Ibrutinib ▼Imbruvica® (Janssen Cilag) en linfoma de células del manto, leucemia linfática crónica y macroglobulinemia de Waldenström
 Nº392
Nº392

Resumen
El ibrutinib es un agente antineoplásico que actúa inhibiendo de forma irreversible y selectiva a la tirosina cinasa de Bruton (BTK), un miembro de las familia de las tirosina cinasas Tec que participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados en la patogenia de diversas neoplasias de linfocitos B. icha inhibición impide la adhesión y migración dependientes de integrinas de los linfocitos B. Ha sido autorizado, como medicamento huérfano, para el tratamiento de pacientes adultos con linfoma de células del manto en recaída o refractario; asimismo, sea autorizado para el tratamiento de pacientes adultos con leucemia linfática crónica que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP53 en pacientes en los que la inmunoquimioterapia no se considera apropiada. Finalmente, también ha sido autorizado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström que han recibido al menos un tratamiento previo, o en tratamiento de primera línea en pacientes en los que la inmunoquimioterapia no se considera apropiada. El ibrutinib viene a asumir un papel relevante en el tratamiento de un conjunto de linfomas que, aunque de muy baja prevalencia (al fin y al cabo, estamos ante un medicamento huérfano), actualmente no tienen un buen tratamiento especialmente en aquellos casos refractarios o recidivantes tras quimio e inmunoterapia. Desde el punto de vista farmacológico, nos encontramos no ante el enésimo inhibidor de tirosina cinasas, sino ante el primer inhibidor selectivo de un tipo muy especial que está particularmente implicado en la génesis de estos linfomas, la tirosina cinasa de Bruton (BTK); lo cual, unido a una cómoda administración oral única al día, nos lleva a considerar al ibrutinib como un medicamento portador de una innovación importante.
ASPECTOS FISIOPATOLÓGICOS
Los linfomas son neoplasias del sistema linfoide que constituyen un grupo heterogéneo de enfermedades neoplásicas definidas por aspectos morfológicos, inmunofenotípicos y genéticos, que tienen su origen en los sistemas mononuclear fagocítico y linfático. Clásicamente, los linfomas se clasificaban en dos grandes grupos, la enfermedad de Hodgkin, que representa el 15-20% de los casos, y los linfomas no Hodgkin. Actualmente, se acepta la clasificación de la Organización Mundial de la Salud (OMS), en la que se definen 3 categorías de neoplasias linfoides: el linfoma de Hodgkin y, dentro de los linfomas no Hodgkin, los de origen B o T/NK. Los linfomas de Hodgkin consisten en una proliferación, localizada o diseminada, de células tumorales que se originan en el sistema linforreticular y que afecta principalmente los ganglios linfáticos y la médula ósea.
Los linfomas no-Hodgkin (LNH) incluyen a todos los linfomas que no encajan dentro de la definición de linfoma de Hodgkin; por tanto, son neoplasias linfoides que pueden presentar fenotipo de células B ó T/NK. Los LNH representan el 4-5% de los nuevos casos de cáncer diagnosticados al año, ocupando el quinto lugar en frecuencia; los de linfocitos B representan el 80-85% de los LNH y los T el 15-20%, mientras que los de células NK (Natural Killer; citotóxicas) tienen una frecuencia marginal.
Entre los linfomas de células B, los más comunes son el linfoma difuso de células B grandes (30-35%) y el linfoma folicular (20-25%); menos prevalentes son el linfoma de tejido linfoide asociado a mucosas (TLAM) (7-10%), el linfoma linfocítico pequeño o leucemia linfocítica crónica (6-8%), el linfoma de células del manto (5-7%), el linfoma de Burkitt (2-3%) y el linfoma mediastínico (tímico) de células B grandes (2-3%). Menos del 2% de los linfomas de células B corresponden al linfoma linfoplasmacítico (macroglobulinemia de Waldenström), el nodal de células B de la zona marginal, el esplénico de zona marginal, el extranodal de células B de zona marginal, el intravascular de células grandes B, el de efusión primaria y la granulomatosis linfomatoide.
Por su parte, los linfomas de células T, que suponen aproximadamente el 12% de todos los LNH, se clasifican en: linfoma extranodal T, linfoma cutáneo de las células T (Síndrome de Sézary y Micosis fungoide), linfoma anaplásico de células grandes y linfoma angioinmunoblástico de las células T. La incidencia de linfomas de células NK es notablemente inferior.
En las últimas décadas se ha registrado un aumento en las tasas de incidencia y de mortalidad de los linfomas no Hodgkin, principalmente en países industrializados. Específicamente, se ha observado un aumento especialmente acusado de las tasas de incidencia de los linfomas no-Hodgkin en España e Italia. El aumento afecta a todos los grupos de edad adulta, aunque el mayor aumento se registra en los sectores de edad más avanzada de la población. En España, según datos del Instituto Nacional de Estadística, murieron en 2013 un total de 4.832 personas (en 2008, fueron 4.451) por tumores malignos del tejido linfático, excepto leucemias, de las que un 53% eran varones y un 47% mujeres.
Los linfomas no hodgkinianos pueden aparecer en cualquier edad de la vida, pero la mediana de presentación se sitúa en torno a los 50 años, siendo más frecuentes en los varones. Tanto en las neoplasias linfoides B como en las T se distinguen dos tipos de transformación neoplásica, uno que se origina a partir de las células precursoras y el otro a partir de las células periféricas.
La etiopatogenia de los LNH varía en los distintos tipos, pero presentan factores de riesgo comunes, tales como la existencia de un sistema inmune debilitado (ya sea por una enfermedad hereditaria o tras un trasplante de órganos), edad elevada, antecedentes familiares, exposición a agentes tóxicos (herbicidas) e infecciones por algunos virus (virus linfotrópico de células T del ser humano tipo 1 –HTLV-1–, virus de la inmunodeficiencia humana –VIH–, virus de Epstein-Barr –EBV–) y bacterias (Helicobacter pylori).
