TERAPIA ANTINEOPLÁSICA Y AGENTES INMUNOMODULADORES. Inmunosupresores: otros. | |
| Indicaciones autorizadas: Tratamiento en combinación con dexametasona de pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento. | |
| VALORACIÓN GLOBAL: INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar. | ♣ ♣ |
| Novedad clínica: Mejora la eficacia clínica del tratamiento farmacológico estándar | ⇑ |
| Novedad toxicológica: Mejora el perfil toxicológico con relación a la terapia farmacológica estándar. | ⇑ |
| FÁRMACOS RELACIONADOS REGISTRADOS ANTERIORMENTE EN ESPAÑA | |||
|---|---|---|---|
| Fármaco | Medicamento® | Laboratorio | Año |
| Lenalidomida | Revlimid | Celgene | 2007 |
| Pomalidomida | Imnovid | Celgene | 2014 |
| COSTE DIRECTO DEL MEDICAMENTO | ||
|---|---|---|
| Fármaco | Dosis | Coste mensual |
| Pomalidomida | 4 mg/24 h* | 9.730,15 € |
* Durante 21 días, en ciclos de 28.
Artículos relacionados
-
31 Mar 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2026Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
23 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