Los linfomas se pueden clasificar por la célula maligna de origen: centro germinal o no centro germinal; también tenemos la zona del manto y la zona marginal. También es muy importante la manera en que se infiltra el ganglio, por ejemplo: el linfoma difuso de célula grande (LDCG) es un linfoma de linfocitos B de tamaño grande (por su estado de maduración) que infiltra el ganglio de forma difusa y puede infiltrarlo tanto en el centro germinal como fuera del mismo (no centro germinal).
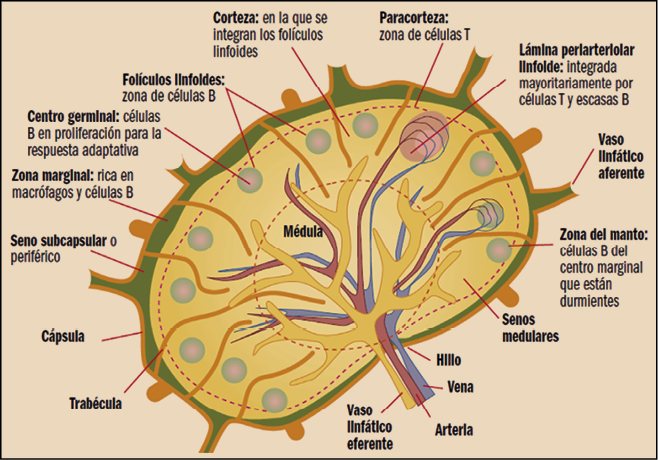
El linfoma de células del manto (LCM) afecta a los linfocitos B en una región de los ganglios linfáticos llamada “zona del manto’’ y representa el 6% de los linfomas no-Hodgkin. Su incidencia se estima en alrededor de 1/100.000. Su incidencia aumenta con la edad, alcanzando un máximo entre los 70 y 79 años, con una notable predominancia masculina (4:1). En el momento del diagnóstico, la mayoría de los pacientes presentan una forma diseminada de la enfermedad. Frecuentemente se asocia con adenopatía generalizada (90%), afectación gastrointestinal (60%) y medular (55-80%). Puede aparecer fiebre y una alteración del estado general (fatiga, pérdida de apetito, pérdida de peso).
El linfoma de células del manto está causado por una translocación cromosómica t(11;14) (q13; q32), que yuxtapone el gen CCND1 al gen que codifica para la cadena pesada de las inmunoglobulinas, conduciendo a una expresión anormalmente alta de ciclina D1, un regulador del ciclo celular, en el núcleo de las células linfoides. El tratamiento del linfoma de células del manto se basa en una quimioterapia intensiva, combinada con anticuerpos monoclonales. En los pacientes más jóvenes, habitualmente se indica un trasplante de células madre autólogas.
No hay, por el momento, ningún tratamiento curativo del LCM, salvo aquellos pacientes que presenten periodos prolongados de supervivencia sin enfermedad tras un trasplante de alogénico de células madre. Entre aquellos con recaídas, la media de esperanza de vida es de 1 a 2 años tras los tratamientos de rescate, frecuentemente asociados a una importante toxicidad.
La leucemia linfática crónica o leucemia linfocítica crónica (LLC) se considera un linfoma de bajo grado, caracterizado por la acumulación de linfocitos B maduros en la sangre, médula ósea y órganos linfáticos. En función de la citogenética, tiene mal pronóstico si presenta la traslocación t(11q; v) o las deleciones del(11q) o del(17p); esta última confiere resistencia a la fludarabina. Por el contrario, el pronóstico es favorable si presenta la deleción del(13q) como única anormalidad. Los linfocitos circulantes son morfológicamente similares a los normales pero funcionalmente anormales; expresan marcadores de superficie CD5, CD20 y CD23. La acumulación se inicia frecuentemente en la médula ósea, diseminándose posteriormente hacia los ganglios linfáticos y bazo, pudiendo haber esplenomegalia.
Es la leucemia más frecuente en los países occidentales, constituyendo el 30 % de todas las formas de leucemia y 75 % de las leucemias crónicas. Su incidencia es de 4,2 casos/100.000 habitantes/año, aunque aumenta con la edad, siendo rara antes de los 40 años; en cualquier caso, es evidente que se trata de una enfermedad rara. A la edad de 50 años alcanza los 5 casos y a los 80 años llega a los 30 casos; de hecho, la mediana de la edad en el momento del diagnóstico es 72 años. Existe un predominio en el sexo masculino (2:1).Afecta a más de 300.000 personas en el mundo y a más de 15.000 en España; concretamente, se diagnostican alrededor de 1.800 nuevos casos cada año en nuestro país.
Los pacientes con LLC activa se caracterizan por una acumulación progresiva de linfocitos, a veces con linfadenopatía, hepatoesplenomegalia, anemia y trombocitopenia. Asimismo, produce un estado de inmunosupresión que incrementa el riesgo de infecciones, que en última instancia es la principal causa de muerte en estos pacientes. El subtipo más frecuente de la LLC es la que afecta a células (linfocitos) B, que representa más del 97% de los casos. En restante el 2-3%, la proliferación clonal anormal se produce a partir de células T. También se han incluido otros patrones leucémicos crónicos dentro de la leucemia linfocítica crónica, tales como la leucemia prolinfocítica, la fase leucémica del linfoma cutáneo de células T (síndrome de Sézary), la leucemia de células peludas (tricoleucemia) y el linfoma leucemizado.
El origen de la LLC sigue siendo desconocido, aunque se apuntan varios hipotéticos, como las radiaciones ionizantes, los agentes alquilantes o ciertos productos leucemógenos, que parecen aumentar el riesgo de desarrollar LLC. La acumulación de linfocitos parece deberse un funcionamiento erróneo en la apoptosis (muerte celular programada); no obstante, se han descrito otros mecanismos que posiblemente colaboren de alguna manera en la acción proliferativa, como ciertas interleucinas o sus receptores, como el factor de necrosis tumoral (TNF) o las interleucinas IL-4 e IL-6. Aproximadamente la mitad de los pacientes, y aún más en estadios avanzados, presentan algún tipo de alteración citogenética. La más frecuente (25-30%) es la trisomía del par cromosómico 12; otras alteraciones menos frecuentes afectan a los cromosomas 11, 12, 13, 14 y 17 (Cuéllar, 2015).
Globalmente, la mediana de supervivencia desde el diagnóstico varía entre los 18 meses y más de 20 años, dependiendo de la presencia de factores de riesgo. En pacientes asintomáticos en estadios iniciales, la mediana de supervivencia es de más de 10 años, mientras que en pacientes con enfermedad avanzada, sintomática o progresiva, la mediana de supervivencia oscila entre 18 meses y 3 años. Debe tenerse en cuenta que los pacientes con LLC también son más propensos a desarrollar una segunda neoplasia. Se han descrito tres grupos pronósticos en función de la citogenética, siendo peor para los casos relacionados con una translocación t(11q; v) o una deleción del(11q) o del(17p); particularmente, esta última confiere resistencia a la fludarabina y se considera como de muy alto riesgo. Considerando un pronóstico de gravedad intermedia la trisomía del par 12 (+12), los casos con mejor pronóstico son aquellos cuya anomalía citogenética implica una deleción del(13q).
La terapia no es curativa y, en cualquier caso, no está indicada en los pacientes con LLC hasta que aparezcan síntomas o progrese la enfermedad. El tratamiento específico incluye quimioterapia, corticoides, cirugía y radioterapia, aunque la cirugía o la radioterapia sólo son útiles en situaciones especiales. En cualquier caso, no se ha demostrado que ninguno de los tratamientos actuales prolongue sustancialmente la supervivencia y generalmente se acepta que el sobretratamiento es más peligroso que el infratratamiento.
La macroglobulinemia de Waldenström es un trastorno linfoproliferativo de células B caracterizado por la acumulación de células monoclonales en la médula ósea y los tejidos linfoides periféricos, que está asociado con la sobreproducción de proteínas séricas (inmunoglobulinas M monoclonales, IgM); es actualmente considerado como un linfoma linfoplasmacítico. Esta enfermedad tiene una incidencia global de 1/260.000 personas/año y supone menos del 2% de todos los linfomas no-Hodgkin. La edad media al diagnóstico es de 63-68 años y la enfermedad afecta en doble proporción a hombres que a mujeres. Son características la hepatoesplenomegalia, la linfadenopatía, la hemorragia oronasal, el síndrome de hiperviscosidad y la citopenia. La fatiga relacionada con una anemia normocítica normocrómica es el síntoma que se presenta con más frecuencia. La infiltración visceral es poco frecuente pero puede dirigirse al estómago, intestino delgado, pulmones, glándulas exocrinas o piel, con síntomas como diarrea, esteatorrea y coloración púrpura de la piel. Se dan neuropatías periféricas en hasta el 38% de los pacientes.
Tiene claramente un componente hereditario, aunque todavía no se han identificado los genes de susceptibilidad para esta enfermedad; no obstante, se ha localizado un loci de susceptibilidad en el cromosoma 6p21.3 y en 4q, y la mitad de los pacientes tienen deleciones del6q en las células tumorales. Más del 90% de los pacientes presentan una mutación somática MYD88 (L265P), la cual favorece el crecimiento neoplásico a través de la vía de la tirosina cinasa de Bruton (BTK).
El tratamiento de los linfomas de grado bajo o indolentes no está estandarizado, aunque la terapia inicial suele consistir en el empleo de antineoplásicos de tipo de los agentes alquilantes (ciclofosfamida, bendamustina) o antimetabolitos (fludarabina) en monoterapia o en combinaciones de 2, 3 o 4 agentes: vincristina, ciclofosfamida, doxorubicina y prednisona más rituximab (protocolo CHOP+R), ciclofosfamida, vincristina, prednisona + rituximab (CVP+R), fludarabina, ciclofosfamida, mitoxantrona, rituximab (FCMR) o fludarabina, mitoxantrona, dexametasona, rituximab (FND-R). Este tipo de tratamientos permite alcanzar tasas de respuesta global de hasta un 90%, que son completas hasta en el 60% de los casos primarios. La duración media de la respuesta oscila entre uno y cuatro años. En caso de recaída, la opción más básica consiste en repetir el tratamiento, generalmente añadiendo un escalón más en combinación.
Los receptores CD20 están presentes en la superficie celular de los linfocitos B, tanto normales como malignos (incluyendo las formas maduras, proliferantes y diferenciadas). Actúan como receptores moleculares del antígeno Bp35, una proteína fosforilada responsable de la restricción de la diferenciación de los linfocitos B que es expresada durante las fases más precoces. Esta circunstancia ha conducido al desarrollo de anticuerpos específicos, como el rituximab, capaces de provocar la destrucción selectiva de linfocitos B (aunque sin distinción entre fisiológicos y malignos) como consecuencia de una combinación, en diferentes grados, de efectos celulares de citotoxicidad y fagocitosis mediados por anticuerpos, así como apoptosis independiente de la cascada de caspasas (inducción directa de la muerte celular) y citotoxicidad dependiente del complemento; no obstante, se considera que la citotoxicidad y la fagocitosis celulares mediadas por anticuerpos son los mecanismos antineoplásicos principales de este tipo de antucuerpos anti-CD20.
Ibritumomab es otro anticuerpo monoclonal que une de forma específica al receptor CD20; está ligado a un agente quelante, el tiuxetano, que actúa como anclaje de un radioisótopo del itrio (90Y), un radionúclido emisor de radiación beta (electrones), de baja penetración (5-10 mm) y con una vida media de 64 h. El medicamento está indicado en el tratamiento de pacientes adultos con linfoma no Hodgkin (LNH) folicular de células B CD20+ en recaída o refractario a rituximab. Normalmente, el tratamiento con ibritumomab-tiuxetano-itrio(90) va precedido por un tratamiento con bajas dosis de rituxumab, con el fin de eliminar los linfocitos B circulantes, facilitando con ello la acción más selectiva del ibritumomab-tiuxetano-itrio(90). Se trata, por consiguiente, de un agente inmunoradioterapéutico, en el que la fracción inmunológica (ibritumomab) es un anticuerpo específico para los receptores CD20, presentes en la gran mayoría de los linfocitos B humanos, tanto fisiológicos como malignos. Esta fracción tiene como misión localizar los linfocitos B, que es la población celular cuya malignificación conduce al desarrollo del linfoma folicular no-Hodgkin. Esta forma de inmunoradioterapia con ibritumumab-tiuxetano- 90Y para las fases avanzadas de linfoma folicular no Hodgkin da lugar a tasas relativamente altas de respuesta, superiores al rituxumab, incluso en pacientes refractarios a otros tratamientos antineoplásicos (incluyendo al propio rituxumab), con respuestas que llegan a alcanzar en una minoría de pacientes hasta más de tres años, aunque dada la fase avanzada de desarrollo del linfoma y la refractoriedad a otras terapias, previsiblemente no puede considerarse éste como un parámetro especialmente relevante.
Por su parte, la bendamustina ha demostrado una clara superioridad sobre el clorambucilo en pacientes con leucemia linfocítica crónica intolerantes a la fludarabina, consideraba como tratamiento de referencia (en combinación con rituximab y ciclfosfamida), si bien la toxicidad de bendamustina también es algo mayor que la del clorambucilo; con todo, el balance beneficio/riesgo parece claramente favorable a la bendamustina.
Los análogos de purinas, fludarabina y cladribina, son algo más eficaces y menos tóxicas que los agentes alquilantes, produciendo remisiones más prolongadas, si bien tampoco se han demostrado ventajas sustanciales con respecto a la supervivencia global, al menos en monoterapia. En pacientes previamente no tratados, con la fludarabina se obtiene un 30-40 % de respuestas completas y un 8-20 % de respuestas parciales, con una mediana de duración de la respuesta de más de 2 años y una supervivencia proyectada a los 6 años del 80 %. La respuesta máxima se obtiene en 2-3 meses y, en caso de no conseguirse respuesta, se debe detener el tratamiento. En pacientes con enfermedad refractaria o en recaída tras diversos tratamiento previos, han demostrado respuestas importantes en el 30-60 % de los casos (15-40 % respuestas completas y, adicionalmente, un 15-50 % respuestas parciales). La cladribina actúa tanto sobre linfocitos (proliferantes o en reposo) como monocitos. En pacientes no tratados previamente se ha obtenido un 25 % de respuestas completas y un 60 % de respuestas parciales, con una mediana de duración de la respuesta superior a 8 meses. En pacientes previamente tratados, se ha obtenido un 5 % de respuestas completas y un 40 % de respuestas parciales, con una mediana de duración de la respuesta de 4 meses.
El rituximab ha demostrado eficacia en pacientes con leucemia linfocítica crónica intensamente pretratados (50% de respuesta global). Con posibilidad de retratamiento (44% de respuesta global). Disminución del tamaño del tumor (87% de los pacientes). En combinación con quimioterapia CHOP (95% de respuesta). La combinación de rituximab con fludarabina y ciclofosfamida requiere una función renal normal y un estado general adecuado, además de que la presencia de determinadas anomalías citogenéticas (como la del17p) son predictores de mala respuesta al tratamiento, lo que limita su utilidad. Cuando la fludarabina no sea apropiada, se considera al clorambucilo, o mejor aún a la bendamustina, como una alternativa para la primera línea de tratamiento, especialmente en pacientes de edad avanzada y comorbilidad importante.
El ofatumumab es otro un anticuerpo monoclonal humano (IgG1) que se une específicamente a la proteína CD20 presente en la membrana de los linfocitos B. Fue autorizado para el tratamiento de leucemia linfocítica crónica en pacientes que son refractarios a fludarabina y alemtuzumab. Ofatumumab produce una respuesta objetiva clínicamente relevante, en la que un 49% de los pacientes doblemente refractarios a fludarabina y alemtuzumab tuvieron una respuesta parcial al tratamiento con ofatumumab, con una superviencia media libre de enfermedad de 6,4 meses y una supervivencia global de 13,9, sin que haya diferencias sustanciales entre los pacientes tratados previamente o no con rituximab (Cuéllar, 2014).
El obinutuzumab también es un anticuerpo monoclonal humanizado de tipo IgG1 (κ) que se une específicamente a la cadena extracelular de la proteína CD20, en este caso autorizado para el tratamiento, en combinación con clorambucilo, de pacientes adultos con leucemia linfática crónica (LLC), no tratados previamente y con comorbilidades que les hace no ser adecuados para el tratamiento basado en una dosis completa de fludarabina. El obinutuzumab forma parte del tipo II de anticuerpos anti-CD20 utilizados en terapéutica. Los de tipo I (rituximab, ofatumumab) actúan fundamentalmente mediante un potente efecto citotóxico dependiente del complemento, en tanto que los de tipo II tienen un efecto citotóxico directo mucho más marcado que su efecto dependiente del complemento. Los anticuerpos de tipo I inducen la translocación del CD20 en grandes microdominios lipídicos, fenómeno al que se atribuye su mayor actividad citotóxica dependiente del complemento. Por el contrario, los de tipo II, como el obinutuzumab, son significativamente más potentes que los de tipo I en inducir la apoptosis de células CD20+, tanto normales como malignas. El obinutuzumab parece producir una mayor depleción que el rituximab de linfocitos B, tanto normales como malignos.
La radioterapia se utiliza sólo para reducir adenopatías que causan síntomas compresivos en pacientes refractarios a otros tratamientos. Aunque la irradiación corporal total en pequeñas dosis ha tenido resultados satisfactorios en algunas ocasiones, el uso más habitual es en administración local en zonas de adenopatías, hígado o bazo para obtener una paliación sintomática transitoria.
No hay tratamiento curativo para la macroglobulinemia de Waldenström. Se debe controlar a los pacientes en etapa asintomática. Los tratamientos que se dan a pacientes sintomáticos dependen de muchos factores y pueden incluir agentes alquilantes, cladribina, fludarabina, rituximab y bortezomib. Cuando se necesita un control rápido de la enfermedad o para aquellos candidatos a un trasplante de células madre autólogo, se usa rituximab combinado con ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP) o dexametasona, rituximab y ciclofosfamida (DRC) o terapias basadas en bortezomib. El tiempo de supervivencia media es de 5-6 años después de iniciar el tratamiento, pero la MW puede ser estable o progresar lentamente durante muchos años antes de necesitar de un tratamiento.
Por su parte, el temserolimús y el bortezomib son los únicos fármacos que hasta ahora habían recibido la autorización de comercialización en Europa como medicamento huérfano para la enfermedad refractaria o recidivante. Las formas localizadas de la enfermedad pueden beneficiarse de una radioterapia. Algunos pacientes (30%) presentan una respuesta completa a los tratamientos actuales. Las medias de supervivencia globales son de 3-5 años.
ACCIÓN Y MECANISMO
El ibrutinib es un agente antineoplásico que actúa inhibiendo de forma irreversible y selectiva a la tirosina cinasa de Bruton (BTK), un miembro de las familia de las tirosina cinasas Tec que participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados en la patogenia de diversas neoplasias de linfocitos B. Dicha inhibición impide la adhesión y migración dependientes de integrinas de los linfocitos B.
El medicamento ha sido autorizado para el tratamiento de pacientes adultos con linfoma de células del manto en recaída o refractario; también está indicado para el tratamiento de pacientes adultos con leucemia linfática crónica que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP53 en pacientes en los que la inmuno-quimioterapia no se considera apropiada. Asimismo, también está indicado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström que han recibido al menos un tratamiento previo, o en tratamiento de primera línea en pacientes en los que la inmuno-quimioterapia no se considera apropiada.
ASPECTOS MOLECULARES
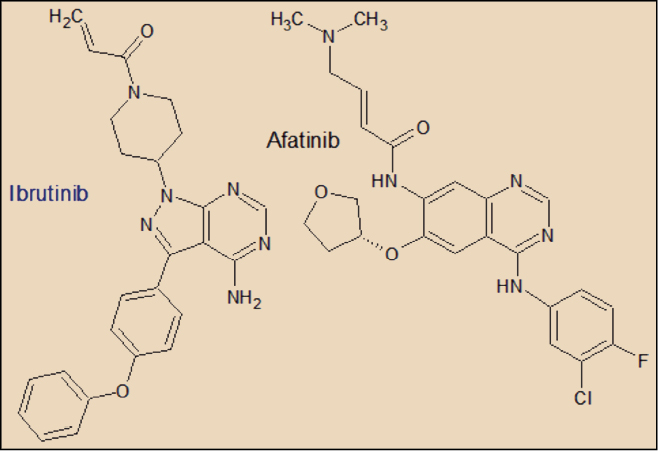
El Ibrutinib está estrechamente relacionado farmacológicamente con otros miembros de la serie de inhibidores de tirosina cinasas. Presenta una evidente relación estructural con el afatanib y el vandetanib.
El ibrutinib se une de forma muy selectiva a un tipo específico de tirosina cinasas, la tirosina cinasa de Bruton (BTK). Se fija específicamente en el hueco enzimático para el ATP, formando un enlace covalente con un resto de cisteína (Cys-481), lo que determina el carácter irreversible del bloqueo.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas del ibrutinib han sido adecuadamente contrastadas en las indicaciones autorizadas mediante ensayos clínicos de fase 2 (información de la eficacia y ampliación de datos de seguridad) y de fase 3 (confirmatorios de eficacia y seguridad).
Linfoma de células del manto
En el estudio de fase 2 (PCYC-1104-CA; EPAR, 2014), abierto, los criterios de inclusión fueron: pacientes con linfoma de células del manto confirmado patológicamente, con sobreexpresión de ciclina D1 o t(11;14), tamaño no inferior a 2 cm y falta de respuesta (al menos parcial) o recaída tras haberla obtenido previamente con al menos un régimen quimioterápico apropiado. La dosis de ibrutinib utilizada fue de 560 mg/24 h por vía oral, hasta la muerte del paciente o aparición de efectos tóxicos inaceptables. La variable primaria de eficacia utilizada en el estudio principal de fase 2 fue la tasa global de respuesta, calculada como la suma de los porcentajes de pacientes con respuesta completa (100%) o parcial (≥50%); las variables secundarias de eficacia fueron la duración de la respuesta, definida como el tiempo medio transcurrido entre la obtención de una respuesta, el tiempo medio hasta obtener respuesta, la supervivencia libre de progresión y la supervivencia global. El estudio fue continuado a largo plazo, con un seguimiento medio de 26,7 meses (Wang, 2015).
En el estudio de fase 3 (Dreyling, 2016), multicéntrico, multinacional, aleatorizado y abierto, los criterios de inclusión fueron: pacientes con linfoma de células del manto confirmado patológicamente, y falta de respuesta o recaída tras haberla obtenido previamente con al menos un régimen inmunoquimioterápico conteniendo rituximab. La variable primaria de eficacia fue la supervivencia libre de progresión.
Leucemia linfocítica crónica
El estudio principal es un ensayo de fase 3 (1112; Byrd, 2014), aleatorizado, multicéntrico y abierto, comparando los efectos del ibrutinib frente a los del ofatumumab (un anticuerpo monoclonal anti-CD20) en pacientes con leucemia linfocítica crónica o linfoma linfocítico pequeño que requiriesen tratamiento y que hubieran recibido al menos uno previamente, no siendo apropiados para un segundo tratamiento con antineoplásicos análogos purínicos.
La dosis de ibrutinib utilizada fue de 420 mg/24 h por vía oral, hasta la muerte del paciente o la aparición de efectos tóxicos inaceptables, mientras que se administraron por vía IV hasta 12 dosis de ofatumumab, a lo largo de seis meses aproximadamente, hasta progresión de la enfermedad, efectos adversos inaceptables o muerte. La variable primaria de eficacia fue la supervivencia libre de progresión, la tasa global de respuesta y la supervivencia global.
Macroglobulinemia de Waldenström
El estudio principal es un ensayo de fase 2 (PCYC-1118E; Treon, 2015), multicéntrico y abierto, con un solo brazo de tratamiento, en pacientes sintomáticos que hubieran recibido al menos tratamiento previamente. La dosis de ibrutinib utilizada fue de 420 mg/24 h por vía oral, hasta la muerte del paciente o aparición de efectos tóxicos inaceptables. La variable primaria de eficacia fue la tasa de respuesta global, incluyendo respuestas completas (RC, resolución de todos los síntomas y normalización de los niveles séricos de inmunoglubina M, IgM), parciales (RP, reducción ≥50% niveles séricos IgM) y menores (RM, reducción ≥25% niveles séricos IgM); como variables secundarias se determinaron la duración de la respuesta, la supervivencia libre de progresión, la supervivencia global y la mejora de los niveles de hemoglobina.
Las características y resultados principales de los ensayos clínicos mencionados anteriormente están recopilados en la tabla 1.
|
Tabla 1. Estudios clínicos de fase 2 y 3 con ibrutinib |
||||
|
Indicación estudiada |
Linfoma de células del manto |
Linfoma de células del manto |
Leucemia linfocítica crónica |
Macroglubulinemia de Waldenström |
|
Tratamiento |
Ibrutinib, 540 mg/24 h (oral) |
Ibrutinib, 560 mg/24 h (oral) Temsirolimús (IV)* |
Ibrutinib, 420 mg/24 h (oral) Ofatumumab (IV)** |
Ibrutinib, 420 mg/24 h (oral) |
|
Tipo de estudio |
Fase 2, abierto |
Fase 3, abierto, aleatorizado |
Fase 3, abierto, aleatorizado |
Fase 2, abierto |
|
Pacientes tratados |
111 |
280 |
391 |
63 |
|
Edad (mediana) |
67,0 años (61% ≥65 años) |
63,0 años (49% ≥65 años) |
||
|
Sexo (Varones) |
68% |
76% |
||
|
Tiempo desde el diagnóstico (mediana) |
42,4 meses (57% ≥36 meses) |
91,3 meses |
73,7 meses |
|
|
Tamaño (% pacientes ≥5 cm) |
39% |
58% |
||
|
Niveles basales de IgM (mediana) |
34,9 g/L |
|||
|
Niveles basales de hemoglobina (mediana) |
10,5 g/L (60% ≤11,0) |
|||
|
Pacientes con afectación de la médula ósea |
60% |
|||
|
Número de regímenes previos (mediana) |
3 |
2 |
2 |
|
|
Pacientes tratados previamente con bortezomib (%) |
43% |
|||
|
Pacientes tratados previamente con quimioinmunoterapia CD-20 |
87% |
|||
|
Citogenética (% pacientes cont(11;14), ciclina D1 o ambos) |
14/69/17% |
|||
|
Citogenética (% pacientes con Del(11q)/Del(17p) |
31/33% |
|||
|
Pacientes con enfermedad avanzada (%) |
72% |
57% |
||
|
Tasa global de respuesta (%; CR/PR/RM) |
67,6% (RC: 20,7; RP: 46,8) |
|
Ibrutinib: 42,6 % (RC: 0; RP: 42,6) Ofatumumab: 4,1% (RC: 0; RP: 4,1) |
90,5% (RC: 0; RP: 73,0; RM: 17,5) |
|
Duración de la respuesta (mediana) |
17,5 meses |
|
|
NE |
|
Supervivencia libre de progresión (mediana) |
13,9 meses |
Ibrutinib: 14,6 meses Temsirolimús: 6,2 meses |
Ibrutinib: NE*** Ofatumumab: 8,1 meses |
NE |
|
Tasa de pacientes libres de progresión (%) |
62,8% (6 meses) 50,6% (12 meses) 41,8% (18 meses) 31,0% (24 meses) |
|
Ibrutinib: 87,8% (6 meses) Ofatumumab: 64,6% (6 meses) Ibrutinib: 65,8% (12 meses) Ofatumumab: 5,9% (12 meses) |
69,1% (24 meses) |
|
Tasa de supervivencia global (%) |
83,5% (6 meses) 64,2% (12 meses) 58,2% (18 meses) 47,0% (24 meses) |
|
Ibrutinib: 94,4% (6 meses) Ofatumumab: 90,2% (6 meses) Ibrutinib; 87,4% (12 meses) Ofatumumab: 81,3% (12 meses) |
95,2% (24 meses) |
|
Variación media de IgM |
|
|
|
-75% |
|
Variación media de hemoglobina |
|
|
|
+31,4% |
|
Pacientes con afectación de la médula ósea (%) |
|
|
|
-58% (de 60 a 25%) |
|
Referencia |
PCYC-1104-CA (EPAR, 2014; Wang, 2015) |
Dreyling, 2016 |
III2 (Byrd, 2014) |
PCYC-1118E (Treon, 2015) |
Nota: Todas las diferencias observadas de las variables con los datos del ibrutinib y, en su caso, los del comparador fueron estadísticamente significativas.
* Infusión IV (30-60’) de 175 mg los días 1, 8 y 15 del primer ciclo de 21 días; luego 75 mg los días 1, 8 y 15 del resto de ciclos.
** La dosis autorizada en la Unión Europea es de 300 mg para la primera perfusión y de 2.000 mg para todas las perfusiones posteriores. El esquema de perfusión es de 8 semanas consecutivas, y transcurrido un periodo de interrupción de 4-5 semanas, dicho esquema pasará a ser de 4 meses consecutivos (por ej. una perfusión cada 4 semanas)
*** No estimado.
Desde el punto de vista de la seguridad, el ibrutinib presenta un perfil toxicológico importante, con frecuentes e relevantes efectos adversos. Aunque la incidencia de los mismos varía según los pacientes (las indicaciones terapéuticas), su espectro es similar. Son frecuentes (>15%): diarrea (50-44%), náuseas (15-30%), estreñimiento (20-25%), vómitos (15-25%), dolor abdominal (15%), fatiga (40-50%), edema periférico (25-30%), astenia (20-30%), infecciones del tracto respiratorio superior (25-30%), anorexia (15-25%), hiperuricemia (15-20%), espasmos musculares (15-20%), mialgia (15%), cefalea (15-20%), disnea (15-25%), tos (20-25%), erupciones exantemáticas (15%) y petequias (15%).
Los efectos graves (grado ≥3) más comunes descritos son neumonía (5-12%), neutropenia (15%), trombocitopenia (10-15%), anemia (2-10%), diarrea (3-5%), dolor abdominal (2-5%). La suspensión del tratamiento como consecuencia de un evento adverso se registró en el 4-11%.
En los estudios directamente comparativos, el ibrutinib ha mostrado una mejor tolerabilidad que el temsirolimús (tasa de eventos adversos del 68 vs. 87%; tasa de descontinuación del tratamiento del 6 vs. 26%). En la comparación con ofatumumab, la tasa global de eventos adversos de grado ≥3 fue superior con ibrutinib (51 vs. 39%), con mayores frecuencias de diarrea (4,1 vs. 1,6%), neutropenia (16,4 vs. 13,6%), neumonía (6,7 vs. 4,7%) y trombocitopenia (5,6 vs. 4,2%), aunque en el caso de la anemia fue inferior (4,6 vs. 7,9%).
ASPECTOS INNOVADORES
El ibrutinib es un agente antineoplásico que actúa inhibiendo de forma irreversible y selectiva a la tirosina cinasa de Bruton (BTK), un miembro de las familia de las tirosina cinasas Tec que participa en la señalización bioquímica del receptor de antígenos (BCR) y del receptor de citocinas de los linfocitos B, implicados en la patogenia de diversas neoplasias de linfocitos B. Dicha inhibición impide la adhesión y migración dependientes de integrinas de los linfocitos B.
Ha sido autorizado, como medicamento huérfano, para el tratamiento de pacientes adultos con linfoma de células del manto en recaída o refractario; asimismo, sea autorizado para el tratamiento de pacientes adultos con leucemia linfática crónica que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP53 en pacientes en los que la inmuno-quimioterapia no se considera apropiada. Finalmente, también ha sido autorizado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström que han recibido al menos un tratamiento previo, o en tratamiento de primera línea en pacientes en los que la inmuno-quimioterapia no se considera apropiada.
La eficacia y la seguridad clínicas del ibrutinib en estas indicaciones han sido contrastadas en varios ensayos clínicos abiertos, tanto de fase 2 como 3, todos ellos de diseño abierto y, en algunos casos sin grupo control (brazo único). En el linfoma de células del manto, las tasas de respuesta objetiva han alcanzado el 67,6%, con tasas de supervivencia libre de progresión a los dos años del 31% y de supervivencia global en ese mismo periodo del 47%. Un estudio directamente comparativo con temsirolimús mostró una clara superioridad, estadísticamente significativa, en la mediana de la supervivencia libre de progresión de 8,4 meses (14,6 vs. 6,2).
Por su parte, en pacientes con leucemia linfocítica crónica se han alcanzado tasas de supervivencia libre de progresión del 88% a los 6 meses y del 66% al año, que contrastan con los respectivos valores obtenidos con ofatumumab (65 y 6%, respectivamente). Esta superioridad viene robustecida por la notable diferencia de las tasas de respuesta objetiva (43 vs. 4%). Finalmente, en la macroglubulinemia de Waldström, el único estudio actualmente disponible muestra también tasas muy elevadas de respuesta objetiva (91%), con un porcentaje del 69% de pacientes libres de progresión a los dos años y una supervivencia global del 95%; todo ello acompañado de notables mejorías en los principales marcadores biológicos de la enfermedad: niveles séricos de IgM (-75%) y hemoglobina (+31%).
El ibrutinib presenta un perfil toxicológico notable, con frecuentes e relevantes efectos adversos. Los más importantes, son neumonía, neutropenia, trombocitopenia, anemia, diarrea y dolor abdominal (2-5%), lo que lleva a la suspensión del tratamiento en el 4-11%. Un 20% de los pacientes experimentan efectos cardiacos (5-10% fibrilación auricular), que se manifiesta fundamentalmente por una significativa actividad sobre el electrocardiograma, acortando el intervalo QTc y alargando el PR, lo que junto con su efecto bradicardizante aconseja un seguimiento de seguridad de esta línea toxicológica.m Asimismo, es importante tener en cuenta que es ibrutinib es metabolizado en el hígado mayoritariamente a través del citocromo CYP3A4, una diana muy común de muchos medicamentos que son susceptibles de inhibirlo, provocando con ello una reducción del aclaramiento hepático del ibrutinib.
Debe valorarse los resultados clínicos del ibrutinib en la macroglobulinemia de Waldström, que multiplican por cuatro o cinco los datos disponibles de supervivencia libre de progresión en registros históricos (EPAR, 2014). Asimismo, son particularmente favorables los datos obtenidos, que muestran respuestas sustanciales incluso en pacientes con mal pronóstico (con mutaciones del17p/TP53) y apoyan su uso en primera línea en pacientes que no puedan ser tratados con inmunoquimioterapia y para los cuales actualmente no se dispone de ninguna opción satisfactoria de tratamiento. Otro tanto cabe decir de los resultados obtenidos en el linfoma de células del manto. En todos los casos, es resaltable las elevadas tasas de respuesta que, aunque de carácter parcial, presentan una notable durabilidad. Igualmente, es destacable que los resultados favorables se distribuyen homogéneamente entre los diversos grupos considerados, por edad, citogenética, tratamientos previos, etc. (Tucker, 2015).
En resumen, el ibrutinib viene a asumir un papel relevante en el tratamiento de un conjunto de linfomas que, aunque de muy baja prevalencia (al fin y al cabo, estamos ante un medicamento huérfano), actualmente no tienen un buen tratamiento especialmente en aquellos casos refractarios o recidivantes tras quimio e inmunoterapia. Desde el punto de vista farmacológico, nos encontramos no ante el enésimo inhibidor de tirosina cinasas, sino ante el primer inhibidor selectivo de un tipo muy especial que está particularmente implicado en la génesis de estos linfomas, la tirosina cinasa de Bruton (BTK); lo cual, unido a una cómoda administración oral única al día, nos lleva a considerar al ibrutinib como un medicamento portador de una innovación importante.
|
VALORACIÓN |
|
|
IBRUTINIB
|
|
|
Grupo Terapéutico (ATC): L01XE. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Otros: inhibidores directos de la proteína cinasa. |
|
|
Indicaciones autorizadas: tratamiento de pacientes adultos con linfoma de células del manto en recaída o refractario. También está indicado para el tratamiento de pacientes adultos con leucemia linfática crónica que han recibido al menos un tratamiento previo, o en primera línea en presencia de deleción del 17p o mutación de TP53 en pacientes en los que la inmuno-quimioterapia no se considera apropiada. Asimismo, también está indicado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström que han recibido al menos un tratamiento previo, o en tratamiento de primera línea en pacientes en los que la inmuno-quimioterapia no se considera apropiada. |
|
|
INNOVACIÓN importante. Aportación sustancial a la terapéutica estándar |
|
|
Novedad clínica: Mejora la eficacia clínica con relación al tratamiento estándar. |
|
|
Novedad molecular: Mecanismo de acción parcialmente diferente, con selectividad hacia dianas específicas implicadas específicamente en la patogenia de las enfermedades para las que se autoriza el medicamento. |
|
|
Novedad toxicológica: Mejora el perfil toxicológico con relación a la terapia estándar. |
|
|
Novedad físico-química: Mejora las características farmacocinéticas, con incidencia en las condiciones de uso. |
|

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Imatinib |
Glivec |
Novartis |
2002 |
|
Erlotinib |
Tarceva |
Roche |
2006 |
|
Sunitinib |
Sutent |
Pfizer |
2007 |
|
Dasatinib |
Sprycel |
Bristol Myers Squibb |
2007 |
|
Sorafenib |
Nexavar |
Bayer |
2007 |
|
Lapatinib |
Tyverb |
Glaxo |
2008 |
|
Nilotinib |
Tasigna |
Novartis |
2008 |
|
Gefitinib |
Iressa |
AstraZeneca |
2010 |
|
Pazopanib |
Votrient |
Glaxo |
2011 |
|
Crizotinib |
Xalkori |
Pfizer |
2014 |
|
Vemurafenib |
Zelboraf |
Roche |
2014 |
|
Dabrafenib |
Tafinlar |
GlaxoSmithKline |
2014 |
|
Axitinib |
Inlyta |
Pfizer |
2014 |
|
Afatinib |
Giotrif |
Boehringer Ingelheim |
2014 |
|
Ruxolitinib |
Jakavi |
Novartis |
2015 |
|
Vandetanib |
Caprelsa |
AstraZeneca |
2015 |
|
Regorafenib |
Stivarga |
Bayer |
2015 |
|
Nintedanib |
Ofev |
Boehringer Ingelheim |
2015 |
|
Ibrutinib |
Imbruvica |
Janssen Cilag |
2016 |
BIBLIOGRAFÍA
Bibliografía
- Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al; RESONATE Investigators.Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014; 371(3): 213-23. doi: 10.1056/NEJMoa1400376.
- Consejo General de Colegios Oficiales de Farmacéuticos.Bot PLUS WEB.https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Pixantrona (Pixuvri®) en linfoma no Hodgkin agresivo y recidivante. Panorama Actual Med 2015; 39(383): 402-6.
- Cuéllar Rodríguez S. Obinutuzumab (Gavyzaro®) en leucemia linfocítica crónica. Panorama Actual Med 2015; 39(389): 1002-7.
- Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al.Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016; 387(10020): 770-8. doi: 10.1016/S0140-6736(15)00667-4.
- European Medicines Agency (EMA). Cyranza®.European Public Assessment Report (EPAR). EMA/596202/2014; EMEA/H/C/002829.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002829/WC500180726.pdf
- Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, at al.Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015; 372(15): 1430-40. doi: 10.1056/NEJMoa1501548.
- Tucker DL, Rule SA.A critical appraisal of ibrutinib in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. Ther Clin Risk Manag. 2015; 11:979-90. doi: 10.2147/TCRM. S73559.
- Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, et al.Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood. 2015; 126(6): 739-45. doi: 10.1182/blood-2015-03-635326.
Artículos relacionados
-
5 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
29 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
6 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
